

Abstract
The title compound, C4H3BrO4, was obtained from a solution of meso-2,3-dibromosuccinic acid and vanadium(IV) oxide. The crystals are isostructural with chloromaleic acid and the molecule has two geometrically different carboxyl groups, one of which has delocalized C—O bonds and is essentially coplanar with the olefinic bond plane [give dihedral angle 15.08 (16)°], whereas the other has a localized C=O bond and forms a dihedral angle of 99.6 (3)° with the C=C bond plane. Two symmetry-independent O—H⋯O hydrogen bonds link the molecules into layers parallel to the bc plane.
Related literature
For the structure of chloromaleic acid, see: Wong et al. (2006 ▶). For the synthesis and structure of 2-bromofumaric acid, see: Fischer (2006 ▶). For the structure and polymorphism of maleic acid, see: Day et al. (2006 ▶). For the structure of 2-methylmaleic acid, see: Batchelor & Jones (1998 ▶).
Experimental
Crystal data
C4H3BrO4
M r = 194.97
Monoclinic,

a = 7.5074 (12) Å
b = 4.9272 (6) Å
c = 16.966 (4) Å
β = 94.213 (12)°
V = 625.9 (2) Å3
Z = 4
Mo Kα radiation
μ = 6.50 mm−1
T = 299 K
0.15 × 0.13 × 0.10 mm
Data collection
Bruker–Nonius KappaCCD diffractometer
Absorption correction: numerical HABITUS (Herrendorf & Bärnighausen, 1997 ▶) T min = 0.325, T max = 0.455
8638 measured reflections
1420 independent reflections
1046 reflections with I > 2σ(I)
R int = 0.064
Refinement
R[F 2 > 2σ(F 2)] = 0.042
wR(F 2) = 0.075
S = 1.16
1420 reflections
84 parameters
H-atom parameters constrained
Δρmax = 0.63 e Å−3
Δρmin = −0.43 e Å−3
Data collection: COLLECT (Nonius, 1999 ▶); cell refinement: DIRAX (Duisenberg, 1992 ▶); data reduction: EVALCCD (Duisenberg et al., 2003 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg, 1999 ▶); software used to prepare material for publication: publCIF (Westrip, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809029602/ya2098sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809029602/ya2098Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O3—H3⋯O4i | 0.82 | 1.80 | 2.617 (4) | 171 |
| O2—H2⋯O1ii | 0.82 | 1.86 | 2.681 (4) | 176 |
Symmetry codes: (i) 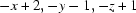 ; (ii)
; (ii)  .
.
Acknowledgments
The Swedish Research Council (VR) is acknowleged for providing funding for the single-crystal diffractometer.
supplementary crystallographic information
Comment
During the ongoing investigation of the bromo-substituted dicarboxylic acids with four carbon atoms, our group tried to prepare and characterize pure acids as well as their metal salts. One of these syntheses involved the reaction of vanadium(IV) oxide with meso-dibromosuccinic acid. It had been shown earlier (Fischer, 2006), that hydrogen bromide can easily be eliminated from racemic 2,3-dibromosuccinic acid, yielding 2-bromofumaric acid. Elimination of hydrogen bromide from meso-2,3-dibromosuccinic acid does not occur as easily. However in the presence of strong bases and at elevated temperature, elimination is observed; it yields 2-bromomaleic acid, whose structure is described here.
While most of bond lengths and angles in the molecule of 2-bromomaleic acid (Fig. 1) are close to those found in 2-bromofumaric acid (Fischer, 2006) and unsubstituted maleic acid (Day et al., 2006), the title compound, in contrast to the aforementioned molecules, is essentially non-planar. In fact, two carboxylic groups in 2-bromomaleic acid form dihedral angle of 77.6 (3)° with each other and only one of them (O3–C4–O4) is almost coplanar with the C2=C3 double bond plane, whereas the second one (O1–C1–O2) forms with the latter plane dihedral angle of 99.6 (3)° The carboxylic group, which is almost coplanar with the olefinic bond, shows much higher degree of delocalization (C4–O3 1.269 (4) and C4–O4 1.254 (5) Å), than the second carboxylic group, bonded to the bromo-substituted carbon atom (C1–O1 1.197 (4) and C1–O2 1.299 (5) °). It is noteworthy that similar geometrical peculiarities were observed in other known structures of monosubstituted maleic acid derivatives, namely in 2-chloromaleic acid (Wong et al., 2006), isostructural with the title compound, and 2-methylmaleic acid (Batchelor & Jones, 1998).
There are two symmetry independent O—H..O bonds (Table 1), one of which involves delocalized carboxyl group and is responsible for formation of dimeric centrosymmetric motives traditional to carboxylic acid crystal structures. Another H-bond involves non-symmetric carboxylic group and further links dimeric aggregates into layers parallel to the bc-plane (Fig. 2).
Experimental
89 mg of VO2 (AlfaAesar, 99%), was added to a solution of 270 mg of meso-dibromosuccinic acid (Sigma Aldrich, 98%) in 4.2 ml of demineralized water. Upon heating to 90°C, vanadium oxide got dissolved, yielding a dark-blue solution, which was put aside for evaporation. Within a week, colourless crystals of the title compound were obtained.
Refinement
H atoms could be located in the Fourier map, however, their isotropic refinement did not yield satisfactory X–H distances. Therefore, H atoms were placed at calculated positions with d(C–H)=0.93 Å, d(O–H)=0.82 Å and included in the subsequent refinement in riding motion approximation with Uiso=1.2Ueq of the carrier atom (1.5 Ueq for hydroxyl H atoms).
Figures
Fig. 1.

Molecular structure of the title compound. Displacement ellipsoids are drawn at the 50% probability level; H atoms are drawn as small circles of arbitrary radius.
Fig. 2.

Crystal packing of the title compound viewed down the a axis. Hydrogen bonds are drawn as dashed lines.
Crystal data
| C4H3BrO4 | F(000) = 376 |
| Mr = 194.97 | Dx = 2.069 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 28 reflections |
| a = 7.5074 (12) Å | θ = 5.6–19.2° |
| b = 4.9272 (6) Å | µ = 6.50 mm−1 |
| c = 16.966 (4) Å | T = 299 K |
| β = 94.213 (12)° | Block, colourless |
| V = 625.9 (2) Å3 | 0.15 × 0.13 × 0.10 mm |
| Z = 4 |
Data collection
| Bruker–Nonius KappaCCD diffractometer | 1046 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.064 |
| φ and ω scans | θmax = 27.5°, θmin = 4.7° |
| Absorption correction: numerical HABITUS (Herrendorf & Bärnighausen, 1997) | h = −9→9 |
| Tmin = 0.325, Tmax = 0.455 | k = −6→6 |
| 8638 measured reflections | l = −17→22 |
| 1420 independent reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | H-atom parameters constrained |
| wR(F2) = 0.075 | w = 1/[σ2(Fo2) + (0.0113P)2 + 1.1107P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.16 | (Δ/σ)max = 0.001 |
| 1420 reflections | Δρmax = 0.63 e Å−3 |
| 84 parameters | Δρmin = −0.43 e Å−3 |
| 0 restraints |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.7961 (5) | 0.0222 (7) | 0.6839 (2) | 0.0317 (8) | |
| C2 | 0.6344 (4) | −0.0621 (8) | 0.6312 (2) | 0.0330 (8) | |
| C3 | 0.6332 (5) | −0.2221 (8) | 0.5700 (2) | 0.0369 (9) | |
| C4 | 0.7955 (5) | −0.3400 (8) | 0.5399 (2) | 0.0369 (9) | |
| Br1 | 0.42183 (5) | 0.08537 (10) | 0.66527 (3) | 0.05260 (18) | |
| O1 | 0.8584 (3) | −0.1231 (6) | 0.73530 (16) | 0.0440 (7) | |
| O2 | 0.8501 (4) | 0.2664 (6) | 0.66922 (17) | 0.0493 (8) | |
| O3 | 0.9456 (3) | −0.2426 (6) | 0.56482 (17) | 0.0466 (7) | |
| O4 | 0.7746 (4) | −0.5294 (6) | 0.49066 (17) | 0.0482 (8) | |
| H3A | 0.5236 | −0.2646 | 0.5439 | 0.044* | |
| H2 | 0.9357 | 0.3049 | 0.7000 | 0.074* | |
| H3 | 1.0256 | −0.3258 | 0.5449 | 0.070* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0278 (18) | 0.031 (2) | 0.036 (2) | 0.0046 (15) | −0.0024 (15) | −0.0024 (17) |
| C2 | 0.0275 (17) | 0.031 (2) | 0.039 (2) | 0.0043 (15) | −0.0055 (14) | 0.0019 (19) |
| C3 | 0.0288 (19) | 0.035 (2) | 0.045 (2) | −0.0016 (16) | −0.0066 (16) | −0.003 (2) |
| C4 | 0.0306 (19) | 0.040 (2) | 0.039 (2) | −0.0025 (16) | −0.0063 (16) | 0.0006 (19) |
| Br1 | 0.0335 (2) | 0.0616 (3) | 0.0615 (3) | 0.0137 (2) | −0.00437 (17) | −0.0122 (3) |
| O1 | 0.0380 (14) | 0.0415 (17) | 0.0498 (17) | −0.0045 (12) | −0.0160 (12) | 0.0119 (14) |
| O2 | 0.0504 (17) | 0.0353 (17) | 0.0578 (19) | −0.0113 (13) | −0.0256 (14) | 0.0076 (15) |
| O3 | 0.0332 (14) | 0.0487 (18) | 0.0578 (19) | −0.0067 (13) | 0.0038 (13) | −0.0141 (16) |
| O4 | 0.0408 (15) | 0.055 (2) | 0.0476 (17) | 0.0022 (13) | −0.0058 (12) | −0.0205 (15) |
Geometric parameters (Å, °)
| C1—O1 | 1.197 (4) | C4—O4 | 1.254 (5) |
| C1—O2 | 1.299 (5) | C4—O3 | 1.269 (4) |
| C1—C2 | 1.512 (5) | C3—H3A | 0.9300 |
| C2—C3 | 1.302 (5) | O2—H2 | 0.8200 |
| C2—Br1 | 1.883 (4) | O3—H3 | 0.8200 |
| C3—C4 | 1.475 (5) | ||
| O1—C1—O2 | 125.6 (3) | O4—C4—O3 | 124.6 (4) |
| O1—C1—C2 | 121.3 (3) | O4—C4—C3 | 117.3 (3) |
| O2—C1—C2 | 112.9 (3) | O3—C4—C3 | 118.1 (4) |
| C3—C2—C1 | 126.5 (3) | C2—C3—H3A | 118.1 |
| C3—C2—Br1 | 121.5 (3) | C4—C3—H3A | 118.1 |
| C1—C2—Br1 | 112.0 (3) | C1—O2—H2 | 109.5 |
| C2—C3—C4 | 123.9 (3) | C4—O3—H3 | 109.5 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O4i | 0.82 | 1.80 | 2.617 (4) | 171 |
| O2—H2···O1ii | 0.82 | 1.86 | 2.681 (4) | 176 |
Symmetry codes: (i) −x+2, −y−1, −z+1; (ii) −x+2, y+1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: YA2098).
References
- Batchelor, E. & Jones, W. (1998). Acta Cryst. C54, 238–240.
- Brandenburg, K. (1999). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Day, G. M., Trask, A. V., Motherwell, W. D. S. & Jones, W. (2006). Chem. Commun. pp. 54-56. [DOI] [PubMed]
- Duisenberg, A. J. M. (1992). J. Appl. Cryst.25, 92–96.
- Duisenberg, A. J. M., Kroon-Batenburg, L. M. J. & Schreurs, A. M. M. (2003). J. Appl. Cryst.36, 220–229.
- Fischer, A. (2006). Acta Cryst. E62, o4190–o4191.
- Herrendorf, W. & Bärnighausen, H. (1997). HABITUS University of Karlsruhe, Germany.
- Nonius (1999). COLLECT Nonius BV, Delft, The Netherlands.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Westrip, S. P. (2009). publCIF. In preparation.
- Wong, A., Pike, K. J., Jenkins, R., Clarkson, G. J., Anupõld, T., Howes, A. P., Crout, D. H. G., Samoson, A., Dupree, R. & Smith, M. E. (2006). J. Phys. Chem. A, 110, 1824–1835. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536809029602/ya2098sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809029602/ya2098Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report