

Abstract
The title compound, C12H15N5OS, was obtained by reaction of 2-(2-(methylthio)pyrimidin-4-yl)-3-oxopropanenitrile with (tetrahydrofuran-3-yl)hydrazine dihydrochloride, and the racemic product was subsequently separated by chiral chromatography (first peak; [α]D 20 = +51.3°). The chiral center at the substituted atom of the tetrahydrofuranyl group has an R-configuration. The pyrimidine and pyrazolyl rings are almost coplanar, their mean planes forming a dihedral angle of 6.4 (1)°. One of the H atoms of the amino group participates in an intramolecular hydrogen bond with the pyrimidine N atom in position 3. The second H atom is involved in an intermolecular hydrogen bond, which links the molecules into an infinite chain.
Related literature
For the structure of a related compound with a methyl-substituted amino group, see: Liu et al. (2009 ▶).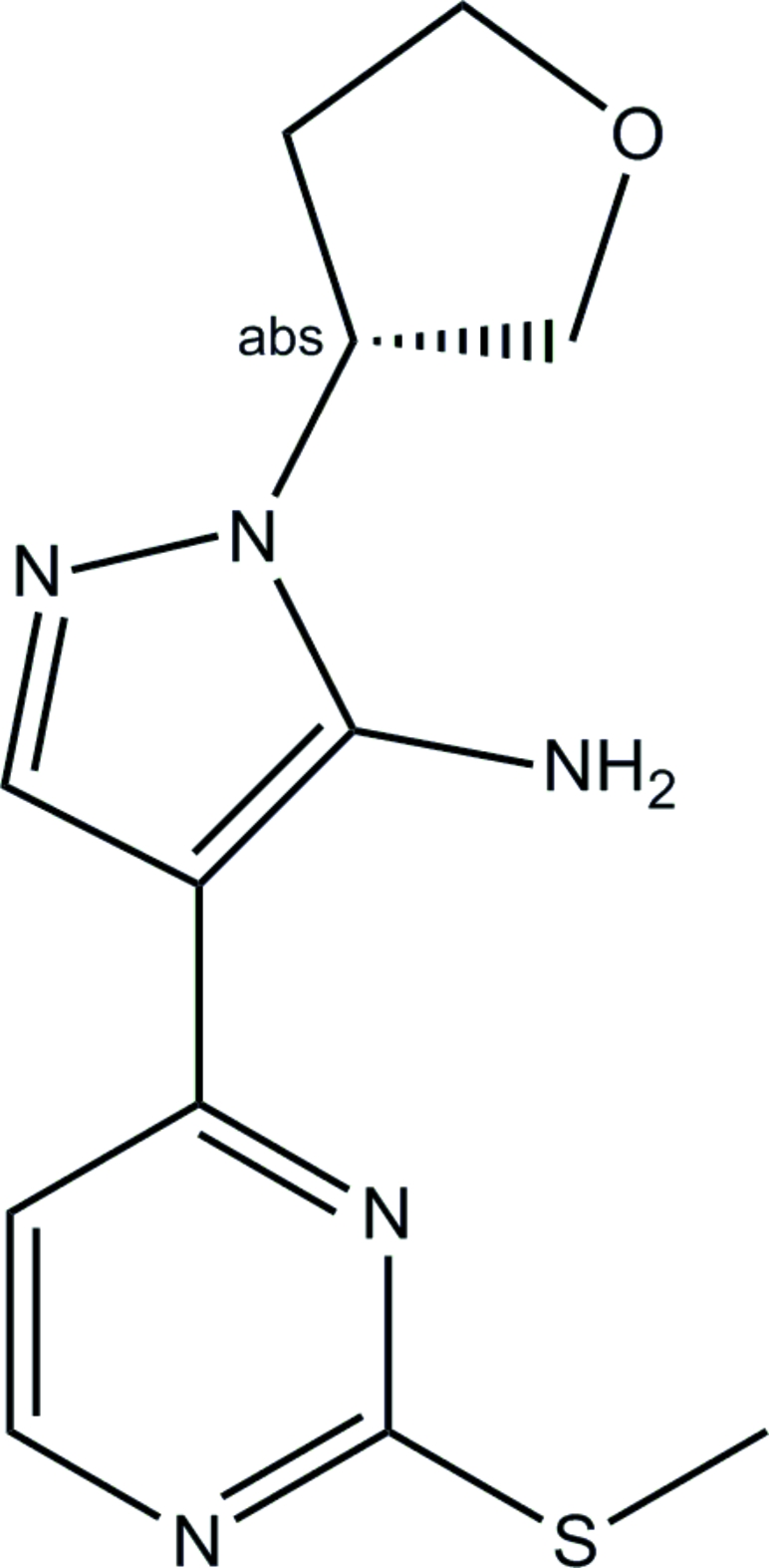
Experimental
Crystal data
C12H15N5OS
M r = 277.35
Orthorhombic,
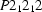
a = 15.479 (2) Å
b = 7.1217 (10) Å
c = 11.7802 (17) Å
V = 1298.6 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.25 mm−1
T = 208 K
0.20 × 0.20 × 0.20 mm
Data collection
Bruker D8 APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2001 ▶) T min = 0.844, T max = 0.952
6570 measured reflections
3025 independent reflections
2844 reflections with I > 2σ(I)
R int = 0.043
Refinement
R[F 2 > 2σ(F 2)] = 0.041
wR(F 2) = 0.115
S = 1.07
3025 reflections
174 parameters
H-atom parameters constrained
Δρmax = 0.24 e Å−3
Δρmin = −0.35 e Å−3
Absolute structure: Flack (1983 ▶), 1180 Friedel pairs
Flack parameter: −0.05 (8)
Data collection: APEX2 (Bruker, 2004 ▶); cell refinement: SAINT (Bruker, 2004 ▶); data reduction: SAINT; program(s) used to solve structure: SIR2004 (Burla et al., 2005 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-32 (Farrugia, 1997 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053680900734X/tk2382sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680900734X/tk2382Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N3—H3A⋯N4i | 0.87 | 2.19 | 2.9731 (19) | 150 |
| N3—H3B⋯N5 | 0.87 | 2.28 | 2.8616 (19) | 124 |
Symmetry code: (i) 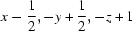 .
.
supplementary crystallographic information
Comment
The title compound was obtained by reaction of 2-(2-(methylthio)pyrimidin-4-yl)-3-oxopropanenitrile with (tetrahydrofuran-3-yl)hydrazine dihydrochloride. The racemic product was then separated with the help of chiral chromatography; the title compound, (I), was collected as the earlier fraction, when eluted with methanol using the Chiralpak column (99% ee; [α]D20 = +51.3°).
The present X-ray study unambiguously established the R configuration of the chiral center at the C3 atom (Fig. 1).
The pyrimidine and pyrazolyl rings lie approximately in one plane; the dihedral angle formed by their mean planes is equal to 6.4 (1)°. The orientation of the tetrahydrofurane ring can be characterized by the dihedral angle 99.6 (1)° formed by the pyrazolyl plane with the C2—C3—C4 plane.
The molecular geometry of (I) is similar to that of related compound with a methyl substituent at the amino group (Liu et al., 2009). However, the crystal packing is substantially different as (I) has one additional H atom capable of H-bond formation. Indeed, while the H3A atom forms an intramolecular H-bond with the N5 atom of the pyrimidine ring similar to that observed in methyl-substituted structure, the H3B atom is involved in intermolecular H-bonding, which links molecules into infinite chains running along the a axis (Fig. 2; Table 2).
Experimental
To a suspension of 2-(2-(methylthio)pyrimidin-4-yl)-3-oxopropanenitrile (13.5 g, 70.0 mmol) in AcOH (100 ml) was added (tetrahydrofuran-3-yl)hydrazine dihydrochloride (12.3 g,70.0 mmol), and the resulting orange mixture was heated at 80°C under nitrogen for 3 h. Acetic acid was removed and the orange solid residue was partitioned between aqueous Na2CO3 (200 ml) and EtOAc (400 ml). The mixture was refluxed for 30 min. The separated organic layer was washed with brine, dried over sodium sulfate and concentrated to give the crude product as a brown gum (16.82 g, 87%). The brown gum (8.32 g) was purified by flash chromatography using 30–70% EtOAc in hexane to afford a yellow solid (5.96 g).
The part of the product thus obtained (4.85 g) was subjected to chiral chromatography on Chiralpak AS—H 21.2 x 250 mm column with 35% MeOH in CO2 at 140 bar as eluent (flow = 55 ml/min; UV detection at 260 nm). Two fractions corresponding to each of the enantiomers (Peak1 and Peak2) were collected and evaporated to dryness; the specific rotation [α]D20 was measured in CH2Cl2 solution and yielded the values of +51.3° and -52.1°, respectively. The enantiomer collected as Peak 1 was recrystallized from EtOAc/hexane to yield colorless single crystals.
Refinement
All H atoms were placed in geometrically calculated positions (C—H 0.94 Å, 0.97 Å, 0.98 Å, and 0.99 Å for aromatic-, methyl-, methylene- and methine-H atoms, respectively; N—H 0.87 Å) and included in the refinement in the riding model approximation. The Uiso(H) values were set to 1.2Ueq of the carrying atom except for 1.5Ueq for methyl-H atoms.
Figures
Fig. 1.

Molecular structure of (I), showing 50% probability displacement ellipsoids and atom numbering scheme. H atoms are drawn as circles with arbitrary small radius.
Fig. 2.

Crystal packing for (I) viewed down the b axis; H-bonds are shown as dashed lines.
Crystal data
| C12H15N5OS | F(000) = 584 |
| Mr = 277.35 | Dx = 1.419 Mg m−3 |
| Orthorhombic, P21212 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2 2ab | Cell parameters from 5004 reflections |
| a = 15.479 (2) Å | θ = 2.6–28.1° |
| b = 7.1217 (10) Å | µ = 0.25 mm−1 |
| c = 11.7802 (17) Å | T = 208 K |
| V = 1298.6 (3) Å3 | Block, yellow |
| Z = 4 | 0.20 × 0.20 × 0.20 mm |
Data collection
| Bruker D8 APEXII CCD area-detector diffractometer | 3025 independent reflections |
| Radiation source: fine-focus sealed tube | 2844 reflections with I > 2σ(I) |
| graphite | Rint = 0.043 |
| phi and ω scans | θmax = 28.1°, θmin = 1.7° |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | h = −20→19 |
| Tmin = 0.844, Tmax = 0.952 | k = −4→9 |
| 6570 measured reflections | l = −15→13 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.041 | w = 1/[σ2(Fo2) + (0.0703P)2 + 0.0815P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.115 | (Δ/σ)max = 0.001 |
| S = 1.07 | Δρmax = 0.24 e Å−3 |
| 3025 reflections | Δρmin = −0.35 e Å−3 |
| 174 parameters | Extinction correction: SHELXL (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.058 (5) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), 1180 Friedel pairs |
| Secondary atom site location: difference Fourier map | Flack parameter: −0.05 (8) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.61092 (15) | 0.3862 (4) | 1.04598 (18) | 0.0592 (6) | |
| H1A | 0.6660 | 0.3550 | 1.0825 | 0.071* | |
| H1B | 0.5758 | 0.4600 | 1.0989 | 0.071* | |
| C2 | 0.62567 (13) | 0.4931 (3) | 0.93597 (14) | 0.0471 (4) | |
| H2A | 0.6760 | 0.5761 | 0.9418 | 0.057* | |
| H2B | 0.5748 | 0.5677 | 0.9154 | 0.057* | |
| C3 | 0.64143 (10) | 0.3354 (3) | 0.85008 (15) | 0.0414 (4) | |
| H3 | 0.6054 | 0.3562 | 0.7818 | 0.050* | |
| C4 | 0.61086 (12) | 0.1571 (3) | 0.91240 (18) | 0.0514 (5) | |
| H4A | 0.5721 | 0.0835 | 0.8639 | 0.062* | |
| H4B | 0.6603 | 0.0785 | 0.9334 | 0.062* | |
| C5 | 0.86860 (11) | 0.3275 (3) | 0.84766 (15) | 0.0456 (4) | |
| H5 | 0.9228 | 0.3341 | 0.8835 | 0.055* | |
| C6 | 0.85782 (9) | 0.3006 (3) | 0.72974 (14) | 0.0355 (4) | |
| C7 | 0.76781 (9) | 0.3006 (2) | 0.71461 (13) | 0.0341 (3) | |
| C8 | 0.92276 (9) | 0.2767 (2) | 0.64376 (13) | 0.0341 (3) | |
| C9 | 1.01122 (10) | 0.2623 (3) | 0.66996 (14) | 0.0427 (4) | |
| H9 | 1.0308 | 0.2671 | 0.7454 | 0.051* | |
| C10 | 1.06733 (10) | 0.2412 (3) | 0.58192 (16) | 0.0466 (4) | |
| H10 | 1.1264 | 0.2289 | 0.5987 | 0.056* | |
| C11 | 0.95801 (10) | 0.2531 (3) | 0.45560 (14) | 0.0371 (3) | |
| C12 | 0.81445 (13) | 0.2818 (3) | 0.31648 (16) | 0.0526 (5) | |
| H12A | 0.7888 | 0.1785 | 0.3583 | 0.079* | |
| H12B | 0.7917 | 0.2833 | 0.2398 | 0.079* | |
| H12C | 0.8006 | 0.3993 | 0.3539 | 0.079* | |
| N1 | 0.73257 (8) | 0.3241 (2) | 0.81741 (12) | 0.0393 (3) | |
| N2 | 0.79452 (9) | 0.3423 (3) | 0.90197 (12) | 0.0488 (4) | |
| N3 | 0.72268 (9) | 0.2815 (3) | 0.61696 (12) | 0.0493 (4) | |
| H3A | 0.6665 | 0.2841 | 0.6182 | 0.059* | |
| H3B | 0.7498 | 0.2667 | 0.5529 | 0.059* | |
| N4 | 1.04322 (9) | 0.2367 (3) | 0.47231 (13) | 0.0453 (4) | |
| N5 | 0.89635 (8) | 0.2704 (2) | 0.53396 (11) | 0.0341 (3) | |
| O1 | 0.56646 (9) | 0.2206 (3) | 1.01145 (14) | 0.0630 (5) | |
| S1 | 0.92940 (3) | 0.25258 (9) | 0.31179 (4) | 0.05191 (18) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0580 (12) | 0.0835 (15) | 0.0362 (10) | 0.0152 (11) | 0.0142 (8) | 0.0072 (10) |
| C2 | 0.0412 (9) | 0.0661 (12) | 0.0340 (8) | 0.0096 (8) | 0.0063 (7) | 0.0034 (8) |
| C3 | 0.0248 (7) | 0.0669 (11) | 0.0325 (8) | 0.0036 (7) | 0.0002 (6) | 0.0017 (8) |
| C4 | 0.0343 (8) | 0.0675 (12) | 0.0523 (11) | −0.0037 (8) | 0.0026 (7) | 0.0040 (10) |
| C5 | 0.0288 (7) | 0.0790 (13) | 0.0290 (7) | −0.0006 (8) | −0.0025 (6) | −0.0024 (8) |
| C6 | 0.0258 (7) | 0.0512 (9) | 0.0293 (7) | −0.0007 (6) | −0.0009 (5) | 0.0000 (7) |
| C7 | 0.0269 (7) | 0.0473 (8) | 0.0282 (7) | 0.0005 (6) | −0.0020 (5) | −0.0004 (6) |
| C8 | 0.0262 (6) | 0.0443 (8) | 0.0320 (7) | −0.0002 (6) | 0.0010 (5) | 0.0008 (6) |
| C9 | 0.0277 (7) | 0.0655 (11) | 0.0350 (7) | 0.0007 (7) | −0.0029 (6) | 0.0006 (9) |
| C10 | 0.0259 (7) | 0.0684 (11) | 0.0456 (9) | 0.0031 (9) | 0.0023 (6) | −0.0035 (9) |
| C11 | 0.0315 (7) | 0.0471 (8) | 0.0327 (7) | −0.0020 (7) | 0.0027 (5) | −0.0045 (8) |
| C12 | 0.0438 (9) | 0.0754 (13) | 0.0385 (9) | 0.0067 (9) | −0.0061 (7) | −0.0041 (10) |
| N1 | 0.0254 (6) | 0.0642 (9) | 0.0282 (6) | 0.0003 (6) | −0.0011 (5) | −0.0007 (6) |
| N2 | 0.0290 (6) | 0.0884 (12) | 0.0290 (7) | −0.0003 (7) | −0.0043 (5) | −0.0034 (8) |
| N3 | 0.0284 (6) | 0.0917 (13) | 0.0278 (6) | 0.0010 (7) | −0.0043 (5) | −0.0065 (8) |
| N4 | 0.0293 (6) | 0.0657 (9) | 0.0408 (7) | −0.0002 (7) | 0.0045 (5) | −0.0076 (9) |
| N5 | 0.0287 (6) | 0.0435 (7) | 0.0300 (6) | −0.0016 (5) | 0.0020 (4) | −0.0026 (6) |
| O1 | 0.0460 (8) | 0.0826 (11) | 0.0604 (9) | 0.0041 (8) | 0.0233 (6) | 0.0185 (8) |
| S1 | 0.0394 (2) | 0.0852 (4) | 0.0311 (2) | −0.0016 (2) | 0.00439 (15) | −0.0075 (2) |
Geometric parameters (Å, °)
| C1—O1 | 1.425 (3) | C7—N1 | 1.339 (2) |
| C1—C2 | 1.520 (3) | C7—N3 | 1.353 (2) |
| C1—H1A | 0.9800 | C8—N5 | 1.357 (2) |
| C1—H1B | 0.9800 | C8—C9 | 1.407 (2) |
| C2—C3 | 1.531 (3) | C9—C10 | 1.361 (2) |
| C2—H2A | 0.9800 | C9—H9 | 0.9400 |
| C2—H2B | 0.9800 | C10—N4 | 1.344 (2) |
| C3—N1 | 1.4644 (18) | C10—H10 | 0.9400 |
| C3—C4 | 1.541 (3) | C11—N5 | 1.3334 (19) |
| C3—H3 | 0.9900 | C11—N4 | 1.339 (2) |
| C4—O1 | 1.428 (3) | C11—S1 | 1.7510 (17) |
| C4—H4A | 0.9800 | C12—S1 | 1.792 (2) |
| C4—H4B | 0.9800 | C12—H12A | 0.9700 |
| C5—N2 | 1.317 (2) | C12—H12B | 0.9700 |
| C5—C6 | 1.412 (2) | C12—H12C | 0.9700 |
| C5—H5 | 0.9400 | N1—N2 | 1.3888 (18) |
| C6—C7 | 1.4046 (19) | N3—H3A | 0.8700 |
| C6—C8 | 1.437 (2) | N3—H3B | 0.8700 |
| O1—C1—C2 | 104.11 (19) | N1—C7—C6 | 106.83 (13) |
| O1—C1—H1A | 110.9 | N3—C7—C6 | 128.32 (15) |
| C2—C1—H1A | 110.9 | N5—C8—C9 | 119.98 (14) |
| O1—C1—H1B | 110.9 | N5—C8—C6 | 117.70 (13) |
| C2—C1—H1B | 110.9 | C9—C8—C6 | 122.32 (14) |
| H1A—C1—H1B | 109.0 | C10—C9—C8 | 117.50 (15) |
| C1—C2—C3 | 102.70 (18) | C10—C9—H9 | 121.2 |
| C1—C2—H2A | 111.2 | C8—C9—H9 | 121.2 |
| C3—C2—H2A | 111.2 | N4—C10—C9 | 123.87 (14) |
| C1—C2—H2B | 111.2 | N4—C10—H10 | 118.1 |
| C3—C2—H2B | 111.2 | C9—C10—H10 | 118.1 |
| H2A—C2—H2B | 109.1 | N5—C11—N4 | 127.69 (15) |
| N1—C3—C2 | 111.55 (15) | N5—C11—S1 | 119.28 (12) |
| N1—C3—C4 | 112.07 (16) | N4—C11—S1 | 113.03 (12) |
| C2—C3—C4 | 103.93 (14) | S1—C12—H12A | 109.5 |
| N1—C3—H3 | 109.7 | S1—C12—H12B | 109.5 |
| C2—C3—H3 | 109.7 | H12A—C12—H12B | 109.5 |
| C4—C3—H3 | 109.7 | S1—C12—H12C | 109.5 |
| O1—C4—C3 | 106.03 (18) | H12A—C12—H12C | 109.5 |
| O1—C4—H4A | 110.5 | H12B—C12—H12C | 109.5 |
| C3—C4—H4A | 110.5 | C7—N1—N2 | 112.28 (13) |
| O1—C4—H4B | 110.5 | C7—N1—C3 | 129.58 (14) |
| C3—C4—H4B | 110.5 | N2—N1—C3 | 118.13 (13) |
| H4A—C4—H4B | 108.7 | C5—N2—N1 | 104.20 (14) |
| N2—C5—C6 | 112.69 (15) | C7—N3—H3A | 120.0 |
| N2—C5—H5 | 123.7 | C7—N3—H3B | 120.0 |
| C6—C5—H5 | 123.7 | H3A—N3—H3B | 120.0 |
| C7—C6—C5 | 104.00 (14) | C11—N4—C10 | 114.37 (14) |
| C7—C6—C8 | 127.19 (14) | C11—N5—C8 | 116.57 (13) |
| C5—C6—C8 | 128.81 (14) | C1—O1—C4 | 105.25 (15) |
| N1—C7—N3 | 124.85 (14) | C11—S1—C12 | 102.76 (8) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3A···N4i | 0.87 | 2.19 | 2.9731 (19) | 150 |
| N3—H3B···N5 | 0.87 | 2.28 | 2.8616 (19) | 124 |
Symmetry codes: (i) x−1/2, −y+1/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: TK2382).
References
- Bruker (2001). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2004). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., De Caro, L., Giacovazzo, C., Polidori, G. & Spagna, R. (2005). J. Appl. Cryst.38, 381–388.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst.32, 837–838.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Liu, Z., Liu, K. K.-C., Elleraas, J., Rheingold, A. L., DiPasquale, A. & Yanovsky, A. (2009). Acta Cryst. E65, o616. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053680900734X/tk2382sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680900734X/tk2382Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report