


Abstract
Sephadex-binding RNA ligands (aptamers) were obtained through in vitro selection. They could be classified into two groups based on their consensus sequences and the aptamers from both groups showed strong binding to Sephadex G-100. One of the highest affinity aptamers, D8, was chosen for further characterization. Aptamer D8 bound to dextran B512, the soluble base material of Sephadex, but not to isomaltose, isomaltotriose and isomaltotetraose, suggesting that its optimal binding site might consist of more than four glucose residues linked via α-1,6 linkages. The aptamer was very specific to the Sephadex matrix and did not bind appreciably to other supporting matrices, such as Sepharose, Sephacryl, cellulose or pustulan. Using Sephadex G-100, the aptamer could be purified from a complex mixture of cellular RNA, giving an enrichment of at least 60 000-fold, compared with a non-specific control RNA. These RNA aptamers can be used as affinity tags for RNAs or RNA subunits of ribonucleoproteins to allow rapid purification from complex mixtures of RNA using only Sephadex.
INTRODUCTION
Affinity tagging of proteins with peptides or proteins such as the FLAG epitope (1), glutathione S-transferase (2), polyhistidine (3,4) or protein A (5) has been used widely for protein research. The fusion proteins containing the affinity tags provide rapid and convenient means of detection, immobilization and purification of the proteins of interest (6). Therefore, it could be of interest to develop affinity tags that can be used with RNA. Such RNA tags might have some advantages over protein tags, especially when RNAs or ribonucleoproteins (RNP) are the main target molecule under study.
In this study RNA affinity ligands to Sephadex were developed using in vitro selection or SELEX (systematic evolution of ligands by exponential enrichment) (7,8). This technique has proven very powerful in discovering RNA and DNA ligands (aptamers) that have affinity for the desired target molecules. RNA aptamers have been developed to recognize a broad spectrum of target molecules ranging from small molecules to more complex macromolecules (reviewed in 9–12). Sephadex, a commonly used matrix in gel filtration, was chosen as the target in this selection because: (i) it is widely used in many laboratories and readily available; (ii) it is relatively inexpensive, making large-scale purification more affordable; (iii) it is very stable and can be regenerated to be used several times; (iv) its structure consists mainly of repeating units of glucose linked via α-1,6 glucosidic bonds, giving it numerous aptamer binding sites and a high binding capacity. RNA aptamers that specifically bind to Sephadex were successfully developed and we show that one selected RNA can be very highly purified from a complex mixture of cellular RNA using Sephadex beads. This study shows the feasibility and potential of using the aptamer as a tag for RNA affinity purification.
MATERIALS AND METHODS
Materials
DNA oligonucleotides were synthesized by the University of Michigan DNA core facility. Sephadex G-100, Sepharose CL-4B and Sephacryl S-500 HR were from Amersham Pharmacia Biotech. Dextran B512 (average mol. wt 10 000), isomaltose, isomaltotriose and isomaltotetraose were from Sigma. Cellulose was from W&R Balston. Pustulan was from Calbiochem. Taq DNA polymerase, Sequenase and T7 RNA polymerase were prepared as described (13–15; D.E.Draper, personal communication).
In vitro selection
Single-stranded deoxyoligonucleotides (0.2 µmol) containing a randomized 40 nt central sequence flanked by defined primer-binding sites were synthesized with the sequence 5′-AGTAATACGACTCACTATAGGGAGTCGACCGACCAGAA(N40)TA-TGTGCGTCTACATCTAGACTCAT-3′. A primer extension reaction of the deoxyoligonucleotides with 0.2 µmol complementary 3′ primer (5′-ATGAGTCTAGATGTAGACGCACATA-3′) was done to generate a double-stranded DNA pool. The reaction was done at 60°C for 4 h, using 200 U Taq DNA polymerase in PCR buffer (10 mM Tris pH 8.3, 50 mM KCl, 1.5 mM MgCl2 and 0.2 mM each dNTP). Approximately 60% of the single-stranded DNA could be converted to double-stranded DNA. The DNA, which contains a T7 RNA polymerase promoter at the 5′-end of the sequence, was then used as a template to generate an RNA library with T7 RNA polymerase. The transcription reaction was done at 37°C for 1 h, in 40 mM Tris pH 8.0, 20 mM MgCl2, 25 mM NaCl, 1 mM spermidine, 20 mM DTT and 4 mM each NTP. The RNA was isolated from a 10% polyacrylamide gel in 8 M urea. The gel slices were soaked in a buffer containing 0.1 M sodium acetate, 1 mM EDTA and 0.2% SDS at 37°C overnight. The RNA was then extracted with phenol/chloroform and precipitated with ethanol. The resulting RNA sequences were 84 nt long [5′-GGGAGUCGACCGACCAGAA(N40)UAUGUGCGUCUACAUCUAGACUCAU-3′] and the calculated complexity of the RNA library was ∼7 × 1016 different sequences.
The RNA library was used to obtain RNA aptamers specific to Sephadex G-100 and selection was done for a total of 11 rounds. In each round of selection RNA (1–10 nmol or 2–10 µM) was incubated with 0.25–1.0 ml of Sephadex beads in Ultrafree-MC centrifugal filter units (Millipore). The binding step was carried out at room temperature for 1 h in binding buffer (50 mM HEPES pH 7.4, 10 mM MgCl2 and 100 mM NaCl). The beads were washed three times with 250 µl of binding buffer for 1 min each in the first six rounds; the washing step was increased in the final round to 12 times for 30 min each with 500 µl of binding buffer in order to increase the stringency. The bound RNA was eluted from Sephadex with elution buffer (8 M urea, 5 mM EDTA) and precipitated with ethanol. The RNA was reverse transcribed with Superscript II (Life Technologies) as described by the manufacturer and the cDNA was amplified with Taq DNA polymerase using 5′ primer (5′-AGTAATACGACTCACTATAGGGAGTCGACCGACCAGAA-3′) and the 3′ primer listed above for 5–10 cycles (95°C for 45 s, 60°C for 1 min, 72°C for 2 min). PCR was then used to generate RNA by in vitro transcription with T7 RNA polymerase. The resulting RNA was isolated from a 10% polyacrylamide gel in 8 M urea and used for subsequent rounds of selection.
The PCR products from the final round were cloned into vector pGEM-T (Promega). Each individual aptamer was generated from plasmid clones by PCR amplification with the 5′ and 3′ primers, followed by in vitro transcription with T7 RNA polymerase. The affinity of each aptamer for Sephadex was tested by Sephadex binding assay (described below). The positive clones were sequenced by dideoxy termination methods and their sequences analyzed.
Binding assays
A Sephadex binding assay was used to determine the affinity of aptamers for Sephadex. Radiolabeled aptamers (2.5–5.0 × 104 c.p.m. or 0.5–1.0 pmol) were incubated with 25 µl of Sephadex G-100 at room temperature for 1 h in the presence of 1 µM tRNA as a carrier to reduce non-specific interaction. The beads were washed three times for 1 min each with 250 µl of binding buffer. The bound RNA was later eluted with elution buffer and its radioactivity counted with a scintillation counter. The percentage of RNA bound to Sephadex over total RNA was determined.
The ability of various sugars (isomaltose, isomaltotriose, isomaltotetraose and dextran B512) to compete with Sephadex G-100 for binding to aptamer D8 was tested using competitive binding assays. Radiolabeled aptamer D8 (2 × 104 c.p.m.) was incubated with 1 µl of Sephadex G-100 beads in the presence of various sugar concentrations (0.1–20 mM) at room temperature for 1 h. The beads were washed and eluted and the percentage of bound RNA determined.
The binding ability of aptamer D8 to various supporting matrices, i.e. Sephadex G-100, Sepharose CL-4B, Sephacryl S-500 HR, cellulose and pustulan, was studied. Radiolabeled aptamer D8 (5 × 104 c.p.m.) was incubated with 100 µl of 12.5–50% suspensions of the above matrices. The reactions were conducted in binding buffer with 1 µM tRNA and incubated at room temperature for 1 h. The matrices were washed and eluted and the percentage of bound RNA determined. A non-specific RNA was also tested as a negative control. The control RNA was derived from pGEM-T containing the 5′ HIV LTR fragment (–310 to +98). The plasmid was cut with SpeI and transcribed with T7 RNA polymerase to generate a 484 nt RNA transcript. The RNA was then tested for binding to the supporting matrices in binding buffer containing 1 µM tRNA as described above.
Affinity purification of RNA aptamers from a complex mixture of RNA
RNA extracts from human cultured cells (HeLa) were prepared using Trizol reagent (Life Technologies) as described by the manufacturer. A sample containing a complex mixture of RNA was generated by combining 10 µg HeLa cellular RNA with radiolabeled aptamer D8 RNA and the control RNA (5 × 104 c.p.m. or ∼30 ng each). Batch purification of the sample was done by incubating the sample with 200 µl of Sephadex G-100 in binding buffer at room temperature for 1 h. The beads were washed three times with 500 µl of binding buffer for 1 min each and then eluted with elution buffer. Samples from the input, unbound, wash and eluate fractions were analyzed as follows: (i) total cellular RNA was monitored by electrophoresis through a 2% agarose gel and staining with ethidium bromide; (ii) aptamer D8 and the control RNA were analyzed on a 10% denaturing polyacrylamide gel in 8 M urea. The gel autoradiograph was visualized using a PhosphorImager system (Molecular Dynamics) and the bands quantitated using IPLab Gel analysis software (Signal Analytics).
RESULTS
In vitro selection and characterization of Sephadex-binding aptamers
RNA aptamers against Sephadex G-100 were selected from an RNA library with a complexity of ∼7 × 1016 different sequences. After 11 rounds of selection PCR fragments from the final round were cloned into plasmids. Fifteen individual aptamer clones were chosen and tested for their binding to Sephadex by Sephadex binding assay. All of them bound efficiently to Sephadex (percent RNA bound to Sephadex ranging from 23 to 60%), compared with a control RNA derived from the original library (1.5%). Sequence analysis of these aptamers showed that they could be classified into two groups based on the consensus sequences (Fig. 1). Their structures were analyzed using an RNA secondary structure prediction program (16). The results did not show any noticeable consensus structure, except that the consensus sequences tended to be in single-stranded regions (data not shown). On average, the aptamers from both groups bound to Sephadex with similar affinity. However, only D8, one of the highest affinity aptamers, was selected for further characterization.
Figure 1.

Sequences of Sephadex-binding aptamers. Only the sequences corresponding to the randomized region in the starting library are shown. The aptamers were classified as group 1 or group 2 based on their consensus sequences (underlined). All of them were unique, except aptamer D8, which appeared twice during screening.
Competitive binding assays using oligosaccharides structurally related to Sephadex
Sephadex is a matrix produced by cross-linking dextran B512 with epichlorohydrin to form beads (17,18). By varying the degree of cross-linking, the beads will have different pore sizes, which make them very useful in size exclusion chromatography. Dextran B512, the base material of Sephadex, contains glucose residues mainly linked together via α-1,6 linkages (95%) and has branches which are linked via α-1,3 glucosidic bonds (5%) (17). The oligosaccharides isomaltose (IM2), isomaltotriose (IM3) and isomaltotetraose (IM4) consist of two, three and four glucose residues linked via α-1,6 glucosidic linkages, respectively (see Discussion), and are structurally similar to Sephadex and dextran B512. Therefore, they were chosen to be tested for their ability to compete with Sephadex for binding to aptamer D8 using competitive binding assays, compared with that of dextran B512. As shown in Figure 2, only dextran B512 can strongly inhibit the binding of aptamer D8 to Sephadex. In contrast, IM2, IM3 and IM4 do not show any significant inhibition, even at concentrations as high as 20 mM. To rule out the possibility that the aptamer might recognize Sephadex at the branch points, which have α-1,3 linkages, dextran B-1355 was tested as a competitor in competitive binding assays. Dextran B-1355 has a branched structure containing a repeating unit of alternating α-1,3 and α-1,6 glucosidic linkages, with up to 35% of α-1,3-linked glucosyl residues (18). However, dextran B-1355 did not show any inhibition even at a concentration as high as 20 mg/ml, whereas at the same concentration 90% of aptamer binding was inhibited when dextran B512 was used as competitor (data not shown). The results suggest that the optimal binding site of aptamer D8 might consist of more than four glucose residues linked via α-1,6 linkages.
Figure 2.

Competitive binding assays showing the ability of various sugars (isomaltose, isomaltotriose, isomaltotetraose and dextran B512) to compete with Sephadex G-100 for binding to aptamer D8. The aptamer was incubated with Sephadex G-100 in the presence of various sugar concentrations ranging from 0.1 to 20 mM. The ratios of the percentage of aptamer binding to Sephadex in the presence of various competitor concentrations over binding in the absence of competitors are shown in the graph.
Binding affinity of aptamer D8 to various supporting matrices
Binding of aptamer D8 and the control RNA to the Sephadex matrix, compared with other supporting matrices, i.e. Sepharose CL-4B, Sephacryl S-500 HR, cellulose and pustulan, were studied. These matrices are mainly carbohydrates or their derivatives. Sepharose CL is agarose (alternating sequences of 1,3-linked β-d-galactopyranose and 1,4-linked α-3,6-anhydro-l-galactopyranose) cross-linked with 2,3-dibromopropanol (19). Sephacryl HR is a composite gel prepared by covalently cross-linking allyl dextran with N,N′-methylene bisacrylamide (19). Cellulose and pustulan (20) are homopolysaccharides consisting of d-glucopyranose linked via β-1,4 and β-1,6 linkages, respectively. As shown in Figure 3, the binding of aptamer D8 is very specific to Sephadex, whereas its binding to other supporting matrices is poor and comparable to that of the control RNA.
Figure 3.

The binding of aptamer D8 to various supporting matrices, compared with that of the control RNA. Radiolabeled RNA of either aptamer D8 or the control RNA was incubated with several matrices, then washed and eluted and the percentage of bound RNA was determined.
Affinity purification of RNA aptamers using Sephadex G-100
To determine the practicality of using aptamer D8 as an affinity purification tool we tested whether the aptamer could be selectively isolated from a complex mixture of RNA using Sephadex. The starting sample was prepared by combining cellular RNA from human tissue culture cells (HeLa) with a trace amount of radiolabeled aptamer D8 and control RNA, which constituted ∼0.6% of the total RNA. Batch purification of the sample with Sephadex G-100 was done and samples from the input, unbound, wash and eluate fractions were analyzed as described in Materials and Methods. In the starting sample radiolabeled D8 and control RNA were present in approximately equal amounts (Fig. 4B). After incubation of the sample with Sephadex G-100 beads the control RNA did not seem to bind to Sephadex, compared with aptamer D8, and most of it eluted in the unbound fraction, reducing the aptamer:control RNA ratio from 1:1 in the starting material to 0.3:1 in the unbound fraction. However, after the beads were washed the control RNA was essentially undetectable in the eluate, whereas ∼30% of aptamer D8 was recovered. Therefore, the ratio aptamer D8:control RNA increased dramatically in the eluate, an estimated purification of at least 60 000-fold. In addition, the eluate contained no detectable cellular RNA, as judged by agarose gel electrophoresis followed by ethidium bromide staining (Fig. 4A), suggesting that affinity purification with Sephadex appeared to have very low background binding. The results from this experiment indicate that the aptamer could be selectively and efficiently isolated from a complex mixture of RNA using Sephadex G-100.
Figure 4.
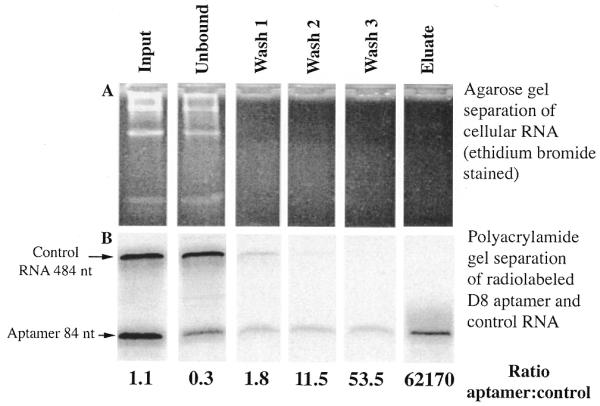
Purification of aptamer D8 from a complex mixture of human cellular RNA using Sephadex G-100. Radiolabeled aptamer D8 RNA and the control RNA (5 × 104 c.p.m. or 30 ng each) were added to 10 µg unlabeled cellular RNA. The sample was incubated with 200 µl of Sephadex beads in 500 µl total volume, washed and eluted as detailed in Materials and Methods. (A) Samples from each fraction, i.e. input, unbound, wash and eluate, were analyzed for total cellular RNA by electrophoresis on a native 2% agarose gel and stained with ethidium bromide. (B) The radiolabeled RNA in the same fractions was analyzed by electrophoresis in a 10% denaturing polyacrylamide gel and visualized with a PhosphorImager system. The ratios of aptamer D8 to the control RNA were calculated to show the enrichment of purification and are shown below each lane.
DISCUSSION
The goal of this study was to develop RNA aptamers that can be used in affinity purification of RNAs or RNPs. Such aptamers or their binding motifs could be inserted into the RNA of interest and later purified using the corresponding aptamer-binding target, which in this study is Sephadex G-100. Aptamer D8 was shown to bind to Sephadex as well as to dextran B512 by competitive binding assay. This cross-binding suggests that the structural integrity of dextran in Sephadex beads does not change dramatically on modification by cross-linking with epichlorohydrin. However, IM2, IM3 and IM4, as well as dextran B-1355, were unable to compete for aptamer D8 in the competitive binding assay, suggesting that the optimal binding site of the aptamer is likely to be longer than four glucose residues. The structures of these various polymers are represented in Figure 5.
Figure 5.

Sephadex structural components. The drawing shows the structure of dextran B512, a homopolysaccharide consisting of d-glucopyranose mainly linked via α-1,6 linkages (95%) and, to a lesser extent, via α-1,3 linkages (5%) at the branch points. Sephadex is prepared by cross-linking dextran B512 with epichlorohydrin. Isomaltose, isomaltotriose and isomaltotetraose are oligosaccharides consisting of two, three and four residues of d-glucopyranose linked via α-1,6 linkages, respectively.
A previous study by Yang et al. (21), which used cellulose as a target for DNA aptamer selection, showed that a subset of these aptamers could bind to cellulose with a binding site as short as two glucose residues linked via β-1,4 glucosidic linkages, because binding to cellulose could be efficiently and specifically competed by a disaccharide, cellobiose [4-O-(β-d-glucopyranosyl)-β-d-glucopyranose]. These findings suggest that the specificity of aptamers for carbohydrate targets, such as dextran or cellulose, results from their ability to recognize differences in linkage configurations (α or β), position of the linkages and the number of monomeric residues in the targets. However, it is not yet known why the dextran RNA aptamer requires a long stretch of glucose residues for specific binding. One explanation is that interactions of the aptamer with many glucose residues might help it bind to dextran more stably. Similar findings have also been observed in the study of anti-dextran antibodies, in which case the antibodies need at least three to seven contiguous glucose residues in dextran for optimal binding (22,23). Alternatively, it is possible that the dextran aptamer recognizes specific determinants in a three-dimensional structure that requires multiple glucosyl residues to form. Another explanation might be the difference in the elution schemes. In the cellulose aptamer selection cellobiose was used in the elution step, whereas an elution buffer containing 8 M urea and 5 mM EDTA, which can efficiently elute bound RNA from the target, was used in this selection. Thus, aptamers that bind to a fewer number of glucose residues were likely to be selected in the former case, whereas aptamers that might bind more tightly and require a longer stretch of glucose would be more favored in the latter case. This finding may be helpful as a guide for future aptamer selections against polysaccharides. If aptamers capable of binding to structurally related oligosaccharides with a specific number of residues were desired, using these oligosaccharides to elute the bound ligands from polysaccharide targets might enable us to obtain such aptamers. However, from the perspective of developing aptamers as an affinity tool we feel that the specificity of the aptamer for a long stretch of dextran might be advantageous, because structurally related mono- or oligosaccharides that might be present in complex cellular mixtures will not interfere with aptamer binding to Sephadex.
The aptamer was also shown to be very specific for the Sephadex matrix; it did not bind to other matrices such as Sepharose, Sephacryl, cellulose and pustulan. Supporting matrices such as Sepharose, Sephacryl and cellulose have been widely used in many purification applications. Because additional purification steps involving different matrices are sometimes required in order to obtain completely purified target molecules, specific binding of an aptamer only to Sephadex would make it less likely that aptamer-tagged RNAs will be trapped and lost in purification steps using different supporting matrices.
To test the practicality of using the aptamer for affinity purification, isolation of the aptamer from a relatively large excess of total cellular RNA was carried out with Sephadex G-100. The aptamer could be selectively purified from a complex mixture of RNA with an enrichment of at least 60 000-fold over the control RNA. Therefore, using the aptamer binding motif as an affinity tag on RNA or an RNA subunit of a RNP particle could allow rapid purification, even on a large scale. Although a denaturing elution buffer containing urea and EDTA was used in this study, it is also possible to specifically elute the aptamer under non-denaturing conditions by extended incubation with soluble dextran B512 (data not shown). This could help preserve the integrity of the target RNA or RNP. This is particularly important if the purified molecules are needed for further functional and structural characterizations.
In summary, anti-Sephadex aptamers provide the means for rapid and powerful purification using the readily available Sephadex matrix as an affinity resin. This could find utility in a variety of applications.
Acknowledgments
ACKNOWLEDGEMENTS
C.S. was supported by a scholarship from the Royal Thai Government and a pre-doctoral fellowship from Horace H. Rackham School of Graduate Studies, University of Michigan. The work was supported by NIH grant GM 29470 to I.J.G. and NIH grant AI 40936 to D.R.E.
References
- 1.Prickett K.S., Amberg,D.C. and Hopp,T.P. (1989) A calcium-dependent antibody for identification and purification of recombinant proteins. Biotechniques, 7, 580–589. [PubMed] [Google Scholar]
- 2.Simons P.C. and Vander Jagt,D.L. (1981) Purification of glutathione S-transferases by glutathione-affinity chromatography. Methods Enzymol., 77, 235–237. [DOI] [PubMed] [Google Scholar]
- 3.Porath J. (1992) Immobilized metal ion affinity chromatography. Protein Expr. Purif., 3, 263–281. [DOI] [PubMed] [Google Scholar]
- 4.Porath J., Carlsson,J., Olsson,I. and Belfrage,G. (1975) Metal chelate affinity chromatography, a new approach to protein fractionation. Nature, 258, 598–599. [DOI] [PubMed] [Google Scholar]
- 5.Stahl S. and Nygren,P.A. (1997) The use of gene fusions to protein A and protein G in immunology and biotechnology. Pathol. Biol., 45, 66–76. [PubMed] [Google Scholar]
- 6.Nilsson J., Stahl,S., Lundeberg,J., Uhlen,M. and Nygren,P.A. (1997) Affinity fusion strategies for detection, purification and immobilization of recombinant proteins. Protein Expr. Purif., 11, 1–16. [DOI] [PubMed] [Google Scholar]
- 7.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 8.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 9.Jayasena S.D. (1999) Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin. Chem., 45, 1628–1650. [PubMed] [Google Scholar]
- 10.Wilson D.S. and Szostak,J.W. (1999) In vitro selection of functional nucleic acids. Annu. Rev. Biochem., 68, 611–647. [DOI] [PubMed] [Google Scholar]
- 11.Hermann T. and Patel,D.J. (2000) Adaptive recognition by nucleic acid aptamers. Science, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- 12.Patel D.J., Suri,A.K., Jiang,F., Jiang,L., Fan,P., Kumar,R.A. and Nonin,S. (1997) Structure, recognition and adaptive binding in RNA aptamer complexes. J. Mol. Biol., 272, 645–664. [DOI] [PubMed] [Google Scholar]
- 13.Engelke D.R., Krikos,A., Bruck,M.E. and Ginsburg,D. (1990) Purification of Thermus aquaticus DNA polymerase expressed in Escherichia coli. Anal. Biochem., 191, 396–400. [DOI] [PubMed] [Google Scholar]
- 14.Tabor S. and Richardson,C.C. (1989) Selective inactivation of the exonuclease activity of bacteriophage T7 DNA polymerase by in vitro mutagenesis. J. Biol. Chem., 264, 6447–6458. [PubMed] [Google Scholar]
- 15.Laing L. (1993) Doctoral dissertation, Johns Hopkins University, Baltimore, MD.
- 16.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 17.Robyt J.F. (1998) Essentials of Carbohydrate Chemistry. Springer, New York, NY.
- 18.Misaki A., Torii,M., Sawai,T. and Goldstein,I.J. (1980) Structure of the dextran of Leuconostoc mesenteroides B-1355. Carbohydr. Res., 84, 273–285. [Google Scholar]
- 19. Pharmacia LKB Biotechnology (1991) Gel Filtration: Principles and Methods, 6th Edn. Pharmacia, Uppsala, Sweden.
- 20.Hellerqvist C.G., Lindberg,B. and Samuelsson,K. (1968) Methylation analysis of pustulan. Acta Chem. Scand., 22, 2736–2737. [Google Scholar]
- 21.Yang Q., Goldstein,I.J., Mei,H.Y. and Engelke,D.R. (1998) DNA ligands that bind tightly and selectively to cellobiose. Proc. Natl Acad. Sci. USA, 95, 5462–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda T. and Kabat,E.A. (1989) Variable region cDNA sequences and antigen binding specificity of mouse monoclonal antibodies to isomaltosyl oligosaccharides coupled to proteins. T-dependent analogues of alpha(1→6)dextran. J. Immunol ., 142, 863–870. [PubMed] [Google Scholar]
- 23.Nashed E.M., Perdomo,G.R., Padlan,E.A., Kovac,P., Matsuda,T., Kabat,E.A. and Glaudemans,C.P. (1990) Binding characteristics of IgA 16.4.12E, a monoclonal antibody with specificity for the nonreducing terminal epitope of alpha-(1→6)-dextrans. Comparisons between IgA hybridoma 16.4.12E and myeloma W3129. J. Biol. Chem., 265, 20699–20707. [PubMed] [Google Scholar]