

Abstract
Genes associated with human microcephaly, a condition characterized by a small brain, include critical regulators of proliferation, cell fate, and DNA repair. We describe a syndrome of congenital microcephaly and diverse defects in cerebral cortical architecture. Genome-wide linkage analysis in two families identified a 7.5 Mb locus on chromosome 19q13.12 containing 148 genes. Targeted high throughput sequence analysis of linked genes in each family yielded > 4000 DNA variants and implicated a single gene, WDR62, as harboring potentially deleterious changes. We subsequently identified additional WDR62 mutations in four other families. MRI and postmortem brain analysis supports important roles for WDR62 in proliferation and migration of neuronal precursors. WDR62 is a WD40 repeat-containing protein expressed in neuronal precursors as well as postmitotic neurons in the developing brain and localizes to the spindle poles of dividing cells. The diverse phenotypes of WDR62 suggest central roles in many aspects of cerebral cortical development.
A first pedigree (LIS-900) with microcephaly with simplified gyri (MCSG) from Mexico consists of distantly related parents with three affected children and one healthy sibling (Fig. 1a). The children showed severe microcephaly, developmental delay, and seizures (see Supplementary Note, Clinical Data) but no other syndromic features. A second family (LIS-2600) from Turkey showed similar clinical features and has been described before1 (Fig. 1b). Subsequent to the identification of the responsible gene, four other families were identified with similar clinical features (Fig. 1c–f and Supplementary Note, Clinical Data). Brain MRI of the six families (Fig. 1g–p), revealed small brains, markedly simplified gyral patterns, corpus callosal abnormalities, plus a diversity of additional cortical malformations including polymicrogyria, schizencephaly, and subcortical heterotopia (arrested neurons), sometimes with asymmetry in the same brain (Supplementary Note, Clinical Data and Supplementary Movie 1).
Figure 1.
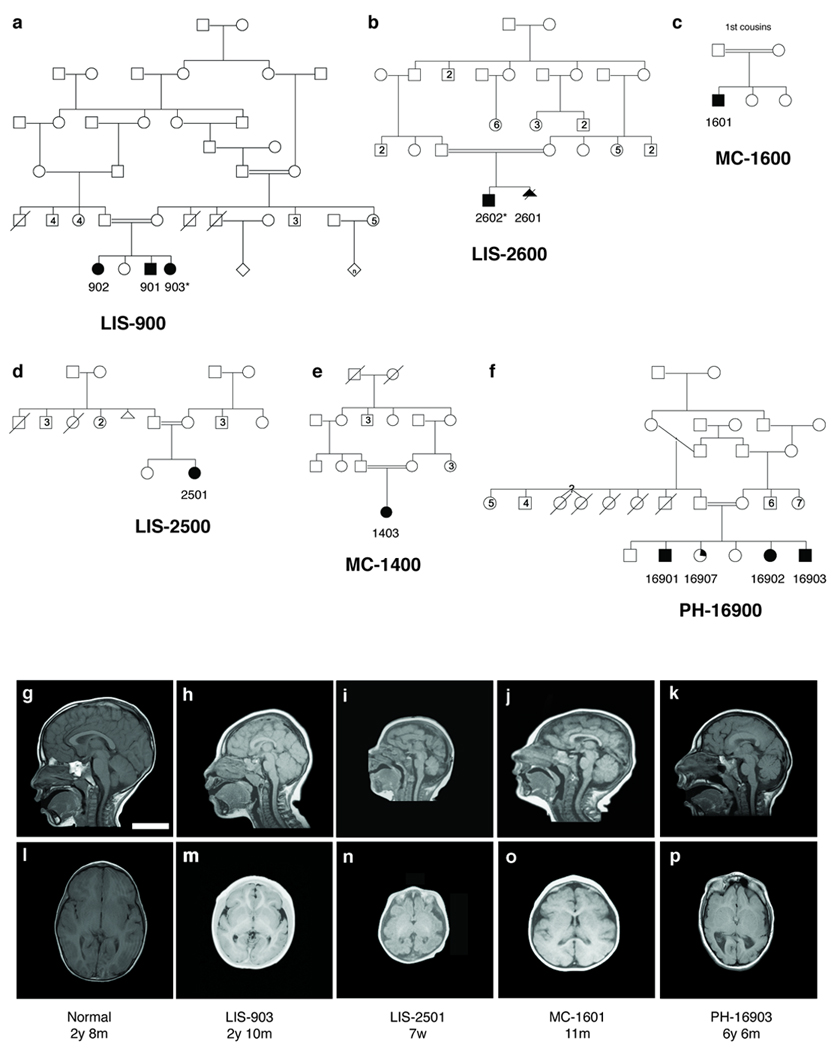
Pedigrees and radiographic findings in six consanguineous families with microcephaly and simplified gyri (MCSG). (a–f) Shaded symbols denote affected individuals. (a) LIS-900, a Mexican family with three affected children. (b) LIS-2600, a Turkish family with two affected children1. (c) LIS-2500, (d) MC-1400, and (e) MC-1600, three Turkish families (not known to be related to LIS-2600 or each other). (f) PH-16900, a Saudi family with three affected children; a fourth child (partial shading) had mild speech delay and articulation and attention difficulties but no brain abnormalities. Whole blood DNA was obtained and analyzed from each nuclear family, with the exception of LIS-2601, who died as a fetus. Asterisks denote the two individuals chosen for targeted high-throughput sequencing. (g–p) MRI features of patients with MCSG, demonstrating the breadth of cortical phenotypes associated with WDR62. Mid-sagittal T1 sections (g–k) and axial T1 sections at the level of the insula (l–p) are shown for LIS-903 (h,m), LIS-2501 (i,n), MC-1601 (j,o), and PH-16903 (k, p) individuals, as well as a control individual (g, l) age-matched to LIS-903. Common findings include microcephaly (h–k, m–p), anomalies of the corpus callosum (small splenium in h and k, absent splenium in i, thick body in k), simplification of the normal gyral pattern (h, i, k, m, n, p) as well as more variable features such as mild asymmetries of cortical size (p), possible heterotopia and cortical clefts (not shown, see Supplementary Note, Clinical Data). Scale bar = 5 cm.
Genome-wide linkage screens of LIS-900 showed a single 11.9 Mb region on chromosome 19q13 (D19S431; D19S217) homozygous in all 3 affected patients and not homozygous in the normal sibling (Supplementary Fig. 1). SNP genotyping of LIS-2600 confirmed the homozygosity and narrowed the interval to ≈ 7.5 Mb (Fig. 2a) (peak combined multipoint LOD score of 3.84, Supplementary Fig. 2). SNP analysis of MC-1400 and MC-1600 confirmed the candidate region but did not reduce it (Fig. 2a).
Figure 2.

Mapping, capture, and sequencing of 148 genes in the MCSG locus. (a) Homozygosity analysis in four pedigrees (LIS-900, LIS-2600, MC-1400 and MC-1600) revealed a 7.5 Mb interval on chr19, bounded by SNP markers rs17581484 and rs4802998. Homozygous SNPs are shown in red or blue, heterozygous SNPs are shown in yellow, and SNPs for which no genotype could be assigned are shown in white. The homozygous region contained 148 annotated UCSC genes. (b) Custom Nimblegen microarrays were designed to target all coding and noncoding regions of the 85 genes in the center of the linkage peak and all exonic regions of the remaining 63 genes (upper green track; the location of probes on the array is shown on the lower track). These were used to capture genomic DNA and generate sequencing libraries from two individuals, LIS-903 and LIS-2602. (c) Libraries were sequenced on a Illumina GA II to an average depth of >200X and completeness of 88–90% (bases covered by >= 10 reads). Depth of sequence coverage over the region is shown.
Because of the high gene density of the linked region, we employed an array capture approach2 followed by high-throughput Illumina sequencing in two affected individuals, LIS-903 and LIS-2602. A custom array was designed with oligonucleotide probes targeting the exons of all 148 predicted UCSC genes in the interval (Fig. 2b) plus non-exonic DNA from a smaller candidate subregion suggested by microsatellite mapping (Supplementary Fig. 2). DNA bound to the array was amplified and used to generate sequencing libraries. 36–40 million paired-end sequence tags (>1.4 Gb of raw sequence) were generated per patient and mapped (hg18) using BWA (Fig. 2c)3. 50–60% of sequence reads were on-target, and average depth of target coverage was 230X and 224X, with 88% and 90% of bases covered by ≥ 10 reads.
Single nucleotide variants and microinsertions/microdeletions were called and filtered for quality and mapping confidence4, revealing >2000 potential sequence variants from each proband (Table 1). Comparison of SNP calls with Affy 5.0 genotype data showed >99.8% concordance. Variants were filtered out if they were present in dbSNP1304 or the 1000 genomes project (1000G), and categorized by their predicted functional effects using GMCC. Of 2310 variants in LIS-903, 262 (11%) were not in dbSNP130 or 1000G, 93 were potentially pathogenic (4.0%); five met both criteria. Of 2570 variants in LIS-2602, 323 (12%) were absent from dbSNP and 1000G, and 99 (3.8%) were potentially pathogenic; seven variants met both criteria. A single gene, WDR62, showed novel, pathogenic mutations in both families, making it a strong candidate (Table 1).
Table 1. Variant filtering identifies WDR62 as the causative gene for MCSG.
Variant filtering identifies a single gene, WDR62, disrupted in two families with MCSG. Variants were filtered to remove common polymorphisms and then annotated to highlight variants with functional splicing or coding implications, reducing the number of candidate mutations in each individual to five in LIS-903 and seven in LIS-2602. Of these candidates, only one gene, WDR62, was disrupted in both families.
| LIS-903 | LIS-2602 | |
|---|---|---|
| Total variants | 2310 | 2570 |
| Rare variants not in dbSNP/1000G | 262 | 323 |
| Pathogenic | 93 | 99 |
| Rare and pathogenic | 5 | 7 |
| Rare, pathogenic and in both families | WDR62 | WDR62 |
Sanger sequencing confirmed WDR62 mutations in the two families, and identified four additional alleles in four other families (Fig. 3a, b, Supplementary Fig. 3). A homozygous single basepair insertion in exon 30 (c.3936_3937insC) in LIS-2602 produced a frameshift mutation (Fig. 3b). All three affected patients in family LIS-900 showed a homozygous single basepair deletion in exon 4 (c.363delT), creating a frameshift mutation (Fig. 3b). Pedigree analysis confirmed appropriate segregation of WDR62 mutations in both families (Fig. 3b). In LIS-2500, the affected individual showed a homozygous mutation one basepair downstream of exon 8 (c.1043+1G>A), disrupting the highly conserved GT consensus splicing sequence (Fig. 3a, Supplementary Fig. 3). In MC-1400, the affected child was homozygous for a 4 bp deletion after exon 23 (c.2867+4_c2867+7delGGTG) (Fig. 3a, Supplementary Fig. 3) that removes the conserved +4 and +5 positions of the intron and is predicted to disrupt the splice donor5 (reference …CAG|gtgggtgtc…; mutation …CAG|gtg----tc…). In MC-1600, the affected child was homozygous for a deletion of 17 bp in the 30th exon (c.3839_3855delGCCAAGAGCCTGCCCTG), causing a frameshift (Fig. 3a, Supplementary Fig. 3). In PH-16900, all three siblings with microcephaly were homozygous for a missense V65M change (c.193G>A, Fig. 3a, Supplementary Fig. 3) that alters a residue conserved in all vertebrates (Supplementary Fig. 4). An unaffected sibling (PH-16907, with mild speech delay and articulation difficulties but normal head circumference and normal MRI) was homozygous for the wildtype allele (Supplementary Fig. 3). Two additional unaffected siblings were homozygous wildtype and heterozygous, respectively (not shown). None of these mutations were found in over 1000 normal chromosomes.
Figure 3.

Six WDR62 mutations reported in association with microcephaly with simplified gyri. (a) Alterations are shown in genomic, coding DNA, and protein contexts. The human WDR62 gene consists of 32 exons shown as boxes, and encodes a protein of 1518 amino acids containing 15 WD40 repeats. Black shaded boxes represent untranslated regions, open boxes represent coding regions, and gray shaded boxes represent alternatively spliced exons. Lines connecting boxes represent introns. The diagram is drawn to scale. Five of the six alleles (from families LIS-900, LIS-2500, LIS-2600, MC-1400, MC-1600, and LIS-2600) disrupt splice sites or cause frameshifts resulting in protein truncations and are likely nulls. The sixth allele, found in PH-16900, is a missense alteration of V65M, a conserved residue. (b) Illustration of the c.3936_3937insC mutation in LIS-2602 by high throughput sequencing, and representative Sanger traces confirming proper segregation. High throughput sequencing data is shown using the Integrated Genome Viewer. Aligned reads are shown as gray tags shaded by quality score, SNPs are identified by the letter code of the substituted base, and the position of the LIS-2602 single basepair insertion is denoted by the letter I. Representative Sanger traces confirm this change in the affected individual and show that both parents are heterozygous for the insertional event. Similarly, representative Sanger traces illustrate the c.363delT mutation in LIS-903 and the carrier status of both parents.
WDR62 encodes a 1518 amino acid protein with multiple WD40 repeats (Fig. 3a)6, but little is known about its function. A proteomic study identified WDR62 as a binding partner of the centrosomal protein CEP1707, which is of interest given the involvement of other centrosomal proteins in microcephaly8–14. Another report identified WDR62 as a binding partner for c-Jun N-terminal kinase (JNK), suggesting that it potentiates JNK activity15. In situ hybridization with a probe to mouse WDR62 showed widespread expression in the developing brain at embryonic day 14.5 (E14.5) (Fig. 4a), with highest expression in forebrain. Expression was seen in the ventricular zone and the cortical plate (Fig. 4b), consistent with roles in progenitor cells and postmitotic neurons.
Figure 4.

WDR62 expression in developing mouse brain and subcellular localization. (a) in situ hybridization of E14.5 mouse brain with antisense probe to mouse WDR62. Sense strand (not shown) showed no specific hybridization. (b) Higher power view. Strong WDR62 message is seen in the ventricular zone, subventricular zone, ventral portion of the intermediate zone, and ganglionic eminences, with some hybridization in the cortical plate as well. (c–h) Confocal microscopy demonstrating WDR62 subcellular localization. (c, d) endogenous WDR62 localization in interphase HeLa cells. (c) anti-WDR62 (green), also stained with anti γ-tubulin (red) and Hoechst for DNA (blue), showing perinuclear localization, surrounding but not overlapping the centrosome. (d) co-staining of interphase HeLa cells with anti-WDR62 and anti-GM130 shows localization of both to the Golgi apparatus near the Hoechst-positive nucleus. (e–g) WDR62 localization in HeLa and HEK cells during M phase. (e) Endogenous WDR62 localizes to the spindle poles in M phase HeLa cells, visualized by double labeling with γ-tubulin. (f) Co-staining with antisera to WDR62 and dynein in HEK cells shows WDR62 at the spindle poles and dynein throughout the spindle. (g) Transfection of C-terminal HA-tagged WDR62 in HEK cells confirms ring-like localization around the centrosome and overlaps with CEP170, another centrosomal protein. (h) Endogenous WDR62 immunoreactivity surrounds LIS1 but does not overlap fully.
WDR62 demonstrates strikingly cell cycle-dependent localization. Immunofluorescence staining of endogenous WDR62 in HeLa cells with an anti-WDR62 antibody revealed punctate, perinuclear expression during interphase, suggesting localization to the Golgi apparatus and confirmed by GM130 immunoreactivity (Fig. 4c, d). In contrast, in HeLa or HEK cells in M phase, WDR62 is found at the spindle poles as demonstrated by double-labeling with gamma-tubulin and dynein (Fig. 4e, f). Close inspection of recombinant HA-tagged WDR62 protein demonstrated subcellular localization closely matching that of CEP170 (Fig. 4g) and surrounding LIS1 (Fig. 4h), a pattern similar to that previously demonstrated for ASPM16,17 and other microcephaly proteins12,13,18,19. Bilguvar and colleagues20 recently described mutations in WDR62 with brain malformations and reported nuclear localization of WDR62 in cortical progenitor cells. While we cannot rule out nuclear localization in some cells, our data, as well as previously published data7 suggest a major centrosomal role.
Postmortem analysis of LIS-2601, an affected sibling in the LIS-2600 family1, suggests roles for WDR62 in neuronal proliferation and migration. The 27-week fetus showed a small cranial cavity enclosing a profoundly small brain (50g, compared to a normal of 127 +/− 20 g for this age and size fetus). The surface of the hemispheres was largely smooth (not shown), with poorly defined Sylvian fissures and few sulci1. The cerebellum appeared remarkably preserved (not shown), whereas the cerebral cortex was severely abnormal (Fig. 5a–g). The outermost cortical layer (layer I) appeared generally normal, but remaining cortical neuronal layers were thinner than normal (Fig. 5b, c). There was an almost complete lack of small- to medium-sized pyramidal neurons in their normal location (layers II and III), suggesting profoundly defective neurogenesis. Neurons located beneath layer I were primarily medium- to large pyramidal neurons, followed by a cell-sparse zone and a third layer of multipolar neurons at the deepest layer of the cortex, an appearance consistent with layers V and VI, respectively (Fig. 5c, d). Neurons in the medium-to-large pyramidal layer retained an immature radial columnar organization and also exhibited abnormal, non-radial clumping (Fig. 5d). In some sections, the pial surface was interrupted by microscopic extrusions of cells into the subarachnoid space, forming heterotopia (Fig. 5e). Heterotopia were also sometimes found in the intermediate zone, arranged in streaks particularly in posterior frontal cortex (Fig. 5f), corresponding to the localization of subcortical heterotopia seen on MRI. There were also clusters (Fig. 5g) of small round cells in the outer subventricular zone (SVZ) suggestive of progenitor cells. These findings support a proliferative defect in brains of patients with WDR62 mutations, potentially affecting the outer SVZ (precursors to upper layer cortical neurons)21 as well as defective neuronal migration, in some ways evocative of the dual roles in proliferation and migration seen for LIS122,23.
Figure 5.

Histopathologic analysis of a 27 week human fetus with MCSG. (a) H&E stained coronal section from the forebrain of a 27 week gestational age fetus with microcephaly with simplified gyri (LIS-2601). Section is at the rostral end of the caudate along the A–P axis. Locations of higher magnification views for additional panels are indicated. (b, c) H&E stained section demonstrating cortical layering. Compared to a (b) 24 week gestational age control fetus, (c) LIS-2601 exhibits an abnormally thin cortical plate (despite being three weeks older), suggestive of a proliferative defect and absence of normal appearing layer II/III cells underneath the molecular layer. The dimensions of the marginal zone are relatively preserved. (d) In addition to a smaller cortical plate, neurons in the cortical plate display abnormal persistence of a radial columnar pattern and disorganized clustering (Kluver-Barrera stain). (e) Occasionally, eruptions of neuroglial cells (arrows) through the pial surface into the subarachnoid space are seen, as are occasional (f) streaky heterotopia in the intermediate zone, and (g) clusters of small, darkly staining cells in the outer subventricular zone that resemble dividing cells. Abbreviations: IZ, intermediate zone; GE, ganglionic eminence; LV, lateral ventricle; bc, body of the caudate; cc, corpus callosum; ctx, cortex; LGE, lateral ganglionic eminence; MGE, medial ganglionic eminence; MZ, marginal zone; CP, cortical plate; SP, subplate; SVZ, subventricular zone; VZ, ventricular zone.
Our results suggest that mutations in WDR62 cause microcephaly, simplified gyral pattern, callosal abnormalities, and a wide range of additional cortical abnormalities, including polymicrogyria, schizencephaly, and subcortical heterotopia. WDR62 lies within the MCPH2 locus24–26 a gene-rich locus that has resisted gene identification for > 10 years, and appears to be allelic to MCPH2 (see accompanying paper). The remarkable diversity of cortical malformations associated with WDR62 mutation suggests that it acts at a critical hub of human cerebral development.
Methods
Human studies
All human studies were reviewed and approved by the IRB of Children’s Hospital, Boston, Beth Israel Deaconess Medical Center, and local institutions.
Genome-wide linkage scans
Family LIS-900 underwent a microsatellite genome-wide linkage screen at the Center for Inherited Disease Research (CIDR) using ~400 markers with a 10 cM average density. Genome-wide screens for Family LIS-2600 were performed at Children’s Hospital Boston using ~400 markers in the ABI linkage mapping set MD v2.5 at ~10 cM average density (Applied Biosystems). In addition, families LIS-900, LIS-2600, LIS-2500, MC-1400 and MC-1600 were genotyped on Affymetrix 5.0 SNP chips at the Broad Institute.
Fine mapping was done using polymorphic microsatellite markers from the ABI linkage mapping set HD v2.5 at 5cM average density (Applied Biosystems) along with additional microsatellite markers identified using the UCSC Human Genome Browser. Single and multipoint lod scores were calculated using Allegro, assuming a recessive mode of disease inheritance, full penetrance, and a disease allele frequency of 0.0001. Nucleotide numbers are provided in reference to genomic coordinates (human genome build hg18), coding DNA coordinates (based on RefSeq mRNA NM_001083961.1, where A of the ATG translational start site is designated bp 1 and positioning within an intron are designated relative to the most 3’ base of the nearest upstream coding exon), or protein coordinates (based UniProt accession number O43379), following HGVS guidelines27.
Microarray sequence capture and sequencing
After flanking recombinant markers defined the candidate interval, a tiled microarray (Roche Nimblegen) was designed to target all exons of the 148 genes in the region, as well as most of the nonrepetitive intronic DNA for the 85 genes in the center of the linkage peak. A total of 3.5 Mb of the ~7.5 Mb region was targeted. From each patient, 20ug of genomic DNA was randomly fragmented to 350–400bp, and the ends were repaired and ligated to Nimblegen gSel adaptors. This library was hybridized to the custom array using the manufacturer’s recommended conditions, and nonbound DNA was washed off. Bound DNA was eluted and subjected to PCR amplification. QPCR demonstrated 355X target enrichment for LIS-903 and 1346X for LIS-2601. Nimblegen-captured DNA fragments were then blunted using T4 DNA Polymerase and Klenow fragment using standard conditions, phosphorylated using T4 DNA kinase, and ligated to form concatamers. The concatamers were then randomly fragmented using a Covaris S2 device, and ends were repaired and ligated to Ilumina paired end sequencing adaptors. Ligated products were gel-purified, and PCR-amplified to enrich for adapter-containing fragments. Finally, the purified DNA was subjected to clustering and 40 cycles of paired-end sequencing using the Illumina GA II. Sequencing and subsequent image processing and basecalling were performed per the manufacturer's recommended conditions.
DNA sequence analysis
High-throughput sequence analysis was performed according to a customized bioinformatic pipeline for tracking sequence data, aligning reads, calculating coverage, calling variants, annotating variants with respect to functional effect, filtering out benign variation, and flagging candidate rare, pathogenic mutations (available on request). Briefly, BWA version 0.5.73 was employed to align short reads to the human genome (reference build hg18). Consensus and variant basecalls were made with SAMtools version 0.1.7, filtered for quality and mapping confidence, and loaded into a MySQL database for storage and further processing, including annotation of the predicted consequences (noncoding, coding synonymous, coding nonsynonymous, frameshift, splice site) of each variant using GMCC28. Candidate mutations were identified by starting with a list of all variants from the targeted homozygous region, removing those present either in dbSNP130 or a pilot 1 release of the 1000G database, and selecting for coding nonsynonymous, frameshift, or splice site changes. Sequence data were visualized using either the UCSC genome browser29 or the Broad Institute Integrated Genome Viewer. All genomic base positions are presented in reference to human genome NCBI build 36 (hg18).
Sequencing of WDR62 in controls
Coding WDR62 exons and at least 50 basepairs of flanking sequence were PCR amplified and submitted for Sanger capillary electrophoresis (Polymorphic DNA Technologies) in accordance with standard methods. Samples included 384 neurologically normal control samples (Coriell) and 124 unaffected individuals from Middle Eastern families with unrelated disorders.
Evidence for four unvalidated WDR62 variants deposited in dbSNP was also reviewed: rs34840924, rs35055110, rs35938064, and rs34734597 are single basepair insertions in WDR62 coding regions that would be predicted to cause frameshifts, but were unvalidated SNPs ascertained from a single study of five individuals (Submitted batch ID xplore_hum_chr19_2, Submitter Population ID Celera_Donors). Sequencing in 508 controls failed to confirm the existence of these variants.
Probe preparation
Plasmid templates for riboprobe synthesis were generated by restriction digestion of mouse WDR62 IMAGE cDNA clone 6405269 to contain a 574 bp sequence corresponding to nucleotide position 4227–4801 of the cDNA. Digoxigenin-labeled sense and antisense RNA probes were synthesized using the DIG RNA Labeling kit (Roche Molecular Biochemicals).
in situ hybridization
In situ hybridization was performed as described30. Sections were hybridized at 65 °C for 12–16 hours in 1–2 µg/ml of riboprobe and then washed. After blocking for 1 h at room temperature, sections were labeled with anti-DIG Fab-AP antibody (1:2000 dilution, Roche) for 2 h at room temperature and then washed and developed in NBT (nitroblue-tetrazolium-chloride) plus BCIP (5-bromo-4-chloro-indolyl-phosphate) at room temperature until ideal intensity.
Immunocytochemistry
HeLa cells were fixed with ice-cold methanol and placed at −20 °C for 5 minutes. The cells were blocked in 1X TBS + 0.15% Tween containing 5% normal donkey serum (v/v) for one hour at room temperature. Cells were then incubated for 3 hours at room temperature in primary antibody diluted in blocking solution. For primary antibodies, we used mouse monoclonal anti-gamma-tubulin (Abcam, ab27074, 1:2000), rabbit polyclonal anti-WDR62 (Bethyl laboratories, A301-560A, 1:500), mouse monoclonal anti-GM130 (BD Transduction Laboratories, 610822), mouse monoclonal anti-dynein heavy chain (Abcam, ab6305, 1:50), rat monoclonal anti-HA (Roche, 3F10, 1:500), rabbit anti-CEP170 (Abcam, ab72505, 1:100), rabbit polyclonal anti-LIS1 (Bethyl laboratories, A300-409A, 1:200). Cells were then washed three times with PBS and incubated with appropriate secondary antibodies against the appropriate species (DyLight, Jackson Labs, 1:200) in blocking solution for 1 hour at room temperature. The cells were then washed twice with blocking solution and incubated with Hoechst 33342 (1:2000, Invitrogen) for 5 minutes at room temperature. Cells were then washed twice with 1X PBS, mounted with Fluoromount-GT, and left at 4 °C overnight to dry. For ICC requiring the use of primary antibodies raised in the same host species, cells were fixed and blocked as normal and then incubated with the first primary antibody for three hours. Cells were then washed three times with blocking solution and incubated with Cy3-conjugated Fab fragments of anti-rabbit IgG (Jackson ImmunoResearch Laboratories) for secondary detection. Cells were then washed four times with blocking solution and incubated with unconjugated Fab fragments of anti-rabbit IgG for two hours. After washing the cells four times with blocking solution, the second primary antibody was applied and detected using the previously described protocol.
Recombinant HA-WDR62
A full-length human WDR62 cDNA (IMAGE Consortium clone 4510905) was obtained and used for site-directed mutagenesis (Quikchange, Agilent) to introduce the HA epitope immediately upstream of the stop codon. WDR62-HA was cloned downstream of the CMV promoter, transfected into HEK cells with lipofectamine, and processed for immunocytochemistry at 24 hours.
Supplementary Material
Acknowledgements
We thank the many families and researchers who participated in this study, Bernard Chang and Annapurna Poduri for expert review of MRI imaging findings, Sofia Lizarraga for contributions to genetic mapping, Megumi Aita and the In Situ Hybridization Core Facility, University of North Carolina Neuroscience Center, Chapel Hill, North Carolina for performing in situ hybridizations, C. G. Woods for helpful discussions and for sharing data prior to publication, Prof. Dr. Andreas von Deimling from the Institut für Pathologie, Heidelberg, for sectioning of the LIS-2601 brain, Adeline Nicholas and Maryam Khurshid for advice regarding immunohistochemistry, Anthony Hill and Lihong Bu at the MRDDRC (Mental Retardation Developmental Disability Research Center) Imaging Core for assistance with confocal microscopy (Grant: NIH-P30-HD-18655 and Fidelity Foundation), and R. Sean Hill for assistance with linkage analysis and helpful discussions. We are indebted to the members of the Microcephaly Collaborative (MC, Supplementary Table 1) for contributing to the cohort from which these families were drawn. TWY was supported by a NIH T32 grant (T32 NS007484-08), the Clinical Investigator Training Program (CITP) at Harvard-MIT Health Sciences and Technology and Beth Israel Deaconess Medical Center in collaboration with Pfizer, Inc. and Merck and Company, Inc., and the Nancy Lurie Marks Junior Faculty MeRIT Fellowship. GHM was supported by the Young Investigator Award of NARSAD as a NARSAD Lieber Investigator. Research was supported by grants from the NINDS (RO1 NSR01-35129) and the Fogarty International Center (R21 NS061772) to CAW, the Dubai Harvard Foundation for Medical Research, and the Manton Center for Orphan Disease Research. Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C and NIH N01-HG-65403. Genotyping at Children’s Hospital Boston is supported by the Intellectual and Developmental Disabilities Research Centers (CHB DDRC, P30 HD18655). Genotyping at the Broad Institute is supported by NHGRI. CAW is an Investigator of the Howard Hughes Medical Institute.
Footnotes
- Human WDR62 mRNA: RefSeq NM_001083961.1
- Human WDR62 Protein: UniProt accession number O43379
- Mouse WDR62 mRNA: IMAGE clone 6405269, GenBank accession BC054747
Author Contributions
TWY helped characterize MCSG syndrome, designed and performed targeted capture experiments, generated sequencing libraries, designed bioinformatics pipelines, analyzed sequencing data, identified WDR62 mutations, designed WDR62 in situ expression studies, helped design and analyze WDR62 immunohistochemistry studies, helped analyze the LIS-2601 postmortem specimen, and wrote the manuscript. GHM helped characterize MCSG syndrome, performed initial genome wide linkage studies, and identified the MCSG locus. DJT designed and performed WDR62 immunohistochemistry studies, helped analyze in situ expression studies, and helped analyze the LIS-2601 postmortem specimen. SKS helped characterize the MCSG syndrome, identified additional families, analyzed SNP and microsatellite genotyping, and narrowed the MCSG interval. LF-S helped characterize MCSG syndrome and wrote the initial clinical description of the LIS-900 family. CMS identified the LIS-2600 family and provided clinical information and the LIS-2601 postmortem specimen. MT identified the MC-1400 and MC-1600 families and provided clinical information. MTM identified the PH-16900 family and provided clinical information. BJB organized clinical information and patient samples. JF sequenced WDR62 in families to confirm high throughput sequencing findings and discover additional alleles. CS helped with bioinformatic pipelines, and generated constructs for WDR62 in situ expression studies. WBD referred the PH-16900 family. RDF analyzed the LIS-2601 postmortem specimen. AJB reviewed MRIs for characterization of the MCSG syndrome. CAW directed the overall research, helped analyze the LIS-2601 postmortem specimen, and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Electronic Database Information
References
- 1.Sergi C, Zoubaa S, Schiesser M. Norman-Roberts syndrome: prenatal diagnosis and autopsy findings. Prenat Diagn. 2000;20:505–509. [PubMed] [Google Scholar]
- 2.Albert TJ, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Durbin R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desmet F-O, et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutchins JRA, et al. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 2010;328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bond J, Woods CG. Cytoskeletal genes regulating brain size. Curr. Opin. Cell Biol. 2006;18:95–101. doi: 10.1016/j.ceb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Rauch A, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 10.Bond J, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nature genetics. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 11.Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 12.Lizarraga SB, et al. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development. 2010;137:1907–1917. doi: 10.1242/dev.040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouprina N, et al. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum Mol Genet. 2005;14:2155–2165. doi: 10.1093/hmg/ddi220. [DOI] [PubMed] [Google Scholar]
- 14.Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. 2009;25:501–510. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wasserman T, et al. A novel c-Jun N-terminal kinase (JNK)-binding protein WDR62 is recruited to stress granules and mediates a nonclassical JNK activation. Mol Biol Cell. 2010;21:117–130. doi: 10.1091/mbc.E09-06-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders RD, Avides MC, Howard T, Gonzalez C, Glover DM. The Drosophila gene abnormal spindle encodes a novel microtubule-associated protein that associates with the polar regions of the mitotic spindle. J Cell Biol. 1997;137:881–890. doi: 10.1083/jcb.137.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Voet M, et al. NuMA-related LIN-5, ASPM-1, calmodulin and dynein promote meiotic spindle rotation independently of cortical LIN-5/GPR/Galpha. Nat Cell Biol. 2009;11:269–277. doi: 10.1038/ncb1834. [DOI] [PubMed] [Google Scholar]
- 18.Faulkner NE, et al. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 19.Bond J, et al. ASPM is a major determinant of cerebral cortical size. Nature Genetics. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 20.Bilguvar K, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010 doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen DV, Lui JH, Parker PRL, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- 22.Gambello MJ, et al. Multiple dose-dependent effects of Lis1 on cerebral cortical development. J Neurosci. 2003;23:1719–1729. doi: 10.1523/JNEUROSCI.23-05-01719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheen VL, et al. Impaired proliferation and migration in human Miller-Dieker neural precursors. Ann. Neurol. 2006;60:137–144. doi: 10.1002/ana.20843. [DOI] [PubMed] [Google Scholar]
- 24.Roberts E, et al. Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 2002;39:718–721. doi: 10.1136/jmg.39.10.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gul A, et al. Genetic studies of autosomal recessive primary microcephaly in 33 Pakistani families: Novel sequence variants in ASPM gene. neurogenetics. 2006;7:105–110. doi: 10.1007/s10048-006-0042-4. [DOI] [PubMed] [Google Scholar]
- 26.Roberts E, et al. The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur J Hum Genet. 1999;7:815–820. doi: 10.1038/sj.ejhg.5200385. [DOI] [PubMed] [Google Scholar]
- 27.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 28.Major JE. Genomic mutation consequence calculator. Bioinformatics. 2007;23:3091–3092. doi: 10.1093/bioinformatics/btm339. [DOI] [PubMed] [Google Scholar]
- 29.Kent WJ, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucker E, et al. Molecular Specification and Patterning of Progenitor Cells in the Lateral and Medial Ganglionic Eminences. Journal of Neuroscience. 2008;28:9504. doi: 10.1523/JNEUROSCI.2341-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.