

Abstract
In the structure of the title compound, [Ag(C12H6N2O2)2]BF4 or [AgL 2]BF4 (L = phendione), the Ag and B atoms are located on twofold rotation axes. The dihedral angle between the two phendione ligands is 36.7 (2)°. The coordination about the AgI center is distorted tetrahedral (τ4 = 0.546). The crystal structure is consolidated by weak C—H⋯O(phendione) and C—H⋯F(BF4 −) interactions. The BF4 − counter-anion is strongly disordered and was modelled with two sets of idealized F atoms.
Related literature
For the different coordination properties of phendione, see: Calderazzo et al. (1999 ▶, 2002 ▶); Calucci et al. (2006 ▶); Galet et al. (2005 ▶); Lei et al. (1996 ▶); Okamura et al. (2006 ▶). For examples with phendione ligands where N and O donors are used simultaneously, see: Fox et al. (1991 ▶); Shavaleev et al. (2003a ▶,b
▶); Ruiz et al. (1999 ▶); Paw & Eisenberg (1997 ▶). Similar structures containing Ag have also been reported by Onuegbu et al. (2007 ▶). For background to phendione chemistry, see: Udeochu et al. (2007 ▶); Onuegbu et al. (2007 ▶). For reference structural data, see: Allen (2002 ▶); Leschke et al. (2002 ▶); Paramonov et al. (2003 ▶); Pallenberg et al. (1997 ▶); Titze et al. (1997 ▶). Details of the τ4 parameter were given by Yang et al. (2007 ▶).
Experimental
Crystal data
[Ag(C12H6N2O2)2]BF4
M r = 615.06
Monoclinic,
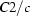
a = 13.2249 (6) Å
b = 12.0115 (17) Å
c = 14.4338 (7) Å
β = 108.481 (5)°
V = 2174.6 (3) Å3
Z = 4
Mo Kα radiation
μ = 1.01 mm−1
T = 200 K
0.44 × 0.37 × 0.28 mm
Data collection
Oxford Diffraction Gemini R diffractometer
Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2007 ▶) T min = 0.856, T max = 1.000 (expected range = 0.646–0.755)
11815 measured reflections
3647 independent reflections
2306 reflections with I > 2σ(I)
R int = 0.024
Refinement
R[F 2 > 2σ(F 2)] = 0.047
wR(F 2) = 0.134
S = 0.98
3647 reflections
204 parameters
32 restraints
H-atom parameters constrained
Δρmax = 1.81 e Å−3
Δρmin = −1.39 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2007 ▶); cell refinement: CrysAlis RED (Oxford Diffraction, 2007 ▶); data reduction: CrysAlis RED; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053680903222X/wm2249sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680903222X/wm2249Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected bond lengths (Å).
| Ag—N1 | 2.356 (2) |
| Ag—N2 | 2.357 (2) |
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C3—H3A⋯O1i | 0.95 | 2.51 | 3.347 (4) | 147 |
| C1—H1A⋯F1Aii | 0.95 | 2.35 | 3.083 (6) | 133 |
| C2—H2A⋯F1B | 0.95 | 2.17 | 2.803 (8) | 123 |
| C10—H10A⋯F2Aiii | 0.95 | 2.24 | 2.859 (5) | 122 |
| C10—H10A⋯F2Biv | 0.95 | 2.28 | 3.065 (4) | 140 |
Symmetry codes: (i) 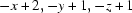 ; (ii)
; (ii) 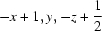 ; (iii)
; (iii) 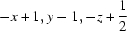 ; (iv)
; (iv) 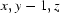 .
.
Acknowledgments
RJB acknowledges the NSF–MRI program (grant No. CHE-0619278) for funds to purchase the X-ray diffractometer.
supplementary crystallographic information
Comment
Phendione (1,10-phenanthroline-5,6-dione) is an excellent ligand that incorporates two functional groups with different coordination properties (Calderazzo et al., 1999, 2002; Calucci et al., 2006; Galet et al., 2005; Lei et al., 1996; Okamura et al., 2006). This well known ligand possesses both the α-diimine and orthoquinone moieties. While phendione usually binds to metals through its imine N atoms, in some cases both the N and O donors are used simultaneously (Fox et al., 1991; Shavaleev et al., 2003a, b; Ruiz et al., 1999; Paw & Eisenberg, 1997). The crystal structures of two complexes of copper and phendione have been determined (Galet et al., 2005;). Similar structures containing Ag have also been reported (Onuegbu et al., 2007). In this paper, as part of our study of phendione chemistry (Udeochu et al., 2007; Onuegbu et al., 2007), we report the synthesis and characterization of the title compound, [AgL2]+(BF4)-, (I).
The structure of (I), shown in Figure 1, is made up of an [Ag(L)2]+ cation and a tetrafluoridoborate anion. The silver atom is coordinated to the two nitrogen atoms of both phendione ligands. Both the Ag atom of the cation and the B atom of the anion lie on a crystallographic twofold rotation axes. The C═O bond lengths in the phendione ligands (1.210 (4) and 1.206 (3) Å) are comparable to those values found in other such complexes (Allen, 2002; Onuegbu et al., 2007). The Ag—N bond lengths (2.356 (2) and 2.357 (2) Å) are similar to those found in related phenanthroline derivatives of silver (Leschke et al., 2002; Paramonov et al., 2003; Pallenberg et al., 1997; Titze et al., 1997).
In (I), the silver cation is in a distorted tetrahedral environment. This is best illustrated by the dihedral angle between the planes of the coordinated ligands which in this case the angle is 36.7 (2)°. This compares with values of 36.8 (2)° found in the analogous perchlorate analog and the values of 32.4° and 70.5° found for other structurally characterized Ag complexes containing the bis(1,10-phenanthroline) core. Another recent parameter for 4-coordinate complexes (τ4, Yang et al., 2007) has been developed to place a structure on the continuum between square planar (0) and tetrahedral (1). For the present structure this value is 0.546.
Copper forms a similar complex with phendione. However, in this case the twofold axis passes between the phendione ligands with a dihedral angle of 44.5° between them.
In the structure of (I), there are weak C—H···O(phendione) and C—H···F(BF4-) interactions (Fig. 2).
Experimental
A flask containing 1,10-phenanthroline hydrate (1.00 g, 5.04 mmol) and potassium bromide (5.95 g, 50.0 mmol) was placed in an ice bath. Concentrated sulfuric acid (20 cm3) was added in small portions, followed by drop-wise addition of concentrated nitric acid (10 cm3). The resulting solution was heated for 2 h at 253–257 K and cooled to room temperature. The solution was then poured into 400 cm3 of water and neutralized with sodium bicarbonate, after which the phendione was extracted with dichloromethane, and recrystallized using a methanol-water mixture.
The title compound was synthesized in an atmosphere saturated with N2. To a solution of tetrakis(acetonitrile)silver(I)tetrafluoridoborate (0.0843 g) in 10 ml of CH3CN, was added drop-wise a solution (10 ml) of CH3CN containing 0.0492 g of phendione. The final yellowish solution was filtered and allowed to slowly evaporate yielding reddish brown crystals of the title compound suitable for X-ray studies.
Refinement
H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms with C—H distances of 0.95 Å and Uiso(H) = 1.2Ueq(C). The tetrafluoridoborate anion is disordered. Two sets of F atoms, constrained to meet the criteria for idealized tetrahedra, were used with occupancy factors of 0.406 (4) and 0.096 (4). The temperature factors for the major component were refined anisotropically and constrained by use of the SIMU and DELU instructions in SHELXL97 (Sheldrick, 2008). In the final difference Fourier there were positive and negative holes of +1.807 and -1.393 eA-3 near the disordered F atoms.
Figures
Fig. 1.

View of (I), [Ag(L)2]+(BF4)-, showing the atom-labelling scheme. The C—H···F interaction is shown as a dashed line. Unlabeled atoms are related by -x, y, 1/2 - z. Displacement ellipsoids are drawn at the 20% probability level. Only the major component of the disordered anion is shown.
Fig. 2.

The molecular packing of (I) viewed approximately along the b axis. Dotted lines indicate the C—H···O and C—H···F interactions. Only the major component of the disordered anion is shown.
Crystal data
| [Ag(C12H6N2O2)2]BF4 | F(000) = 1216 |
| Mr = 615.06 | Dx = 1.879 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 5444 reflections |
| a = 13.2249 (6) Å | θ = 4.6–32.4° |
| b = 12.0115 (17) Å | µ = 1.00 mm−1 |
| c = 14.4338 (7) Å | T = 200 K |
| β = 108.481 (5)° | Prism, colorless |
| V = 2174.6 (3) Å3 | 0.44 × 0.37 × 0.28 mm |
| Z = 4 |
Data collection
| Oxford Diffraction Gemini R diffractometer | 3647 independent reflections |
| Radiation source: fine-focus sealed tube | 2306 reflections with I > 2σ(I) |
| graphite | Rint = 0.024 |
| Detector resolution: 10.5081 pixels mm-1 | θmax = 32.5°, θmin = 4.6° |
| φ and ω scans | h = −19→19 |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2007) | k = −17→17 |
| Tmin = 0.856, Tmax = 1.000 | l = −16→21 |
| 11815 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.047 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.134 | H-atom parameters constrained |
| S = 0.98 | w = 1/[σ2(Fo2) + (0.087P)2] where P = (Fo2 + 2Fc2)/3 |
| 3647 reflections | (Δ/σ)max < 0.001 |
| 204 parameters | Δρmax = 1.81 e Å−3 |
| 32 restraints | Δρmin = −1.39 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| Ag | 0.5000 | 0.21159 (3) | 0.2500 | 0.04327 (15) | |
| O1 | 1.01490 (18) | 0.3415 (2) | 0.5002 (2) | 0.0563 (7) | |
| O2 | 0.99985 (17) | 0.1301 (2) | 0.56180 (17) | 0.0507 (6) | |
| N1 | 0.65530 (18) | 0.3220 (2) | 0.29553 (17) | 0.0304 (5) | |
| N2 | 0.64367 (17) | 0.10772 (19) | 0.35180 (17) | 0.0284 (5) | |
| C1 | 0.6565 (2) | 0.4291 (3) | 0.2713 (2) | 0.0371 (6) | |
| H1A | 0.5941 | 0.4599 | 0.2261 | 0.044* | |
| C2 | 0.7443 (2) | 0.4976 (3) | 0.3086 (2) | 0.0413 (7) | |
| H2A | 0.7414 | 0.5742 | 0.2914 | 0.050* | |
| C3 | 0.8357 (2) | 0.4515 (3) | 0.3713 (2) | 0.0387 (7) | |
| H3A | 0.8980 | 0.4957 | 0.3966 | 0.046* | |
| C4 | 0.8364 (2) | 0.3402 (2) | 0.39734 (19) | 0.0294 (5) | |
| C5 | 0.9324 (2) | 0.2908 (2) | 0.4662 (2) | 0.0356 (6) | |
| C6 | 0.9259 (2) | 0.1693 (3) | 0.4981 (2) | 0.0329 (6) | |
| C7 | 0.82541 (19) | 0.1076 (2) | 0.45309 (18) | 0.0282 (5) | |
| C8 | 0.8176 (2) | −0.0024 (3) | 0.4776 (2) | 0.0373 (7) | |
| H8A | 0.8766 | −0.0393 | 0.5224 | 0.045* | |
| C9 | 0.7227 (2) | −0.0579 (3) | 0.4360 (2) | 0.0404 (7) | |
| H9A | 0.7159 | −0.1346 | 0.4489 | 0.048* | |
| C10 | 0.6377 (2) | 0.0010 (2) | 0.3753 (2) | 0.0370 (6) | |
| H10A | 0.5716 | −0.0365 | 0.3489 | 0.044* | |
| C12 | 0.7426 (2) | 0.2784 (2) | 0.35847 (19) | 0.0255 (5) | |
| C11 | 0.73725 (19) | 0.1602 (2) | 0.38786 (18) | 0.0249 (5) | |
| B | 0.5000 | 0.7040 (2) | 0.2500 | 0.060 (2) | |
| F1A | 0.4655 (3) | 0.6438 (4) | 0.3134 (3) | 0.0308 (13) | 0.406 (4) |
| F2A | 0.4411 (3) | 0.7977 (3) | 0.2246 (4) | 0.107 (3) | 0.406 (4) |
| F3A | 0.60395 (19) | 0.7324 (4) | 0.2940 (3) | 0.113 (3) | 0.406 (4) |
| F4A | 0.4924 (4) | 0.6425 (5) | 0.1696 (3) | 0.0439 (18) | 0.406 (4) |
| F1B | 0.5925 (6) | 0.6663 (3) | 0.2398 (12) | 0.053 (4)* | 0.094 (4) |
| F2B | 0.5000 | 0.8168 (3) | 0.2500 | 0.053 (4)* | 0.189 (8) |
| F3B | 0.4168 (8) | 0.6665 (3) | 0.1746 (7) | 0.053 (4)* | 0.094 (4) |
| F4B | 0.4905 (13) | 0.6663 (3) | 0.3353 (5) | 0.053 (4)* | 0.094 (4) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Ag | 0.02050 (16) | 0.0611 (3) | 0.0386 (2) | 0.000 | −0.00431 (12) | 0.000 |
| O1 | 0.0270 (11) | 0.0649 (16) | 0.0674 (17) | −0.0135 (11) | 0.0012 (11) | −0.0144 (14) |
| O2 | 0.0269 (10) | 0.0765 (16) | 0.0400 (12) | 0.0105 (10) | −0.0016 (9) | 0.0064 (12) |
| N1 | 0.0259 (11) | 0.0366 (12) | 0.0260 (11) | 0.0005 (9) | 0.0045 (9) | 0.0018 (9) |
| N2 | 0.0236 (10) | 0.0309 (12) | 0.0281 (11) | −0.0015 (8) | 0.0043 (8) | −0.0018 (9) |
| C1 | 0.0384 (14) | 0.0394 (16) | 0.0301 (14) | 0.0047 (12) | 0.0061 (12) | 0.0040 (12) |
| C2 | 0.0499 (17) | 0.0374 (16) | 0.0387 (17) | −0.0061 (14) | 0.0171 (14) | 0.0010 (13) |
| C3 | 0.0368 (14) | 0.0429 (17) | 0.0407 (16) | −0.0119 (12) | 0.0185 (13) | −0.0085 (13) |
| C4 | 0.0255 (12) | 0.0387 (15) | 0.0257 (12) | −0.0073 (11) | 0.0104 (10) | −0.0074 (11) |
| C5 | 0.0234 (12) | 0.0477 (17) | 0.0352 (15) | −0.0040 (11) | 0.0087 (11) | −0.0133 (13) |
| C6 | 0.0216 (11) | 0.0519 (16) | 0.0228 (12) | 0.0036 (11) | 0.0037 (9) | −0.0026 (12) |
| C7 | 0.0212 (11) | 0.0392 (15) | 0.0235 (12) | 0.0050 (10) | 0.0062 (9) | −0.0006 (10) |
| C8 | 0.0347 (14) | 0.0453 (17) | 0.0332 (15) | 0.0138 (12) | 0.0126 (12) | 0.0056 (12) |
| C9 | 0.0442 (16) | 0.0338 (15) | 0.0457 (18) | 0.0038 (13) | 0.0180 (14) | 0.0061 (13) |
| C10 | 0.0335 (13) | 0.0398 (16) | 0.0400 (16) | −0.0065 (12) | 0.0150 (12) | −0.0032 (12) |
| C12 | 0.0219 (11) | 0.0349 (14) | 0.0197 (11) | −0.0028 (9) | 0.0067 (9) | −0.0035 (10) |
| C11 | 0.0207 (10) | 0.0310 (13) | 0.0218 (11) | 0.0015 (10) | 0.0048 (8) | −0.0027 (10) |
| B | 0.110 (6) | 0.009 (2) | 0.085 (5) | 0.000 | 0.065 (5) | 0.000 |
| F1A | 0.032 (3) | 0.036 (3) | 0.022 (2) | −0.022 (2) | 0.006 (2) | −0.0135 (18) |
| F2A | 0.189 (9) | 0.027 (3) | 0.124 (7) | 0.049 (3) | 0.077 (6) | 0.031 (3) |
| F3A | 0.141 (7) | 0.044 (4) | 0.170 (8) | −0.033 (4) | 0.071 (6) | −0.051 (5) |
| F4A | 0.046 (4) | 0.051 (3) | 0.027 (3) | 0.023 (3) | 0.001 (2) | 0.003 (2) |
Geometric parameters (Å, °)
| Ag—N1 | 2.356 (2) | C5—C6 | 1.541 (5) |
| Ag—N1i | 2.356 (2) | C6—C7 | 1.480 (4) |
| Ag—N2i | 2.357 (2) | C7—C8 | 1.381 (4) |
| Ag—N2 | 2.357 (2) | C7—C11 | 1.396 (3) |
| O1—C5 | 1.210 (4) | C8—C9 | 1.379 (4) |
| O2—C6 | 1.206 (3) | C8—H8A | 0.9500 |
| N1—C12 | 1.329 (3) | C9—C10 | 1.380 (4) |
| N1—C1 | 1.335 (4) | C9—H9A | 0.9500 |
| N2—C10 | 1.335 (4) | C10—H10A | 0.9500 |
| N2—C11 | 1.338 (3) | C12—C11 | 1.489 (4) |
| C1—C2 | 1.385 (4) | B—F1A | 1.354 (2) |
| C1—H1A | 0.9500 | B—F2A | 1.352 (2) |
| C2—C3 | 1.377 (4) | B—F3A | 1.362 (2) |
| C2—H2A | 0.9500 | B—F4A | 1.352 (2) |
| C3—C4 | 1.389 (4) | B—F1B | 1.355 (2) |
| C3—H3A | 0.9500 | B—F2B | 1.355 (2) |
| C4—C12 | 1.402 (4) | B—F3B | 1.356 (2) |
| C4—C5 | 1.466 (4) | B—F4B | 1.355 (2) |
| N1—Ag—N1i | 111.52 (12) | C8—C7—C11 | 119.5 (2) |
| N1—Ag—N2i | 158.56 (8) | C8—C7—C6 | 119.8 (3) |
| N1i—Ag—N2i | 70.42 (8) | C11—C7—C6 | 120.7 (2) |
| N1—Ag—N2 | 70.42 (8) | C9—C8—C7 | 118.8 (3) |
| N1i—Ag—N2 | 158.56 (8) | C9—C8—H8A | 120.6 |
| N2i—Ag—N2 | 116.08 (11) | C7—C8—H8A | 120.6 |
| C12—N1—C1 | 118.6 (2) | C8—C9—C10 | 118.3 (3) |
| C12—N1—Ag | 117.40 (18) | C8—C9—H9A | 120.8 |
| C1—N1—Ag | 123.46 (19) | C10—C9—H9A | 120.8 |
| C10—N2—C11 | 118.4 (2) | N2—C10—C9 | 123.5 (3) |
| C10—N2—Ag | 124.49 (19) | N2—C10—H10A | 118.3 |
| C11—N2—Ag | 117.15 (17) | C9—C10—H10A | 118.3 |
| N1—C1—C2 | 123.3 (3) | N1—C12—C4 | 122.1 (2) |
| N1—C1—H1A | 118.3 | N1—C12—C11 | 117.4 (2) |
| C2—C1—H1A | 118.3 | C4—C12—C11 | 120.4 (2) |
| C3—C2—C1 | 118.0 (3) | N2—C11—C7 | 121.4 (2) |
| C3—C2—H2A | 121.0 | N2—C11—C12 | 117.4 (2) |
| C1—C2—H2A | 121.0 | C7—C11—C12 | 121.2 (2) |
| C2—C3—C4 | 119.6 (3) | F2A—B—F4A | 110.07 (10) |
| C2—C3—H3A | 120.2 | F2A—B—F1A | 109.81 (10) |
| C4—C3—H3A | 120.2 | F4A—B—F1A | 109.81 (9) |
| C3—C4—C12 | 118.2 (3) | F1A—B—F1B | 113.8 (5) |
| C3—C4—C5 | 120.2 (2) | F2B—B—F1B | 109.52 (10) |
| C12—C4—C5 | 121.6 (3) | F2A—B—F4B | 108.2 (5) |
| O1—C5—C4 | 123.2 (3) | F2B—B—F4B | 109.52 (10) |
| O1—C5—C6 | 119.0 (3) | F1B—B—F4B | 109.52 (10) |
| C4—C5—C6 | 117.9 (2) | F4Ai—B—F3B | 109.1 (6) |
| O2—C6—C7 | 122.5 (3) | F2B—B—F3B | 109.43 (10) |
| O2—C6—C5 | 119.3 (3) | F1B—B—F3B | 109.43 (10) |
| C7—C6—C5 | 118.1 (2) | F4B—B—F3B | 109.41 (10) |
| N1i—Ag—N1—C12 | 152.8 (2) | O2—C6—C7—C11 | 172.5 (3) |
| N2i—Ag—N1—C12 | −116.1 (2) | C5—C6—C7—C11 | −3.7 (4) |
| N2—Ag—N1—C12 | −4.37 (18) | C11—C7—C8—C9 | 0.6 (4) |
| N1i—Ag—N1—C1 | −18.6 (2) | C6—C7—C8—C9 | 178.9 (3) |
| N2i—Ag—N1—C1 | 72.5 (3) | C7—C8—C9—C10 | −3.2 (4) |
| N2—Ag—N1—C1 | −175.8 (2) | C11—N2—C10—C9 | 1.0 (4) |
| N1—Ag—N2—C10 | −176.9 (3) | Ag—N2—C10—C9 | −179.2 (2) |
| N1i—Ag—N2—C10 | 83.9 (3) | C8—C9—C10—N2 | 2.5 (5) |
| N2i—Ag—N2—C10 | −19.1 (2) | C1—N1—C12—C4 | −2.2 (4) |
| N1—Ag—N2—C11 | 2.92 (18) | Ag—N1—C12—C4 | −174.04 (19) |
| N1i—Ag—N2—C11 | −96.3 (3) | C1—N1—C12—C11 | 177.1 (2) |
| N2i—Ag—N2—C11 | 160.7 (2) | Ag—N1—C12—C11 | 5.3 (3) |
| C12—N1—C1—C2 | −0.2 (4) | C3—C4—C12—N1 | 2.4 (4) |
| Ag—N1—C1—C2 | 171.1 (2) | C5—C4—C12—N1 | −179.6 (2) |
| N1—C1—C2—C3 | 2.2 (5) | C3—C4—C12—C11 | −176.9 (2) |
| C1—C2—C3—C4 | −2.0 (4) | C5—C4—C12—C11 | 1.1 (4) |
| C2—C3—C4—C12 | −0.2 (4) | C10—N2—C11—C7 | −3.7 (4) |
| C2—C3—C4—C5 | −178.3 (3) | Ag—N2—C11—C7 | 176.42 (18) |
| C3—C4—C5—O1 | −2.8 (5) | C10—N2—C11—C12 | 178.4 (2) |
| C12—C4—C5—O1 | 179.2 (3) | Ag—N2—C11—C12 | −1.4 (3) |
| C3—C4—C5—C6 | 175.6 (3) | C8—C7—C11—N2 | 3.0 (4) |
| C12—C4—C5—C6 | −2.4 (4) | C6—C7—C11—N2 | −175.3 (2) |
| O1—C5—C6—O2 | 5.7 (5) | C8—C7—C11—C12 | −179.3 (2) |
| C4—C5—C6—O2 | −172.7 (3) | C6—C7—C11—C12 | 2.4 (4) |
| O1—C5—C6—C7 | −177.9 (3) | N1—C12—C11—N2 | −2.6 (4) |
| C4—C5—C6—C7 | 3.6 (4) | C4—C12—C11—N2 | 176.7 (2) |
| O2—C6—C7—C8 | −5.8 (4) | N1—C12—C11—C7 | 179.5 (2) |
| C5—C6—C7—C8 | 178.0 (3) | C4—C12—C11—C7 | −1.1 (4) |
Symmetry codes: (i) −x+1, y, −z+1/2.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3A···O1ii | 0.95 | 2.51 | 3.347 (4) | 147 |
| C1—H1A···F1Ai | 0.95 | 2.35 | 3.083 (6) | 133 |
| C2—H2A···F1B | 0.95 | 2.17 | 2.803 (8) | 123 |
| C10—H10A···F2Aiii | 0.95 | 2.24 | 2.859 (5) | 122 |
| C10—H10A···F2Biv | 0.95 | 2.28 | 3.065 (4) | 140 |
Symmetry codes: (ii) −x+2, −y+1, −z+1; (i) −x+1, y, −z+1/2; (iii) −x+1, y−1, −z+1/2; (iv) x, y−1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: WM2249).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Calderazzo, F., Marchetti, F., Pampaloni, G. & Passarelli, V. (1999). J. Chem. Soc. Dalton Trans. pp. 4389–4396.
- Calderazzo, F., Pampaloni, G. & Passarelli, V. (2002). Inorg. Chim. Acta, 330, 136–142.
- Calucci, L., Pampaloni, G., Pinzino, C. & Prescimone, A. (2006). Inorg. Chim. Acta, 359, 3911–3920.
- Fox, G. A., Bhattacharya, S. & Pierpont, C. G. (1991). Inorg. Chem.30, 2895–2899.
- Galet, A., Munoz, M. C., Agusti, G., Martinez, V., Gaspar, A. B. & Real, J. A. (2005). Z. Anorg. Allg. Chem.631, 1985–1987.
- Lei, Y., Shi, C. & Anson, F. C. (1996). Inorg. Chem.35, 3044–3049.
- Leschke, M., Rheinwald, G. & Lang, H. (2002). Z. Anorg. Allg. Chem.628, 2470–2477.
- Okamura, R., Fujihara, T., Wada, T. & Tanaka, K. (2006). Bull. Chem. Soc. Jpn, 79, 106–112.
- Onuegbu, J., Butcher, R. J., Hosten, C., Udeochu, U. C. & Bakare, O. (2007). Acta Cryst. E63, m2309–m2310.
- Oxford Diffraction. (2007). CrysAlis CCD and CrysAlis RED Oxford Diffraction Ltd, Abingdon, England.
- Pallenberg, A. J., Marschner, T. M. & Barnhart, D. M. (1997). Polyhedron, 16, 2711–2719.
- Paramonov, S. E., Kuzmina, N. P. & Troyanov, S. I. (2003). Polyhedron, 22, 837–841.
- Paw, W. & Eisenberg, R. (1997). Inorg. Chem.36, 2287–2293. [DOI] [PubMed]
- Ruiz, R., Caneschi, A., Gatteschi, D., Gaspar, A. B., Real, J. A., Fernandez, I. & Munoz, M. C. (1999). Inorg. Chem. Commun.2, 521–523.
- Shavaleev, N. M., Moorcraft, L. P., Pope, S. J. A., Bell, Z. R., Faulkner, S. & Ward, M. D. (2003a). Chem. Commun. pp. 1134–1135. [DOI] [PubMed]
- Shavaleev, N. M., Moorcraft, L. P., Pope, S. J. A., Bell, Z. R., Faulkner, S. & Ward, M. D. (2003b). Chem. Eur. J.9, 5283–5291. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Titze, C., Kaim, W. & Zalis, S. (1997). Inorg. Chem.36, 2505–2510.
- Udeochu, U., Jimerson, T., Vivoni, A., Bakare, O. & Hosten, C. M. (2007). J. Phys. Chem. A, 111, 3409–3415. [DOI] [PubMed]
- Yang, L., Powell, D. R. & Houser, R. P. (2007). Dalton Trans. pp. 955–964. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S160053680903222X/wm2249sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680903222X/wm2249Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report