

Abstract
The centrosymmetric title compound, C8H8I2, was prepared by metathesis from the dibromo analogue. In the crystal structure, weak C—H⋯I interactions link the molecules into stacks down the b axis. The structure is further stabilized by short I⋯I contacts [3.8433 (2) Å], forming undulating sheets in the (101) plane.
Related literature
For the synthesis, see: Moore & Stupp (1986 ▶); Kida et al. (2005 ▶). For related structures, see: Basaran et al. (1992 ▶); Fun et al. (2009 ▶); Jones & Kus (2007 ▶); Zhang et al. (2007 ▶). For applications of dihalo-p-xylenes in living radical polymerization processes, see: Samakande et al., (2007 ▶); Asandei et al. (2008 ▶). For other polymer applications, see: Leir & Stark (1989 ▶); Hochberg & Schulz (1993 ▶). For additional applications of dihalo-p-xylenes, see: Le Baccon et al. (2001 ▶); Sobransingh & Kaifer (2006 ▶); Song et al. (2008 ▶); Au et al. (2009 ▶). For details of halogen⋯halogen interactions, see: Pedireddi et al. (1994 ▶) and for reference structural data, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C8H8I2
M r = 357.94
Monoclinic,
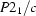
a = 9.0978 (3) Å
b = 4.5982 (2) Å
c = 11.2793 (3) Å
β = 99.808 (1)°
V = 464.96 (3) Å3
Z = 2
Mo Kα radiation
μ = 6.69 mm−1
T = 89 K
0.21 × 0.15 × 0.03 mm
Data collection
Bruker APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2006 ▶) T min = 0.410, T max = 0.818
8198 measured reflections
1674 independent reflections
1538 reflections with I > 2σ(I)
R int = 0.026
Refinement
R[F 2 > 2σ(F 2)] = 0.013
wR(F 2) = 0.033
S = 1.06
1674 reflections
62 parameters
All H-atom parameters refined
Δρmax = 0.51 e Å−3
Δρmin = −0.51 e Å−3
Data collection: APEX2 (Bruker 2006 ▶); cell refinement: APEX2 and SAINT (Bruker 2006 ▶); data reduction: SAINT; program(s) used to solve structure: SIR92 (Altomare et al., 1993 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶) and TITAN (Hunter & Simpson, 1999 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and Mercury (Macrae et al., 2006 ▶); software used to prepare material for publication: SHELXL97, enCIFer (Allen et al., 2004 ▶), PLATON (Spek, 2009 ▶) and publCIF (Westrip, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809021151/hb2998sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809021151/hb2998Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C4—H42⋯I1i | 1.02 (2) | 3.12 (2) | 3.9774 (16) | 141.8 (16) |
Symmetry code: (i) 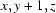 .
.
Acknowledgments
We thank the New Economy Research Fund (grant No. UOO-X0808) for support of this work and the University of Otago for the purchase of the diffractometer.
supplementary crystallographic information
Comment
Dihalo-p-xylenes are extensively used in polymer science (Leir & Stark, 1989; Hochberg & Schulz, 1993; Samakande et al., 2007) and as a versatile synthon for –CH2—C6H4—CH2– connective units in other areas of chemistry (Le Baccon et al., 2001; Song et al., 2008; Au et al., 2009). The bulk of this work utilizes the commercially available α,α'-dibromo-p-xylene, but the diiodo- derivative offers additional reactivity (Moore & Stupp, 1986; Sobransingh & Kaifer, 2006; Asandei et al., 2008). Our interest in the title compound, (I), Fig. 1, is as one of the components in xylene bridged electroactive gels. In these, the chemical links are quaternary amines formed by reaction of the alkylhalogen termini with amine residues in the other gel component.
The molecule lies about an inversion centre located at the centroid of the benzene ring. The C1···C4 atoms lie in a plane (r.m.s. deviation 0.01 Å) and the C—C and C—I distances in the molecule are unremarkable (Allen et al., 1987). This structure is the fourth in a series of XCH2C6H4CH2X molecules; X = F, II (Fun et al., 2009); Cl, III (Basaran et al., (1992); Br, IV (Jones & Kus, 2007; Zhang et al., 2007). All four molecules are closely isostructural with only the C—halogen bond distance distinguishing them. Indeed the CH2C6H4CH2 fragment of the title compound overlays with corresponding portions of the related molecules with r.m.s. deviations, 0.032 Å for II, 0.013 Å for III and 0.007 Å for IV respectively (Macrae et al., 2006).
In the crystal structure, weak C—H···I interactions and short I···I contacts, 3.8433 (2) Å, form undulating sheets in the 101 plane, Fig. 2. Each I atom interacts with two adjacent iodine atoms (symmetry operations 2 - x, -1/2 + y, 1/2 - z and 2 - x, 1/2 + y, 1/2 - z). The I···I contacts observed here fit with the type II designation of halogen···halogen interactions proposed previously (Pedireddi et al. 1994). The packing arrangement for I is closely similar to that observed for IV, (Jones & Kus, 2007; Zhang et al., 2007). I, III and IV all crystallize in the space group P21/c with unit cells that differ only in a small but significant increase in volume as the size of the halogen increases.
Experimental
The title compound was prepared by a combination of the methods of Moore & Stupp (1986) and Kida et al. (2005). Thus α,α'-dibromo-p-xylene (1.32 g, 5 mmol) was refluxed for 7 h with sodium iodide (2.25 g, 15 mmol) in acetone (25 ml). The solution was allowed to cool overnight, and the resulting yellow plates of (I) that developed were rinsed gently with water to remove sodium bromide and air dried. Confirmation of the metathesized (iodo) product was by microanalysis, mass spectroscopy and diagnostic tests. 1H and 13C NMR spectra are distinct from those of the dibromo precursor.
Refinement
All H-atoms were located in a difference Fourier map and refined freely.
Figures
Fig. 1.

The structure of (I) with displacement ellipsoids for the non-hydrogen atoms drawn at the 50% probability level. Unlabelled atoms are generated by the symmetry operation (1–x, –y, 1–z).
Fig. 2.

Crystal packing for (I) viewed down the b axis with hydrogen bonds and short I···I contacts drawn as dashed lines.
Crystal data
| C8H8I2 | F(000) = 324 |
| Mr = 357.94 | Dx = 2.557 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 5590 reflections |
| a = 9.0978 (3) Å | θ = 2.3–33.0° |
| b = 4.5982 (2) Å | µ = 6.69 mm−1 |
| c = 11.2793 (3) Å | T = 89 K |
| β = 99.808 (1)° | Rectangular plate, pale yellow |
| V = 464.96 (3) Å3 | 0.21 × 0.15 × 0.03 mm |
| Z = 2 |
Data collection
| Bruker APEXII CCD area-detector diffractometer | 1674 independent reflections |
| Radiation source: fine-focus sealed tube | 1538 reflections with I > 2σ(I) |
| graphite | Rint = 0.026 |
| ω scans | θmax = 33.4°, θmin = 3.7° |
| Absorption correction: multi-scan (SADABS; Bruker, 2006) | h = −13→13 |
| Tmin = 0.410, Tmax = 0.818 | k = −5→7 |
| 8198 measured reflections | l = −16→16 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.013 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.033 | All H-atom parameters refined |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0145P)2 + 0.1407P] where P = (Fo2 + 2Fc2)/3 |
| 1674 reflections | (Δ/σ)max = 0.001 |
| 62 parameters | Δρmax = 0.51 e Å−3 |
| 0 restraints | Δρmin = −0.51 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.62504 (16) | 0.1521 (3) | 0.47504 (13) | 0.0138 (2) | |
| C2 | 0.63784 (17) | −0.0405 (3) | 0.57245 (14) | 0.0154 (3) | |
| C3 | 0.51429 (17) | −0.1903 (3) | 0.59710 (14) | 0.0154 (3) | |
| C4 | 0.75717 (18) | 0.3189 (4) | 0.45106 (15) | 0.0190 (3) | |
| I1 | 0.880515 (9) | 0.07951 (2) | 0.331669 (8) | 0.01438 (4) | |
| H2 | 0.729 (3) | −0.073 (4) | 0.620 (2) | 0.021 (6)* | |
| H41 | 0.841 (3) | 0.354 (5) | 0.524 (2) | 0.031 (6)* | |
| H3 | 0.523 (2) | −0.330 (5) | 0.6678 (19) | 0.022 (5)* | |
| H42 | 0.733 (3) | 0.509 (5) | 0.405 (2) | 0.021 (5)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0142 (6) | 0.0109 (6) | 0.0180 (6) | −0.0017 (5) | 0.0078 (5) | −0.0032 (5) |
| C2 | 0.0133 (6) | 0.0159 (7) | 0.0174 (7) | 0.0007 (5) | 0.0038 (5) | −0.0010 (5) |
| C3 | 0.0175 (6) | 0.0126 (6) | 0.0175 (6) | 0.0006 (5) | 0.0070 (5) | 0.0012 (5) |
| C4 | 0.0191 (7) | 0.0150 (7) | 0.0258 (8) | −0.0039 (5) | 0.0124 (6) | −0.0051 (6) |
| I1 | 0.01367 (5) | 0.01628 (6) | 0.01473 (5) | −0.00037 (3) | 0.00680 (3) | 0.00092 (3) |
Geometric parameters (Å, °)
| C1—C3i | 1.396 (2) | C3—C1i | 1.396 (2) |
| C1—C2 | 1.401 (2) | C3—H3 | 1.01 (2) |
| C1—C4 | 1.489 (2) | C4—I1 | 2.1907 (15) |
| C2—C3 | 1.386 (2) | C4—H41 | 1.04 (2) |
| C2—H2 | 0.92 (2) | C4—H42 | 1.02 (2) |
| C3i—C1—C2 | 118.88 (13) | C1i—C3—H3 | 118.7 (12) |
| C3i—C1—C4 | 120.68 (14) | C1—C4—I1 | 111.52 (10) |
| C2—C1—C4 | 120.42 (14) | C1—C4—H41 | 116.4 (13) |
| C3—C2—C1 | 120.64 (14) | I1—C4—H41 | 100.5 (13) |
| C3—C2—H2 | 119.1 (14) | C1—C4—H42 | 114.9 (13) |
| C1—C2—H2 | 120.3 (14) | I1—C4—H42 | 101.8 (13) |
| C2—C3—C1i | 120.48 (14) | H41—C4—H42 | 109.8 (19) |
| C2—C3—H3 | 120.9 (12) | ||
| C3i—C1—C2—C3 | −0.1 (2) | C3i—C1—C4—I1 | −91.95 (15) |
| C4—C1—C2—C3 | 178.24 (14) | C2—C1—C4—I1 | 89.71 (15) |
| C1—C2—C3—C1i | 0.1 (2) |
Symmetry codes: (i) −x+1, −y, −z+1.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C4—H42···I1ii | 1.02 (2) | 3.12 (2) | 3.9774 (16) | 141.8 (16) |
Symmetry codes: (ii) x, y+1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB2998).
References
- Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004). J. Appl. Cryst.37, 335–338.
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Altomare, A., Cascarano, G., Giacovazzo, C. & Guagliardi, A. (1993). J. Appl. Cryst.26, 343–350.
- Asandei, A. D., Chen, Y., Simpson, C., Gilbert, M. & Moran, I. W. (2008). Polymer Preprints (Am. Chem. Soc. Div. Polym. Chem.), 49, 489–490.
- Au, R. H. W., Fraser, C. S. A., Eisler, D. J., Jennings, M. C. & Puddephat, R. J. (2009). Organometallics, 28, 1719–1729.
- Basaran, R., Dou, S.-Q. & Weiss, A. (1992). Ber. Bunsenges. Phys. Chem.96, 1688–1690.
- Bruker (2006). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst.30, 565.
- Fun, H.-K., Kia, R., Patil, P. S. & Dharmaprakash, S. M. (2009). Acta Cryst. E65, o459. [DOI] [PMC free article] [PubMed]
- Hochberg, G. C. & Schulz, R. C. (1993). Polym. Int.32, 309–317.
- Hunter, K. A. & Simpson, J. (1999). TITAN2000 University of Otago, New Zealand.
- Jones, P. G. & Kus, P. (2007). Z. Naturforsch. Teil B, 62, 725–731.
- Kida, T., Kikuzawa, A., Higashimoto, H., Nakatsuji, Y. & Akashi, M. (2005). Tetrahedron, 61, 5763–5768.
- Le Baccon, M., Chuburu, F., Toupet, L., Handel, H., Soibinet, M., Dechamps-Olivier, I., Barbier, J.-P. & Aplincourt, M. (2001). New J. Chem.25, 1168–1174.
- Leir, C. M. & Stark, J. E. (1989). J. Appl. Polym. Sci.38, 1535–1547.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst.39, 453–457.
- Moore, J. S. & Stupp, S. I. (1986). Macromolecules, 19, 1815–1824.
- Pedireddi, V. R., Reddy, D. S., Goud, B. S., Craig, D. C., Rae, A. D. & Desiraju, G. R. (1994). J. Chem. Soc. Perkin Trans. 2, pp. 2353–2360.
- Samakande, A., Sanderson, R. D. & Hartmann, P. C. (2007). Synth. Commun.37, 3861–3872.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sobransingh, D. & Kaifer, A. E. (2006). Org. Lett.8, 3247–3250. [DOI] [PubMed]
- Song, Z., Weng, X., Weng, L., Huang, J., Wang, X., Bai, M., Zhou, Y., Yang, G. & Zhou, X. (2008). Chem. Eur. J.14, 5751–5754. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Westrip, S. P. (2009). publCIF In preparation.
- Zhang, M., Su, P., Meng, X.-G. & Xu, X.-M. (2007). Acta Cryst. E63, o951–o952.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809021151/hb2998sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809021151/hb2998Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report