

Abstract
The Telomeric Repeat Amplification Protocol (TRAP) and its modified versions (including ours, TP-TRAP) change the size and/or the ratio of the telomerase products in the amplification stage of the assay. Based on our recently published method we developed a new TRAP. This method ensures that the number of telomeric repeats present in the original telomerase products does not change on PCR amplification. The usefulness of the method was proved with amplification of chemically synthesized telomerase products and a newly designed telomerase substrate oligonucleotide. This is the first report in which the PCR products directly reflect the size distribution of telomerase products generated by the enzyme.
INTRODUCTION
Telomerase is a ribonucleoprotein that elongates the telomeres in eukaryotic cells. It catalyzes the synthesis of telomeric repeats (5′-GGTTAG-3′ in vertebrates), using its RNA component as template (1,2). A number of recent studies have indicated that telomerase expression is associated with cell immortalization and tumorigenesis (3,4). Telomerase is a potentially useful tumor marker and is a novel target for chemotherapy (5). A PCR-based assay, the Telomeric Repeat Amplification Protocol (TRAP), has been developed to measure telomerase activity in a limited number of cells (3). The original protocol does not allow one to estimate the processivity of the enzyme because the reverse primer (CX) can bind anywhere along the telomeric repeats during the amplification process. Therefore, PCR products may be generated which are shorter or longer than the original telomerase products due to staggered annealing of the primers. Addition of a tag sequence at the 5′-end of the reverse primer prevents PCR-mediated elongation of the telomerase products (6–8), but shorter products are still generated, at least in the very first cycles of amplification. Thus the final PCR products do not directly represent the distribution of the original telomerase products. We have recently developed a new PCR-based protocol for amplification of telomeric repeats that is suitable for the quantitation of telomerase activity and also hampers elongation of the primer telomerase products on amplification (9). In this method we used two reverse primers and a highly specific PCR protocol.
We describe here an amplification protocol which does not change the distribution of the starting telomerase products during amplification. Thus it is suitable for studying the mechanism of telomerase-catalyzed reactions and the effects of various compounds or oligonucleotide substrates on processivity of the enzyme.
MATERIALS AND METHODS
Chemicals and oligonucleotides
Chemicals were purchased from Sigma Aldrich and Amersham Pharmacia and were analytical grade or better. The following oligonucleotides were synthesized by standard phosphoramidite chemistry on a Pharmacia Gene Assembler Plus oligonucleotide synthesizer and purified by polyacrylamide gel electrophoresis in our laboratory: CX, 5′-(CCCTTA)3CCCTAA-3′; TS, 5′-AATCCGTCGAGCAGAGTT-3′; MTS, 5′-AGCATCCGTCGAGCAGAGTT-3′; GTS, 5′-GGTGGGGCGAGCAGAGTT-3′; MTSR6, 5′-AGCATCCGTCGAGCAGAGTTAG(GGTTAG)6-3′; RP, 5′-TAGAGCACAGCCTGTCCGTG-3′; RPC3, 5′-TAGAGCACAGCCTGTCCGTG(CTAACC)3-3′; RPC3g, 5′-TAGAGCACAGCCTGTCCGTG(CTAACC)3GG-3′. The following oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA): MTSR1, 5′-AGCATCCGTCGAGCAGAGTTAGGGTTAG-3′; MTSR2, 5′-AGCATCCGTCGAGCAGAGTTAG(GGTTAG)2-3′; MTSR3, 5′-AGCATCCGTCGAGCAGAGTTAG(GGTTAG)3-3′; MTSR9, 5′-AGCATCCGTCGAGCAGAGTTAG(GGTTAG)9-3′.
Preparation of cell extracts and partial purification of human telomerase
Partially purified human telomerase (HL-60 DEAE) was prepared from HL-60 cells as described previously (9). Crude cell extracts from HeLa, HL-60 and endothelial cells were prepared by the CHAPS lysis method (3). The human umbilical vein endothelial cells were kindly provided by Dr György Balla and Dr József Balla. Protein concentrations were determined by Bio-Rad protein assay (Bio-Rad Laboratories).
Detection of telomerase activity
The telomerase-mediated extension reaction and subsequent PCR amplification were conducted using our previously described protocol (9) with minor modifications. In brief, the TRAP reaction mixture (48 µl) contained 20 mM Tris–HCl pH 8.3, 1.5 mM MgCl2, 63 mM KCl, 1 mM EGTA, 0.1 mg/ml BSA, 0.005% Tween 20, 50 µM dATP, dGTP and dTTP, 20 µM dCTP, 5 µCi [α-32P]dCTP (3000 Ci/mmol; Isotope Institute, Hungary), 25 pmol MTS primer and telomerase (cell extract or partially purified enzyme). After 30 min incubation at 30°C for telomerase-mediated primer extension, the mixtures were heated to 90°C for 3 min to inactivate telomerase activity. During this treatment 2 µl of reverse mixture [3 U Taq polymerase (Sigma) and 25 pmol RP and 0.5 pmol RPC3g primers] was added for hot-start PCR and then the samples were subjected to 29 cycles of PCR amplification under the following conditions: the first and second PCR cycles consisted of 94°C for 30 s, 55°C for 60 s and 72°C for 90 s; the subsequent cycles consisted of 94°C for 30 s, 63°C for 30 s and 72°C for 30 s. Half of the PCR products were electrophoresed on 8% non-denaturing polyacrylamide gels. The gels were then dried and autoradiography was performed for 24 or 48 h.
For the heat pretreatment control 5 µl of extract was heated at 95°C for 5 min. When the TS/CX primers were used the PCR was performed under the following conditions: 94°C for 30 s, 50°C for 30 s and 72°C for 90 s for 27 or 29 cycles (3).
RESULTS AND DISCUSSION
The most important features of our recently published TP-TRAP method are the use of two reverse primers instead of one and a highly specific PCR protocol (9). In this report the same reverse primer set was used except that the RPC3 primer was extended with a non-complementary dinucleotide (GG) at its 3′-end, designated RPC3g. When RPC3g anneals to the 3′-end of a telomerase product its 5′-end (20 nt) is copied, yielding a 20 nt long non-telomeric overhang on the product of telomerase. In this step of amplification the telomerase product primes DNA synthesis on the 5′-end of RPC3g. In subsequent cycles the overhang serves as a primer binding site for the reverse primer (RP), present in large excess. If RPC3g anneals at any other sites on the telomeric repeats, both 3′-ends (telomerase primer products and RPC3g) are mismatched, thus Taq DNA polymerase cannot elongate them. The mechanism of amplification is outlined in Figure 1.
Figure 1.

The outline of PCR amplification of a telomerase product with MTS/RPC3g/RP oligonucleotides. This amplification protocol does not change the length of the telomerase product, except that of the overhang (20 nt). The arrows indicate the directions of primer elongation. The key steps of the process are binding of oligonucleotide RPC3g to the telomerase product (step 2) and elongation of the telomerase product by Taq polymerase on the 5′-end of RPC3g (step 3).
To prove this assumption a 58mer oligonucleotide, an artificial telomerase product (MTSR6), was amplified. When RPC3 and RP primers were used as reverse primers, four bands appeared, as expected. When RPC3g was used instead of RPC3, only one band was generated, in agreement with the proposed mechanism (Fig. 2A–C). Using the new primer set the efficiency of PCR amplification was lower because in the initial stage of PCR the extra GG prevents elongation of one of the strands and because of possible competition between the primers (RPC3g and RP). Therefore, the number of PCR cycles was increased from 27 to 29 in subsequent experiments.
Figure 2.
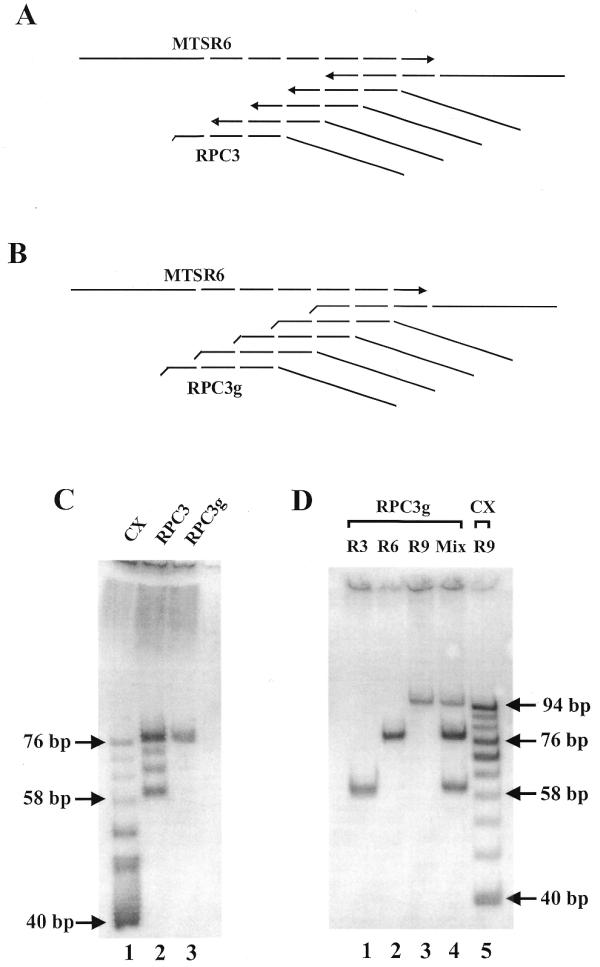
Amplification of synthetic telomerase products. (A) Possible annealing sites of RPC3 on MTSR6. (B) Possible annealing sites of RPC3g on MTSR6. DNA synthesis directions are indicated with arrows. (C) Amplification of 0.2 amol MTSR6 with different primer combinations (27 PCR cycles). Lane 1, reverse primer CX (25 pmol), forward primer TS (25 pmol); lane 2, reverse primers RPC3 (0.5 pmol) and RP (25 pmol), forward primer MTS (25 pmol); lane 3, reverse primers RPC3g (0.5 pmol) and RP (25 pmol), forward primer MTS (25 pmol). The generated PCR products start at 40 (TS/CX primers) or 60 bp (MTS/RPC3/RP primers) and display 6 bp periodicity. (D) Amplification of 0.1 amol artificial telomerase products, containing three, six and nine complete telomeric repeats, alone and in combination with the new primer set, MTS/RPC3g/RP (29 PCR cycles): lane 1, MTSR3; lane 2, MTSR6; lane 3, MTSR9; lane 4, a mixture of MTSR3, MTSR6 and MTSR9 at 0.1 amol each. As a molecular size control 0.1 amol MTSR9 was amplified with 25 pmol TS and 25 pmol CX primers (lane 5).
In the next experiment three chemically synthesized telomerase products (MTSR3, MTSR6 and MTSR9) were amplified, individually and in a mixture, with RPC3g and RP reverse primers. This set of oligonucleotides represents a wide range of possible telomerase products. MTSR9 was also amplified with the TS and CX primers (3), in order to generate a size marker ladder. Gel electrophoretic analysis of the products is shown in Figure 2D. When the artificial telomerase products were amplified separately, only single bands were detected. The sizes of the products are in agreement with the proposed amplification mechanism (60, 78 and 96 nt). When an equimolar mixture of the three oligonucleotides was amplified, three bands were formed with the same electrophoretic mobility as the single ones. This result suggests that the presence of oligonucleotides (MTSR3, MTSR6 and MTSR9) with different chain lengths in the same reaction mixture does not change the mechanism of amplification. Therefore, it is presumed that amplification of the original telomerase products (a mixture of oligonucleotides with various chain lengths) will follow the above proposed mechanism. The only difference among the original telomerase products is their sizes (the number of telomeric repeats attached). Presumably, amplification under the same conditions (i.e. in the same reaction mixture) does not change the quantitative ratio of the primer products. When a high number of PCR cycles are used the various telomerase products may compete for primers in the later stages of amplification. This competition may modify the quantitative ratio of the final products.
Gel electrophoretic analysis of the telomerase products is presented in Figure 3A. MTSR3 and MTSR9 were amplified and used as controls, besides heat-inactivated enzymes. The partially purified enzyme and the crude cell extracts (from HL-60 and HeLa) produced the expected DNA ladder with 6 nt increments. The shortest product in each case was 6 nt shorter than the MTSR3 band. This observation was further examined by amplification of 0.1 amol MTSR1, MTSR2 and MTSR3, containing one, two or three complete telomeric repeats (Fig. 3B). No amplification product was formed with MTSR1 under our assay conditions. In this case only 11 nt of RPC3g are fully complementary to MTSR1 (Fig. 3C). At the 3′-end of MTSR2 and MTSR3 17 and 18 nt are complementary to RPC3g, respectively (Fig. 3C). In both cases amplification products were obtained. The MTSR2 and longer telomerase products formed a stable double helix with RPC3g under our assay conditions, supporting amplification with the MTS/RPC3g/RP primer set. The results are in agreement with the observation presented in Figure 3A. Possible elongation of MTSR3 is also shown (Fig. 3C), forming a 12 nt long double helix between MTSR3 and RPC3g (MTSR3+RPCg*). Such a complex is not formed under our assay conditions, including a hot-start, as proved by the results presented in Figures 2D and 3A. These experiments clearly prove that only those telomerase products which are elongated with two complete telomeric repeats, i.e. with 14 nt (2+12), could be amplified with the MTS/RPC3g/RP primer set.
Figure 3.
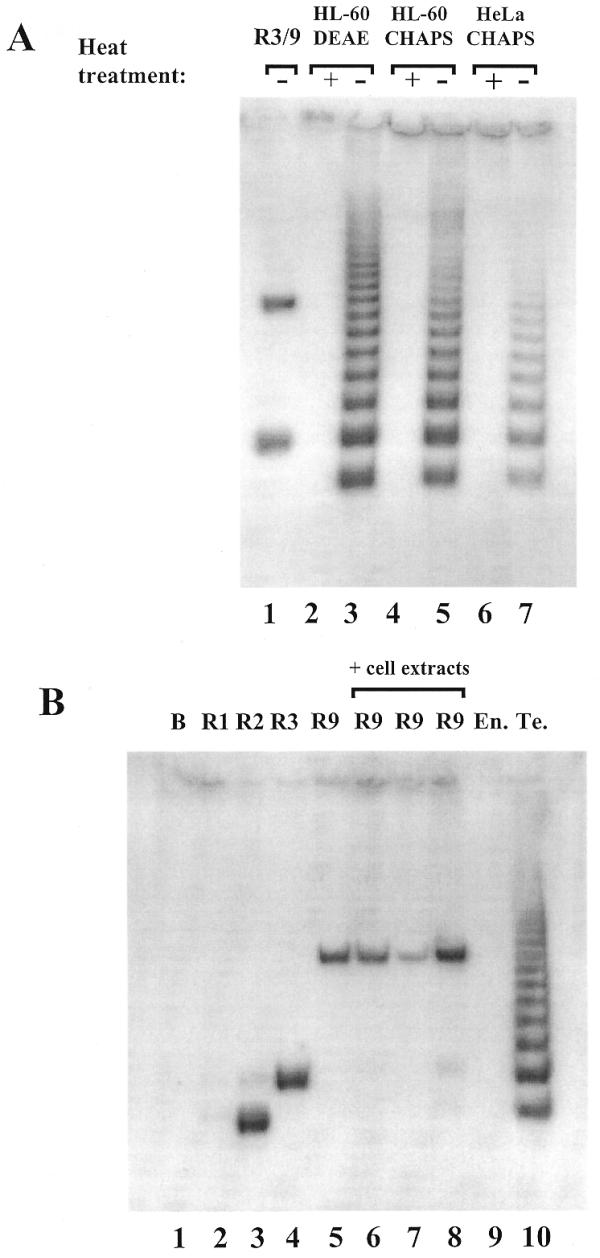

(A) Detection of telomerase activity with the new primer combination, MTS/RPC3g/RP. Partially purified enzyme (HL-60 DEAE) containing 100 ng protein (lanes 2 and 3) and crude extracts from HL-60 (lanes 4 and 5) and HeLa (lanes 6 and 7) cells containing 1000 ng protein were used as telomerase enzymes. Heat-inactivated samples acted as controls (lanes 2, 4 and 6). As a positive control, 0.1 amol MTSR3 and 0.1 amol MTSR9 were amplified (lane 1). (B) Amplification of MTSR1, MTSR2, MTSR3 and MTSR9 in the presence of different cell extracts. Lane 1, no template was added (blank); lanes 2–4, 0.1 amol MTSR1, MTSR2 and MTSR3, respectively, were amplified; lane 5, amplification of 0.1 amol MTSR9. Before PCR amplification 0.1 amol MTSR9 was incubated with 100 ng heat-inactivated telomerase enzyme (HL-60 DEAE) (lane 6), 1000 ng crude cell extract (HL-60 CHAPS) (lane 7) or 2000 ng extracts from primer endothelial cells (lane 8). As control reactions 2000 ng endothel cell extract (lane 9) and 100 ng partially purified telomerase enzyme (HL-60 DEAE) (lane 10) were used without MTSR9. (C) Annealing RPC3g with artificial telomerase products. The structures shown in the first two schemes (MTSR3+RPC3g and MTSR2+RPC3g) can support amplification with the MTS/RPC3g/RP primer set. The structures shown in the third (MTSR1+RPC3g) and fourth (MTSR3+RPC3g*) schemes are not stable under the assay conditions, producing no amplified products.
MTSR9 was amplified in the presence of heat-inactivated partially purified human telomerase, heat-inactivated HL-60 cell extract and cell extract prepared from human umbilical vein endothelial cell cultures. The endothelial cell extract did not show any telomerase activity in this assay (Fig. 3B). The cell extracts did not have any significant effects on amplification of MTSR9, although some slight differences in the intensity of the bands could be detected (Fig. 3B). These control experiments were further evidence for the specificity of the amplification method introduced in this paper.
It should be noted that we obtained 10–15 repeats after 30 min incubation at 30°C using the new amplification protocol. In the conventional (without amplification) telomerase assay human telomerase can synthesize 65–70 repeats under optimal conditions (2). The differences in the number of telomeric repeats synthesized in vitro depends on a number of parameters, including the sequence of the oligonucleotide substrate and the concentration of dNTPs. Most PCR-based assays use an oligonucleotide substrate that is non-telomeric, to ensure a specific primer binding site for subsequent PCR amplification and to avoid primer dimer formation. Conventional assays may use an oligonucleotide with telomeric repeats. Morin has shown that the use of primers with telomeric repeats or primers with a G-rich upstream region yielded longer telomerase products than TS-like non-telomeric substrates (10). To study the effectiveness of our new protocol we have synthesized an 18mer telomerase substrate primer (GTS) which contains a G-rich region at its 5′-end. A longer version of the GTS primer was published earlier as an αα thalassemia break point sequence (10). The products generated by initiation of the two telomerase substrate oligonucleotides (MTS and GTS) at various temperatures were compared by our new protocol (Fig. 4). Indeed, longer telomerase products were obtained with the GTS than the MTS primer, as expected. We could not demonstrate decreased processivity of the enzyme at 37°C, as shown by others (11). However, the catalytic activity of human telomerase increased with increasing temperature. Both the longer telomerase products obtained with the GTS primer and the increased catalytic activity at higher temperature suggest that our new amplification protocol can be successfully utilized in studies of the human telomerase enzyme.
Figure 4.
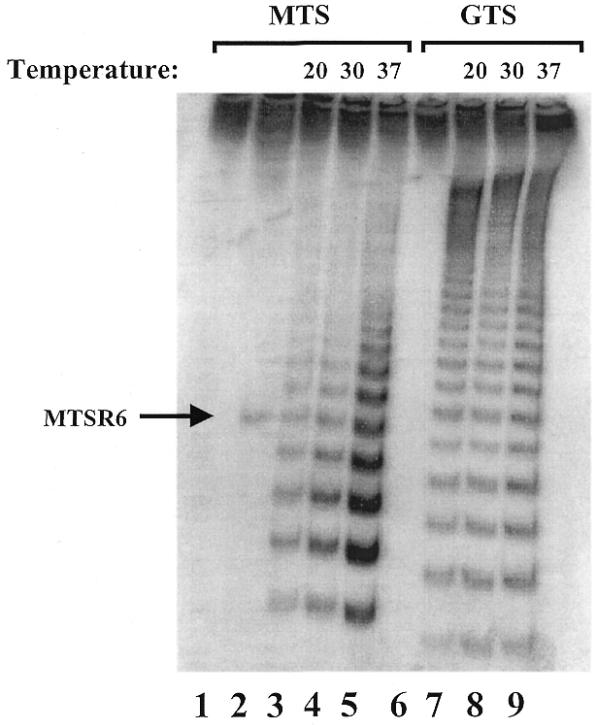
Determination of the processivity of telomerase with two different oligonucleotide substrates. Telomerase-mediated primer extension was conducted for 30 min at the indicated temperature, using two different telomerase substrate primers: 25 pmol MTS primer (lanes 1–5) and 25 pmol GTS primer (lanes 6–9). As a negative control no telomerase enzyme was added (lanes 1 and 6); as a positive control 0.2 amol MTSR6 was used (lane 2).
In summary, we have introduced a new TRAP which first attaches a tag sequence to the original telomerase products then amplifies them, using the tag sequence as a reverse primer binding site. This is the only method for high fidelity PCR amplification of telomerase products developed so far.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Lisa A. Bryce (University of Glasgow) and Dr Joan E. MacDiarmid (SUNY at Buffalo) for proofreading the manuscript. This work was supported by research grant OTKA TO 20128 and in part by FKFP 1043/97 (Hungary).
References
- 1.Greider C.W. and Blackburn,E.H. (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell, 43, 405–413. [DOI] [PubMed] [Google Scholar]
- 2.Morin G.B. (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell, 49, 521–529. [DOI] [PubMed] [Google Scholar]
- 3.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L.C., Covielo,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 4.Holt S.E. and Shay,J.W. (1999) Role of telomerase in cellular proliferation and cancer. J. Cell Physiol., 180, 10–18. [DOI] [PubMed] [Google Scholar]
- 5.McKenzie K.E., Umbricht,C.B. and Sukumar,S. (1999) Applications of telomerase research in the fight against cancer. Mol. Med. Today, 5, 114–122. [DOI] [PubMed] [Google Scholar]
- 6.Krupp G., Kühne,K., Tamm,S., Klapper,W., Heidorn,K., Rott,A. and Parwaresch,R. (1997) Molecular basis of artifacts in the detection of telomerase activity and a modified primer for a more robust ‘TRAP’ assay. Nucleic Acids Res., 25, 919–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim N.W. and Wu,F. (1997) Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res., 25, 2595–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tatematsu K., Nakayama,J., Danbara,M., Shionoya,S., Sato,H., Omine,M. and Ishikawa,F. (1996) A novel quantitative ‘stretch PCR assay’, that detects a dramatic increase in telomerase activity during the progression of myeloid leukemias. Oncogene, 13, 2265–2274. [PubMed] [Google Scholar]
- 9.Szatmari I., Tökés,S., Dunn,C.B., Bardos,T.J. and Aradi,J. (2000) Modified telomeric repeat amplification protocol: a quantitative radioactive assay for telomerase without using electrophoresis. Anal. Biochem., 282, 80–88. [DOI] [PubMed] [Google Scholar]
- 10.Morin G.B. (1991) Recognition of a chromosome truncation site associated with alpha-thalassaemia by human telomerase. Nature, 353, 454–456. [DOI] [PubMed] [Google Scholar]
- 11.Sun D., Hurley,H.L. and Hoff,D.D. (1998) Telomerase assay using biotinylated-primer extension and magnetic separation of the products. Biotechniques, 25, 1046–1051. [DOI] [PubMed] [Google Scholar]