

Abstract
In the title compound, C16H16BrN, the benzene rings are inclined to each other with a dihedral angle between their mean planes of 50.5 (3)° and the C=C bond adopts a cis conformation.
Related literature
For background information on photonic materials, see: He et al. (2008 ▶). For related systems of stilbene, see: Moreno-Fuquen et al. (2008 ▶, 2009 ▶). For literature related to the synthesis, see: Maryanoff & Reitz (1989 ▶).
Experimental
Crystal data
C16H16BrN
M r = 302.21
Monoclinic,
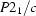
a = 14.804 (2) Å
b = 6.0962 (5) Å
c = 15.2106 (10) Å
β = 95.331 (9)°
V = 1366.8 (2) Å3
Z = 4
Mo Kα radiation
μ = 2.99 mm−1
T = 123 K
0.38 × 0.25 × 0.10 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2002 ▶) T min = 0.457, T max = 0.749
8606 measured reflections
2986 independent reflections
1533 reflections with I > 2σ(I)
R int = 0.064
Refinement
R[F 2 > 2σ(F 2)] = 0.075
wR(F 2) = 0.213
S = 0.99
2986 reflections
165 parameters
H-atom parameters constrained
Δρmax = 0.94 e Å−3
Δρmin = −0.54 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 1997 ▶); software used to prepare material for publication: PARST95 (Nardelli, 1995 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536809020492/hg2518sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809020492/hg2518Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
RMF is grateful to the Spanish Research Council (CSIC) for the use of a free-of-charge licence to the Cambridge Structural Database (Allen, 2002 ▶). RMF also thanks the Universidad del Valle, Colombia, for partial financial support.
supplementary crystallographic information
Comment
The present work is part of a structural study of molecular complexes based on the matrix of stilbene which can be used as non-linear optical material (He et al., 2008). Our research group has developed the study of other related systems of stilbene (Moreno-Fuquen et al., 2008; Moreno-Fuquen et al., 2009). Though the present molecular system is centrosymmetric, information about its crystal structure is very important to the study of the general behavior of stilbenes because crystallographic information of stilbene systems is still rather small. The main objective of the present work is to present the molecular and crystal structure of the 4-dimethylamino-4'-bromostilbene (DMBS) and to analyse the conformational structure of the system. A perspective view of the molecule of the title compound, showing the atomic numbering scheme, is given in Fig. 1. The benzene rings ot the title structure are inclined to each other showing a dihedral angle between their mean planes of 50.5 (3)°. The phenyl rings are twisted out of the ethylene bond plane, and are defined by the the torsion angles C5—C4—C7=C8 and C7=C8—C9—C10. The dimethylamino group forms a dihedral angle of 8.6 (7)° with respect to its phenyl ring. The title molecule shows a torsion angle C4 C7 C8 C9 of 7.1 (15)° indicating the existence of a great repulsion between the aromatic rings. These values allow to define its conformation structure as cis. The title system does not observe the formation of intermolecular hydrogen bonds.
Experimental
By means of Wittig reaction (Maryanoff & Reitz, 1989), the 4-dimethylamino-benzyl-triphenylphosphonium iodide was prepared. The title stilbene was obtained by the reaction of equimolar quantities of phosphonium salt and 4-bromo benzaldehyde (0.03 mol) in THF solution. The mixture was maintained with stirring under argon atmosphere. The reaction mixture was kept at 273 K and it was dropped with a solution of tert-butanol and potassium tert-butoxide. Crystals of medium quality but suitable for single-crystal X-ray diffraction were grown in chloroform. An attempt was made to improve the quality of the crystals without success. Thin layer chromatography (TLC) was used to confirm the structure of the individual compounds. IR spectra were recorded on a Shimadzu FT—IR 8400 spectrophotometer.
N-(p-chlorophenyl)maleimide. Yellow crystals; yield 60%; mp 354 (1) K. IR (KBr) 2884 cm-1 (C—H), 3433 cm-1 (=C—H), 1609 cm-1 (C=C), 815 cm-1 (C=Br).
Refinement
All H-atoms were located from difference maps and then they were treated as riding atoms [Caro—H= 0.93 A° and Csp3—H= 0.96 A°, Uiso(H)= 1.2Ueq(Caro), Uiso(H)= 1.5Ueq(Csp3)).
Figures
Fig. 1.

An ORTEP-3 (Farrugia, 1997) plot of the DMBS compound, with the atomic labelling scheme. The shapes of the ellipsoids correspond to 50% probability contours of atomic displacement and, for the sake of clarity, H atoms are shown as spheres of arbitrary radius.
Crystal data
| C16H16BrN | F(000) = 616 |
| Mr = 302.21 | Dx = 1.469 Mg m−3 |
| Monoclinic, P21/c | Melting point: 354(1) K |
| Hall symbol: -P 2ybc | Mo Kα radiation, λ = 0.71073 Å |
| a = 14.804 (2) Å | Cell parameters from 2824 reflections |
| b = 6.0962 (5) Å | θ = 2.7–29.0° |
| c = 15.2106 (10) Å | µ = 2.99 mm−1 |
| β = 95.331 (9)° | T = 123 K |
| V = 1366.8 (2) Å3 | Slab, yellow |
| Z = 4 | 0.38 × 0.25 × 0.10 mm |
Data collection
| Bruker APEXII CCD diffractometer | 2986 independent reflections |
| Radiation source: fine-focus sealed tube | 1533 reflections with I > 2σ(I) |
| graphite | Rint = 0.064 |
| φ and ω scans | θmax = 27.0°, θmin = 2.7° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 2002) | h = −18→18 |
| Tmin = 0.457, Tmax = 0.749 | k = −7→7 |
| 8606 measured reflections | l = −19→19 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.075 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.213 | H-atom parameters constrained |
| S = 0.99 | w = 1/[σ2(Fo2) + (0.1119P)2] where P = (Fo2 + 2Fc2)/3 |
| 2986 reflections | (Δ/σ)max < 0.001 |
| 165 parameters | Δρmax = 0.94 e Å−3 |
| 0 restraints | Δρmin = −0.54 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.36239 (6) | −0.41740 (14) | 0.35579 (5) | 0.0487 (4) | |
| N1 | −0.0563 (6) | 0.4693 (11) | 0.6162 (5) | 0.055 (2) | |
| C1 | 0.3671 (6) | −0.2184 (13) | 0.4545 (5) | 0.043 (2) | |
| C2 | 0.4151 (6) | −0.2818 (16) | 0.5312 (5) | 0.055 (2) | |
| H2 | 0.4433 | −0.4217 | 0.5367 | 0.066* | |
| C3 | 0.4210 (6) | −0.1319 (16) | 0.6019 (5) | 0.055 (2) | |
| H3 | 0.4562 | −0.1695 | 0.6552 | 0.066* | |
| C4 | 0.3768 (5) | 0.0699 (13) | 0.5960 (5) | 0.0394 (19) | |
| C5 | 0.3297 (6) | 0.1216 (13) | 0.5151 (5) | 0.046 (2) | |
| H5 | 0.3014 | 0.2613 | 0.5089 | 0.055* | |
| C6 | 0.3215 (6) | −0.0195 (14) | 0.4429 (5) | 0.050 (2) | |
| H6 | 0.2871 | 0.0176 | 0.3892 | 0.061* | |
| C7 | 0.3859 (6) | 0.2246 (15) | 0.6700 (5) | 0.050 (2) | |
| H7 | 0.4442 | 0.2300 | 0.7017 | 0.060* | |
| C8 | 0.3242 (6) | 0.3591 (14) | 0.6993 (5) | 0.047 (2) | |
| H8 | 0.3483 | 0.4566 | 0.7442 | 0.056* | |
| C9 | 0.2261 (6) | 0.3859 (12) | 0.6758 (4) | 0.042 (2) | |
| C10 | 0.1699 (6) | 0.2157 (13) | 0.6391 (4) | 0.039 (2) | |
| H10 | 0.1963 | 0.0776 | 0.6278 | 0.047* | |
| C11 | 0.0804 (6) | 0.2435 (12) | 0.6198 (5) | 0.041 (2) | |
| H11 | 0.0458 | 0.1253 | 0.5936 | 0.049* | |
| C12 | 0.0344 (6) | 0.4455 (11) | 0.6371 (5) | 0.0369 (18) | |
| C13 | 0.0914 (6) | 0.6088 (12) | 0.6775 (4) | 0.042 (2) | |
| H13 | 0.0663 | 0.7459 | 0.6924 | 0.050* | |
| C14 | 0.1825 (6) | 0.5720 (12) | 0.6956 (5) | 0.043 (2) | |
| H14 | 0.2178 | 0.6859 | 0.7242 | 0.052* | |
| C15 | −0.1121 (6) | 0.3046 (14) | 0.5665 (6) | 0.054 (2) | |
| H15A | −0.0868 | 0.2745 | 0.5105 | 0.081* | |
| H15B | −0.1742 | 0.3600 | 0.5547 | 0.081* | |
| H15C | −0.1127 | 0.1692 | 0.6012 | 0.081* | |
| C16 | −0.1007 (6) | 0.6780 (14) | 0.6312 (6) | 0.055 (2) | |
| H16A | −0.0761 | 0.7393 | 0.6880 | 0.082* | |
| H16B | −0.1661 | 0.6543 | 0.6319 | 0.082* | |
| H16C | −0.0898 | 0.7805 | 0.5837 | 0.082* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.0677 (7) | 0.0433 (5) | 0.0349 (4) | 0.0116 (4) | 0.0043 (3) | −0.0004 (4) |
| N1 | 0.080 (7) | 0.033 (4) | 0.053 (4) | 0.003 (4) | 0.010 (4) | −0.008 (3) |
| C1 | 0.068 (6) | 0.037 (5) | 0.025 (4) | 0.000 (4) | 0.010 (4) | 0.008 (3) |
| C2 | 0.057 (6) | 0.069 (6) | 0.038 (5) | 0.022 (5) | 0.001 (4) | 0.001 (5) |
| C3 | 0.047 (6) | 0.085 (7) | 0.032 (4) | 0.014 (5) | −0.001 (4) | 0.005 (5) |
| C4 | 0.037 (5) | 0.051 (5) | 0.030 (4) | −0.010 (4) | 0.002 (3) | −0.001 (4) |
| C5 | 0.061 (6) | 0.043 (5) | 0.036 (4) | 0.003 (4) | 0.014 (4) | 0.006 (4) |
| C6 | 0.064 (6) | 0.062 (6) | 0.025 (4) | 0.019 (5) | 0.004 (4) | 0.015 (4) |
| C7 | 0.058 (6) | 0.059 (6) | 0.031 (4) | −0.012 (5) | 0.000 (4) | 0.008 (4) |
| C8 | 0.066 (7) | 0.052 (5) | 0.024 (4) | −0.011 (5) | 0.011 (4) | 0.001 (4) |
| C9 | 0.075 (7) | 0.030 (5) | 0.020 (3) | −0.006 (4) | 0.007 (3) | 0.004 (3) |
| C10 | 0.065 (6) | 0.036 (5) | 0.019 (3) | −0.008 (4) | 0.014 (3) | −0.007 (3) |
| C11 | 0.068 (7) | 0.031 (4) | 0.026 (4) | −0.017 (4) | 0.012 (4) | 0.001 (3) |
| C12 | 0.055 (6) | 0.026 (4) | 0.031 (4) | 0.004 (4) | 0.014 (3) | 0.003 (3) |
| C13 | 0.082 (7) | 0.021 (4) | 0.024 (4) | −0.003 (4) | 0.007 (4) | −0.004 (3) |
| C14 | 0.075 (7) | 0.030 (4) | 0.024 (4) | −0.011 (4) | 0.005 (4) | −0.004 (3) |
| C15 | 0.068 (7) | 0.038 (5) | 0.055 (5) | −0.003 (4) | 0.007 (4) | 0.000 (4) |
| C16 | 0.069 (7) | 0.045 (5) | 0.049 (5) | 0.006 (4) | 0.001 (4) | −0.009 (4) |
Geometric parameters (Å, °)
| Br1—C1 | 1.927 (8) | C8—H8 | 0.9500 |
| N1—C12 | 1.359 (11) | C9—C14 | 1.353 (11) |
| N1—C16 | 1.460 (10) | C9—C10 | 1.412 (10) |
| N1—C15 | 1.465 (11) | C10—C11 | 1.342 (11) |
| C1—C2 | 1.364 (10) | C10—H10 | 0.9500 |
| C1—C6 | 1.391 (11) | C11—C12 | 1.443 (10) |
| C2—C3 | 1.408 (12) | C11—H11 | 0.9500 |
| C2—H2 | 0.9500 | C12—C13 | 1.408 (11) |
| C3—C4 | 1.392 (11) | C13—C14 | 1.371 (11) |
| C3—H3 | 0.9500 | C13—H13 | 0.9500 |
| C4—C5 | 1.393 (10) | C14—H14 | 0.9500 |
| C4—C7 | 1.465 (11) | C15—H15A | 0.9800 |
| C5—C6 | 1.392 (11) | C15—H15B | 0.9800 |
| C5—H5 | 0.9500 | C15—H15C | 0.9800 |
| C6—H6 | 0.9500 | C16—H16A | 0.9800 |
| C7—C8 | 1.334 (12) | C16—H16B | 0.9800 |
| C7—H7 | 0.9500 | C16—H16C | 0.9800 |
| C8—C9 | 1.472 (12) | ||
| C12—N1—C16 | 120.5 (7) | C10—C9—C8 | 123.1 (7) |
| C12—N1—C15 | 123.1 (7) | C11—C10—C9 | 121.9 (8) |
| C16—N1—C15 | 115.9 (7) | C11—C10—H10 | 119.1 |
| C2—C1—C6 | 124.3 (8) | C9—C10—H10 | 119.1 |
| C2—C1—Br1 | 117.8 (6) | C10—C11—C12 | 122.7 (7) |
| C6—C1—Br1 | 117.9 (6) | C10—C11—H11 | 118.7 |
| C1—C2—C3 | 117.4 (8) | C12—C11—H11 | 118.7 |
| C1—C2—H2 | 121.3 | N1—C12—C13 | 124.4 (7) |
| C3—C2—H2 | 121.3 | N1—C12—C11 | 121.4 (7) |
| C4—C3—C2 | 121.9 (7) | C13—C12—C11 | 114.2 (8) |
| C4—C3—H3 | 119.0 | C14—C13—C12 | 120.7 (7) |
| C2—C3—H3 | 119.0 | C14—C13—H13 | 119.7 |
| C3—C4—C5 | 116.8 (7) | C12—C13—H13 | 119.7 |
| C3—C4—C7 | 120.7 (7) | C9—C14—C13 | 125.0 (7) |
| C5—C4—C7 | 122.4 (8) | C9—C14—H14 | 117.5 |
| C6—C5—C4 | 123.8 (8) | C13—C14—H14 | 117.5 |
| C6—C5—H5 | 118.1 | N1—C15—H15A | 109.5 |
| C4—C5—H5 | 118.1 | N1—C15—H15B | 109.5 |
| C1—C6—C5 | 115.7 (7) | H15A—C15—H15B | 109.5 |
| C1—C6—H6 | 122.2 | N1—C15—H15C | 109.5 |
| C5—C6—H6 | 122.2 | H15A—C15—H15C | 109.5 |
| C8—C7—C4 | 129.5 (8) | H15B—C15—H15C | 109.5 |
| C8—C7—H7 | 115.2 | N1—C16—H16A | 109.5 |
| C4—C7—H7 | 115.2 | N1—C16—H16B | 109.5 |
| C7—C8—C9 | 132.6 (8) | H16A—C16—H16B | 109.5 |
| C7—C8—H8 | 113.7 | N1—C16—H16C | 109.5 |
| C9—C8—H8 | 113.7 | H16A—C16—H16C | 109.5 |
| C14—C9—C10 | 115.4 (8) | H16B—C16—H16C | 109.5 |
| C14—C9—C8 | 121.3 (7) | ||
| C6—C1—C2—C3 | −2.3 (14) | C14—C9—C10—C11 | 4.5 (10) |
| Br1—C1—C2—C3 | 177.7 (6) | C8—C9—C10—C11 | 178.7 (7) |
| C1—C2—C3—C4 | 2.7 (13) | C9—C10—C11—C12 | −1.8 (10) |
| C2—C3—C4—C5 | −2.9 (12) | C16—N1—C12—C13 | 3.1 (11) |
| C2—C3—C4—C7 | −178.7 (8) | C15—N1—C12—C13 | 173.9 (7) |
| C3—C4—C5—C6 | 2.8 (12) | C16—N1—C12—C11 | −177.6 (7) |
| C7—C4—C5—C6 | 178.5 (8) | C15—N1—C12—C11 | −6.8 (11) |
| C2—C1—C6—C5 | 2.1 (13) | C10—C11—C12—N1 | 179.5 (7) |
| Br1—C1—C6—C5 | −177.8 (6) | C10—C11—C12—C13 | −1.1 (10) |
| C4—C5—C6—C1 | −2.4 (12) | N1—C12—C13—C14 | −179.3 (7) |
| C3—C4—C7—C8 | −142.8 (9) | C11—C12—C13—C14 | 1.3 (9) |
| C5—C4—C7—C8 | 41.7 (13) | C10—C9—C14—C13 | −4.4 (10) |
| C4—C7—C8—C9 | 7.1 (15) | C8—C9—C14—C13 | −178.8 (7) |
| C7—C8—C9—C14 | −160.7 (9) | C12—C13—C14—C9 | 1.5 (11) |
| C7—C8—C9—C10 | 25.4 (12) |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HG2518).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Bruker (2007). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- He, T., Wang, C., Pan, X., Yang, H. & Lu, G. (2008). Dyes Pigments, 82, 47–52.
- Maryanoff, B. E. & Reitz, A. B. J. (1989). Chem. Rev.89, 863–927.
- Moreno-Fuquen, R., Aguirre, L. & Kennedy, A. R. (2008). Acta Cryst. E64, o2259. [DOI] [PMC free article] [PubMed]
- Moreno-Fuquen, R., Dvries, R., Theodoro, J. & Ellena, J. (2009). Acta Cryst. E65, o1371. [DOI] [PMC free article] [PubMed]
- Nardelli, M. (1995). J. Appl. Cryst.28, 659.
- Sheldrick, G. M. (2002). SADABS. University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536809020492/hg2518sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809020492/hg2518Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report