

Abstract
Insulin resistance arises from the inability of insulin to act normally in regulating nutrient metabolism in peripheral tissues. Increasing evidence from human population studies and animal research has established correlative as well as causative links between chronic inflammation and insulin resistance. However, the underlying molecular pathways are largely unknown. In this report, we show that many inflammation and macrophage-specific genes are dramatically upregulated in white adipose tissue (WAT) in mouse models of genetic and high-fat diet-induced obesity (DIO). The upregulation is progressively increased in WAT of mice with DIO and precedes a dramatic increase in circulating-insulin level. Upon treatment with rosiglitazone, an insulin-sensitizing drug, these macrophage-originated genes are downregulated. Histologically, there is evidence of significant infiltration of macrophages, but not neutrophils and lymphocytes, into WAT of obese mice, with signs of adipocyte lipolysis and formation of multinucleate giant cells. These data suggest that macrophages in WAT play an active role in morbid obesity and that macrophage-related inflammatory activities may contribute to the pathogenesis of obesity-induced insulin resistance. We propose that obesity-related insulin resistance is, at least in part, a chronic inflammatory disease initiated in adipose tissue.
Introduction
Insulin resistance is defined as a decreased response of the peripheral tissues to insulin action. Individuals with insulin resistance are predisposed to developing type 2 diabetes mellitus (T2DM). Increasingly, insulin resistance has been recognized as the integral feature of the so-called metabolic syndrome, which includes glucose intolerance, insulin resistance, obesity, hypertriglyceridemia, low HDL cholesterol, hypertension, and accelerated atherosclerosis.
Growing evidence has pointed to a correlative and causative relationship between inflammation and insulin resistance/T2DM. The proinflammatory cytokine TNF-α has been demonstrated to mediate insulin resistance as a result of obesity in many rodent obesity models (1, 2). TNF-α was overexpressed in white adipose tissue (WAT) in obese and insulin-resistant states; mice lacking the TNF-α ligand or the p55 TNF receptor were partially protected from obesity-induced insulin resistance (3–6). Recently, the chemokine monocyte chemotactic protein-1 (MCP-1) was also shown to impair adipocyte insulin sensitivity (7).
In recent years, a large number of human population studies have linked insulin resistance to systemic inflammation (8, 9). For example, in one recent report, the acute-phase response was studied in Caucasian subjects with T2DM. There was a significant graded increase of serum sialic acid, a marker of the acute-phase response; α-1 acid glycoprotein; IL-6; and urinary albumin-excretion rate among three groups, with the lowest levels in nondiabetic subjects, intermediate levels in T2DM patients without metabolic syndrome, and the highest levels in T2DM patients with metabolic syndrome (10). In a larger study, the relation of C-reactive protein (CRP), fibrinogen, and white cell count to components of insulin resistance syndrome was evaluated in the nondiabetic population of the Insulin Resistance Atherosclerosis Study (n = 1,008). CRP, a predictor of cardiovascular events in previous reports, was found to be independently related to insulin insensitivity (11).
Salicylates, including sodium salicylate and aspirin, are used to treat inflammatory conditions such as rheumatic fever and rheumatoid arthritis. Historically, it has also been known that high doses of salicylates are able to lower blood glucose concentrations (12). It was recently shown that reduced signaling through the IKKβ pathway, a key pathway in tissue inflammation, either by salicylate-based inhibitors or decreased IKKβ expression, is accompanied by improved insulin sensitivity in vivo (13, 14). Mice deficient of JNK1, another key mediator of inflammatory responses, were shown to have improved systemic insulin sensitivity (15). Thus, key genes in inflammation-signaling pathways are causatively linked to insulin responsiveness.
To explore the molecular mechanisms underlying obesity and insulin resistance, we performed extensive transcriptional profiling studies using multiple tissues taken from mice with genetic or diet-induced obese mice. In addition, we tracked the transcriptional regulation of several representative macrophage and inflammation genes in WAT of these mouse models and determined the specific cell types in which they are expressed. We also detailed the morphological differences between wild-type and ob/ob WAT by various methods. Based on our data, we hypothesize that macrophage-related inflammatory activities in WAT play an active role in obesity-induced insulin resistance.
Methods
Mouse models.
Male mice of the genetically obese/diabetic models ob/ob, db/db, agouti, and tubby in the C57BL/6J strain, along with littermate controls, were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA). These mice were fed standard chow (Farmer’s Exchange, Framingham, Massachusetts, USA). For diet-induced obese (DIO) studies, starting at 4–5 weeks of age, wild-type C57BL/6J mice from The Jackson Laboratory were put on diets containing 10%, 45%, or 60% kcal from fat (Research Diets Inc., New Brunswick, New Jersey, USA). For transcriptional profiling studies, mice with early-onset genetic obesity (ob/ob and db/db) were sacrificed at 15 weeks of age; mice with late-onset genetic obesity (agouti and tubby) were sacrificed at 25 weeks of age; mice with diet-induced obesity (45% kcal from fat) were sacrificed at 20 weeks of age after 16 weeks of high-fat diet. Animals used to track progression to obesity were sacrificed after 0, 3, 6, 8, 11, 16, and 26 weeks on high-fat diet (60% kcal from fat). Tissues and body fluids collected for this work were epididymal WAT, liver, skeletal muscle (gastrocnemius and quadriceps), lung, spleen, and blood.
For rosiglitazone treatment, 8-week-old male ob/ob mice from The Jackson Laboratory were acclimated for 1 week. Rosiglitazone or vehicle (sterile water) was orally gavaged once a day at a dose of 15 mg/kg for 28 consecutive days. At the end of the study, mice were sacrificed by CO2 inhalation, and epididymal fat pads were excised for RNA extraction. All animal experiments were approved by the Institutional Animal Care and Use Committee of Millennium Pharmaceuticals Inc.
Tissue collection, total-RNA preparation, and quantitative PCR.
Mice were sacrificed by CO2 inhalation. Tissues were collected and frozen immediately in liquid nitrogen. For total-RNA preparation, tissues were ground into fine powder in liquid nitrogen and homogenized in Trizol solution (Invitrogen, Carlsbad, California, USA). RNA was isolated according to the manufacturer’s instructions. For WAT, the surface oil layer was removed prior to chloroform extraction to ensure better RNA quality. TaqMan real-time quantitative PCR was performed and analyzed according to the manufacturer’s instructions (Applied Biosystems, Foster City, California, USA).
Isolation of adipocytes, stromal-vascular cells, and primary macrophages.
Isolation of adipocytes and stromal-vascular cells was performed as described previously (16). Five days before macrophage isolation, male C57BL/6J mice were injected with 2 ml of BBL thioglycolate culture medium (Becton Dickinson and Co., Sparks, Maryland, USA). Mice were sacrificed by CO2 inhalation, and the peritoneal cavities were washed with cold sterile PBS to collect macrophages. After removal of red blood cells, macrophages were resuspended in RPMI containing 10% heat-inactivated FBS and plated into 10-cm dishes. After 3 hours of incubation, cells were washed three times with PBS and Trizol solution was added for RNA extraction.
Immunohistochemistry and in situ hybridization.
Fresh frozen sections of WAT were cut for in situ hybridization. Probes were generated by RT-PCR from mRNA isolated from WAT with gene-specific primers tailed with T3 (sense) and T7 (antisense) promoter sequences. The hybridization was performed as described previously (17).
At the time of sacrifice, portions of WAT were either flash frozen in liquid nitrogen or fixed in 4% paraformaldehyde or Bouin’s fix (Poly Scientific R&D Corp., Bay Shore, New York, USA). After dehydration with increasing concentrations of ethanol, tissues were incubated in xylenes and then embedded in paraffin at 60°C. Seven-micron sections were cut on a microtome and mounted on slides. Samples were dewaxed, rehydrated, stained with toluidine blue O, destained, and dehydrated. Mounting solution and coverslips were added.
Paraffin sections of WAT were also stained with anti-mouse F4/80 antibody (Serotec Inc., Raleigh, North Carolina, USA). Primary antibody was biotinylated. Prior to incubation with antibody, sections were blocked with biotin and avidin. Endogenous peroxidase activity was blocked by pretreatment with 0.06% H2O2 in methanol. Endogenous Fc receptor was blocked with 20% FCS in PBS. Sections were incubated in primary antibodies (1:100 dilution) for 1 hour and then washed with PBS/0.05% Tween-20. HRP-conjugated streptavidin (DAKO Corp., Carpinteria, California, USA) was added and incubated for 30 minutes, and then slides were washed with PBS/0.05% Tween-20. Finally, peroxidase substrate diaminobenzidine (Vector Laboratories Inc., Burlingame, California, USA) was added and incubated for 15 minutes. Slides were rinsed and counterstained with Mayer’s hematoxylin. Mounting solution and coverslips were added.
Isolated stromal-vascular cells were seeded in chamber slides (Nalge Nunc International, Naperville, Illinois, USA) at a concentration of 5 × 105 per chamber in DMEM containing 20% FCS (HyClone Laboratories, Logan, Utah, USA) and 20 ng/ml recombinant mouse G-CSF (R&D Systems Inc., Minneapolis, Minnesota, USA) for overnight culture. To prepare for fixation, the cells were rinsed five times with PBS+ (PBS supplemented with 1 mM MgCl2 and 0.1 mM CaCl2) and fixed in PBS+ containing 3% paraformaldehyde for 30 minutes at room temperature. Cells were then incubated in PBS+ containing 50 mM NH4Cl for 5 minutes at room temperature and rinsed three times with PBS+.
Immunohistochemistry on stromal-vascular cells was performed as described below. After fixing with paraformaldehyde, stromal-vascular cells were used for immunohistochemical staining with anti-mouse F4/80 antibody. Primary antibody was diluted 1:50 for the experiments. Alkaline phosphatase–conjugated secondary antibody (Serotec Inc.) was used (1:300 dilution). Levamisole was used to block endogenous alkaline phosphatase activity. Alkaline phosphatase substrate was purchased from Vector Laboratories Inc. Slides were counterstained with Mayer’s hematoxylin. Mounting solution and coverslips were added.
Oil red O was used to stain lipids in fixed stromal-vascular cells. Cells were washed three times in PBS+ and then once in 60% isopropanol. Oil red O was added, and the cells were incubated for 10 minutes at room temperature. Cells were washed once in 60% isopropanol and three times in PBS+. Slides were rinsed and counterstained with Mayer’s hematoxylin. Mounting solution and coverslips were added.
Results
Expression levels of genes in inflammatory pathways are significantly upregulated in WAT of obese mice.
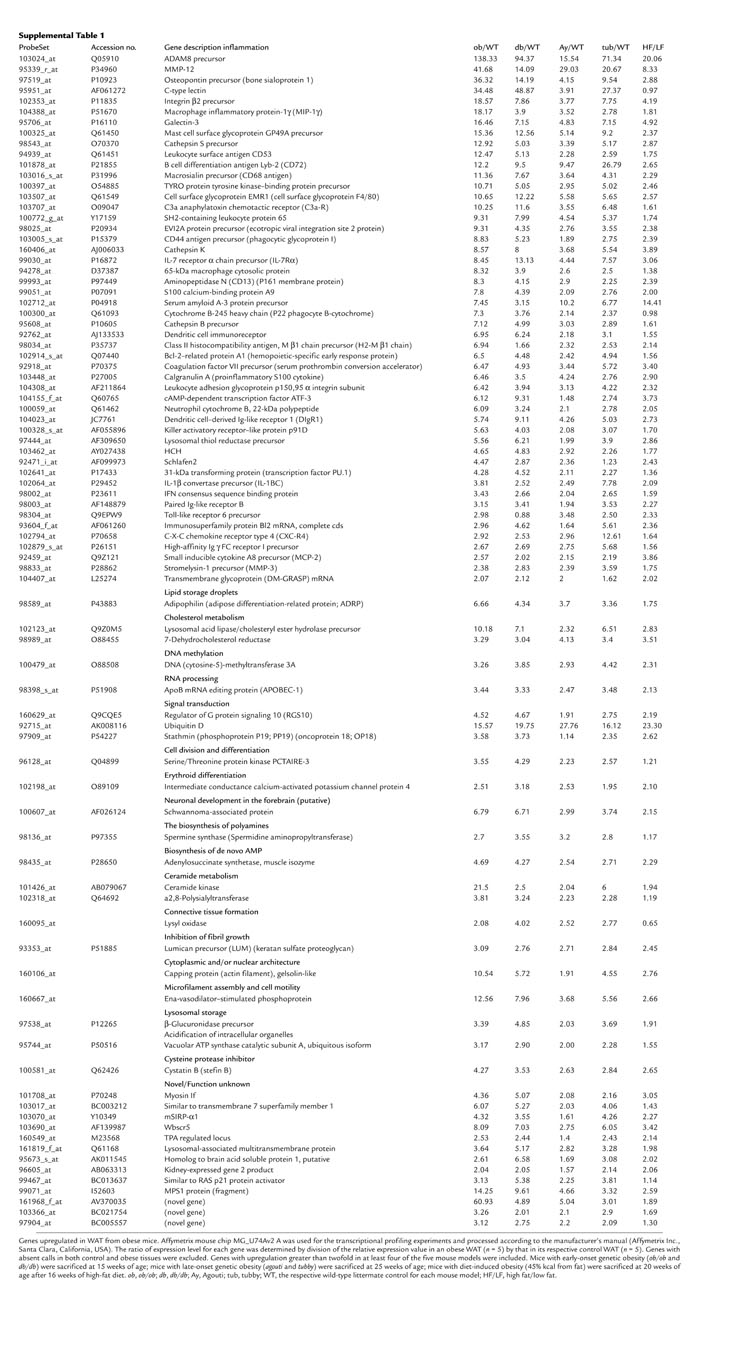
To study obesity and obesity-induced insulin resistance, we performed global transcriptional profiling studies with various tissues (WAT, brown adipose tissue, muscle, liver, stomach, hypothalamus, small intestine, and pancreas) taken from genetically obese mice, including ob/ob, db/db, tubby, agouti, and DIO mice. Notably, we found that many of the most significantly upregulated genes in WAT were not known to be involved in adipocyte biology; instead, they could be broadly categorized as macrophage- or inflammation-related genes. Of the genes upregulated more than twofold in at least four of these five models, 59% (50/85) could be counted as inflammation genes, as determined by their known functions. The remaining genes were involved in diverse molecular pathways, including fat storage, cholesterol metabolism, DNA modification, transcription, cell division, signal transduction, and unknown functions (see Supplemental Table 1 for details; http://www.jci.org/cgi/content/full/112/12/1821/DC1). These results were consistent with an earlier study on leptin-deficient ob/ob mice (18); however, with multiple models of genetic and diet-induced obesity, our data suggest that the inflammatory response is a general phenomenon of the obese state, independent of the availability of the leptin protein. We also noticed that this phenomenon was WAT-specific and was not observed in any other tissues we profiled.
To understand this apparent inflammatory response in WAT in detail and to explore its role in obesity and insulin resistance, we focused our follow-up studies on two genetic mouse models (ob/ob and db/db) and the diet-induced obesity model. We chose the following six macrophage or inflammation genes for further study: ADAM8, a disintegrin-like metalloproteinase strongly expressed in monocytic lineage (19); macrophage inflammatory protein-1α (MIP-1α), a gene derived from mononuclear cells and involved in the acute inflammatory state in the recruitment and activation of polymorphonuclear leukocytes (20); MCP-1, which is a member of the small inducible cytokines family and which plays a role in the recruitment of monocytes to sites of injury and infection (21); macrophage antigen-1 (MAC-1) (CD11b), an integrin found predominantly on monocytes, macrophages, neutrophils, and NK cells (22); F4/80, a classical macrophage-restricted surface glycoprotein (23); and CD68 (macrosialin), a heavily glycosylated transmembrane protein expressed specifically in macrophages and macrophage-related cells (24).
As shown in Figure 1a, the mRNA levels of these six genes were consistently and significantly upregulated in WAT of ob/ob, db/db, and DIO mice that had been on a high-fat diet for 16 weeks, confirming the microarray experiments. Regulation for three genes was further confirmed by Northern blot analysis using independent samples from the same experiment (Figure 1b). The fold increase for each gene among different obesity models was not directly comparable. DIO mice were studied separately from the other two genetic models. The DIO mice used here were 20 weeks old, fed on a high-fat diet (60% kcal from fat) for 16 weeks, with an average body weight of about 60 g. Each obese model was compared with its respective lean control. The absolute expression levels of the genes of interest were lower in the lean controls of the DIO model than in the wild-type controls of the genetic models because of the difference in diets (10% low-fat diet for the former vs. 16% regular chow for the latter). This resulted in an exaggerated fold change for the DIO model. Excluding the diet factor and experimental variations, however, it was still apparent that different genes were upregulated differently among obese models even when the expression levels were normalized, as shown in Figure 1b. For example, ADAM8 was most dramatically upregulated in DIO mice fed on 60% high-fat diet. In short, upregulation of inflammation genes is observed in WAT of genetic and DIO mouse models, although the expression levels for each gene appear to be regulated differently from one model to another. Both diet and lack of leptin-signaling pathways might have played a role here. Consistent with this possibility, regulation of inflammation genes was less dramatic with DIO mice fed with a 45% fat diet (Supplemental Table 1, http://www.jci.org/cgi/content/full/112/12/1821/DC1). The absence of leptin in genetically obese mice might even be protective, since leptin has been reported to increase cholesterol ester synthesis in cultured macrophages (25).
Figure 1.

(a) The transcriptional regulation of inflammation genes in the WAT of mice with genetic or diet-induced obesity/diabetes by quantitative RT-PCR (TaqMan). For comparison, the expression level of these genes in lean mice was arbitrarily set at 1; error bars represent ± SE. LF, low fat (10% fat); HF, high fat (60% fat). ob/ob and db/db mice and appropriate controls (n = 5 per group) were obtained from The Jackson Laboratory, fed a standard chow diet (Farmer’s Exchange), and sacrificed at 15 weeks of age. DIO mice (C57BL/6J; The Jackson Laboratory) were obtained at 4 weeks of age and placed on the designated diet of 60% kcal from fat (Research Diets Inc.) for 16 weeks (n = 10 per group). y axes show arbitrary units representing relative expression levels of mRNAs. (b) Confirmation of inflammation-gene regulation by Northern blot analysis. An independent set of animals from the same experiment was used.
To determine whether the upregulation of these genes occurs prior to the development of systematic insulin resistance, which is characterized by hyperinsulinemia, we tracked the expression levels of these genes in WAT of mice with high-fat diet–induced obesity at multiple time points for 26 weeks. The body weight increased steadily over this period, as did the fasting blood glucose level, although the latter remained within the normal range (<120 mg/dl) until sometime after 16 weeks (Figure 2a). Meanwhile, we observed an increase in expression of some of these inflammation genes as early as 3 weeks on high-fat diet (Figure 2b). Around 16 weeks on high-fat diet, a much more dramatic upregulation of these transcripts occurred, which correlated closely with a marked increase in fasting blood insulin levels (Figure 2). It appears that the adipose inflammatory response increases with an increase of adiposity, prior to the increase of fasting insulin level, but intensifies at the onset of hyperinsulinemia.
Figure 2.

The transcriptional regulation of inflammation genes in progressively obese and insulin-resistant mice. Time is shown as weeks on diet. (a) Changes in body weight, fasting blood glucose, and fasting plasma insulin over 26 weeks on diets. *Statistically significant difference (P < 0.05, Student’s t test). (b) mRNA expression of ADAM8, MIP-1 For comparison, the expression level of these genes in lean mice was arbitrarily set at 1; error bars represent ± SE. C57BL/6J mice were obtained from The Jackson Laboratory and started on diets of 10% fat (low fat; LF) or 60% fat (high fat; HF) (Research Diets Inc.) at 4 weeks of age (0-week time point). Animals were sacrificed after the specified number of weeks on the diet (0 weeks, n = 10 per group; 3 weeks, n = 3 per group; 6 weeks, n = 10 per group; 8 weeks, n = 3 per group; 11 weeks, n = 3 per group; 16 weeks, n = 10 per group; 26 weeks, n = 10 per group). y axes show arbitrary units representing relative expression levels of mRNAs.
On the other hand, in muscle and liver, two other important tissues for insulin action, the mRNA expression of these genes was barely detectable and essentially unchanged in these obese mice (data not shown). To find out whether similar inflammatory activities are present in other tissues rich in macrophages, we compared expression of ADAM8, MIP-1α, MCP-1, MAC-1, F4/80, and CD68 in lung, spleen, and WAT in ob/ob and db/db mice with that in their lean littermate controls. Overall, there was little change in these genes in lung and spleen in the obese state (Figure 3). In the progression study, after 26 weeks on high-fat diet, a significant upregulation of CD68 was observed in the liver, although the absolute expression level was much lower compared with that in fat.
Figure 3.

The transcriptional regulation of ADAM8, MIP-1α, MCP-1, MAC-1, F4/80, and CD68 in macrophage-rich tissues (WAT, lung, and spleen) of ob/ob and db/db mice. For comparison, the gene expression level in lean mice was arbitrarily set at 1; error bars represent ± SE. y axes show arbitrary units representing relative expression levels of mRNAs.
The expression of inflammation genes is restricted to, or enriched in, macrophages.
To sort out the cell types in which these genes were expressed, we separated cells in WAT into two fractions: adipocytes and stromal-vascular cells. As shown in Figure 4, TaqMan analysis revealed that the inflammation genes under investigation were predominantly expressed in the stromal-vascular fraction, where macrophages might reside. These genes were expressed at much lower levels in the adipocyte fraction. Notably, TNF-α and IKKβ, two inflammation genes linked to obesity-induced insulin resistance by loss-of-function studies, were also more abundant in the stromal-vascular fraction. To control for the quality of RNA from adipocytes, leptin, an adipocyte-specific hormone, was used as a marker to show that the fractionation was done properly. The expression pattern of the inflammation genes was further examined in many tissues including adipose and primary macrophages isolated from the peritoneal cavity of normal mice. Consistent with existing evidence, these genes were expressed at the highest levels in the primary-macrophage population, followed by WAT, demonstrating that WAT is normally enriched in macrophages (data not shown).
Figure 4.

The mRNA expression of ADAM8, MIP-1α, MCP-1, MAC-1, F4/80, and CD68 in stromal-vascular and adipocyte fractions. For quality control, leptin was examined in these samples. To compare with the expression of known adipose inflammation genes, TNF-α and IKKβ expression was also examined. For genes predominantly expressed in the stromal-vascular fraction, the expression in adipocytes was set at 1; error bars represent ± SE. For leptin, the expression in stromal-vascular cells was set at 1. y axes show arbitrary units representing relative expression levels of mRNAs.
Since the stromal-vascular fraction from fat contains several cell types including preadipocytes, it was important to determine which cells within the stromal-vascular fraction significantly express these inflammation genes. To address this question, two cultured preadipocyte cell lines, 3T3-L1 and 3T3-F442A, were stimulated to undergo adipogenesis. TaqMan analysis showed that ADAM8, MIP-1α, F4/80, and MAC-1 were not detectable in either preadipocytes or adipocytes. CD68 had only a very low level of expression in adipocytes, while MCP-1 showed moderate expression in preadipocytes (data not shown).
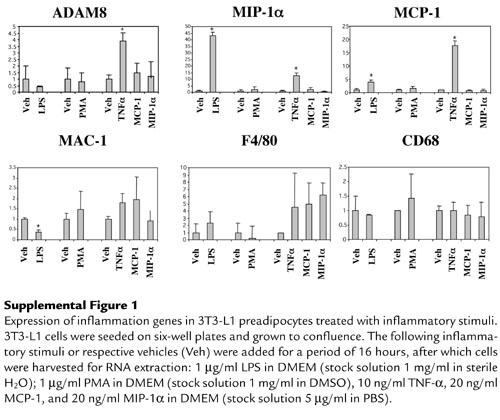
Recent evidence has indicated that preadipocytes have the potential to “transdifferentiate” into macrophages (26). To explore the possibility that preadipocytes could express these inflammation and macrophage genes in an inflammatory environment, we used cytokines upregulated in the obese state, TNF-α, MIP-1α, and MCP-1, as well as general inflammatory stimuli (LPS and PMA) to stimulate 3T3-L1 preadipocytes for 16 hours. While treatment with these inflammatory stimuli did not increase the expression of macrophage-marker genes (MAC-1, F4/80, and CD68) in 3T3-L1 preadipocytes, TNF-α did significantly stimulate the expression of ADAM8, MIP-1α, and MCP-1 in 3T3-L1 preadipocytes (Supplemental Figure 1, http://www.jci.org/cgi/content/full/112/12/1821/DC1). This suggests that obesity-related inflammatory stimuli can trigger preadipocytes to increase expression of some, but not all, inflammation genes under certain conditions.
Morphological changes of WAT in obesity.
The apparent upregulation of gene expression could be a consequence of both increased transcription in existing macrophages and increased macrophage infiltration into WAT of obese mice. To determine the source of this signal, adipose tissue sections from wild-type and ob/ob mice were compared. The morphological differences were striking. Besides the well-documented size difference of adipocytes from wild-type and ob/ob mice, there were clusters of what appeared to be small, nucleated cells in the interstitial spaces between adipocytes in ob/ob fat tissues, in contrast to the near absence of such cells in the control-mouse sections (Figure 5a). As the obese mice aged (5 months), these cell clusters became more prevalent and adipocytes showed early features of lipolysis manifested by multifocal cell shrinkage (Figure 5b). In situ hybridization and immunohistochemical staining using F4/80 RNA probes and antibody illustrated that F4/80 expression was restricted to these clusters of small, nucleated cells (Figure 5, c and d). Similar results were seen with other inflammation genes as well, confirming the findings with F4/80 (data not shown). Lack of costaining with platelet-endothelial cell adhesion molecule (PECAM), a vascular endothelium marker, excluded involvement of endothelial cells (data not shown).
Figure 5.

Histological comparison between wild-type and ob/ob WAT and stromal-vascular cells. For each panel, the wild type at ×100 is seen at the left, ob/ob at ×100 in the middle, and ob/ob at ×400 at the right. (a) WAT morphological differences at 3 months (toluidine blue O on paraffin sections). Note the presence of nucleated stromal cells in the high magnification of the ob/ob type at the right. (b) WAT morphological differences at 5 months (toluidine blue O on paraffin sections). The stromal multinucleated cells have increased in the ob/ob type seen at the right, with early features of lipolysis in the ob/ob adipocytes manifested by multifocal cell shrinkage. (c) WAT at 3 months probed with F4/80 antisense RNA (in situ hybridization on fresh frozen sections). (d) WAT at 3 months immunostained with anti–F4/80 antibody (immunohistochemistry on paraffin sections, brown staining). (e) Primary stromal-vascular cells from 5-month-old mice, immunostained with anti–F4/80 antibody (red staining). (f) Primary stromal-vascular cells from 5-month-old mice stained with oil red O.
To further determine that these cells were not preadipocytes, stromal-vascular fractions from wild-type and ob/ob adipose tissues were seeded on slides and stained with anti–F4/80 antibody. Most of the stromal cells from wild-type WAT appeared to be fibroblasts, possibly preadipocytes, and were F4/80 negative. In the stromal-vascular fraction from ob/ob mice, on the other hand, there were numerous F4/80–positive cells. Many of these cells contained multiple nuclei, reminiscent of giant cells (Figure 5e). Giant cells are formed by the fusion of multiple macrophages and are seen in instances of chronic inflammation and granuloma (27). In mice at 5 months of age, it was determined (by positive F4/80 staining) that 2% of wild-type and 33% of ob/ob stromal-vascular cells were macrophages. As shown in Figure 5f, F4/80–positive cells also contained many oil red O–staining vesicles, indicating intracytoplasmic lipid accumulation. This observation is consistent with histiocytic (phagocytic) activity. We also stained the stromal cells for the T cell–specific marker CD3, the B cell–specific marker CD72, and the granulocyte-specific marker Ly6G (data not shown). Of the four cell types tested, only macrophages were observed in WAT. From this, we conclude that there is significant macrophage accumulation in WAT during the development of obesity.
Rosiglitazone downregulates the mRNA level of the inflammation genes.
PPARγ is a member of the nuclear hormone receptor family. It is the decisive transcription factor regulating adipocyte differentiation (28, 29). PPARγ agonists, including troglitazone, pioglitazone, and rosiglitazone, are a new and highly efficacious class of insulin-sensitizing drugs known as thiazolidinediones (TZDs). To explore the relationship of insulin resistance and inflammation in adipose tissue from a different perspective, we treated ob/ob mice with rosiglitazone, a PPARγ agonist. As shown in Figure 6, ADAM8, MAC-1, F4/80, and CD68 showed statistically significant reduction in expression levels after the treatment. MIP-1α and MCP-1 expression also trended down but did not reach statistical significance.
Figure 6.

The expression of ADAM8, MIP-1α, MCP-1, MAC-1, F4/80, and CD68 in ob/ob WAT treated with rosiglitazone. Twelve-week-old ob/ob mice were treated with either 15 mg/kg rosiglitazone (Rosi) or vehicle (sterile water; Veh) for 28 consecutive days (n = 10 in each group). For comparison, the expression level in rosiglitazone-treated ob/ob mice was arbitrarily set at 1; error bars represent ± SE; *P < 0.05. y axes show arbitrary units representing relative expression levels of mRNAs.
DISCUSSION
Increasing evidence has established correlative and causative links between chronic inflammation and insulin resistance. To further explore the molecular pathways underlying obesity-induced insulin resistance in general, we have studied mouse models of genetic and high-fat diet–induced obesity/diabetes. We have found that many macrophage-specific or -enriched genes, exemplified by ADAM8, MIP-1α, MCP-1, MAC-1, F4/80, and CD68, are dramatically upregulated in WAT of obese mice. In WAT, these genes are predominantly expressed in the stromal-vascular fraction where macrophages reside. Additional histological data provide evidence for a significant increase of macrophage numbers. We have also demonstrated, through immunohistochemical methods, that macrophages, not granulocytes or lymphocytes, are the cells involved in adipose inflammation. Because we used multiple obesity models, our results suggest that macrophage inflammatory response in WAT is a general phenomenon associated with morbid fat-mass expansion.
Macrophage infiltration is the likely explanation for the increase of macrophage numbers, since it is well established that extravasations of leukocytes from circulation lead to accumulation of immune cells at the site of inflammation. The observed upregulation of chemotaxin genes, such as MIP-1α and MCP-1, supports the conclusion. It was previously reported that stromal-vascular cells from obese mice express more TNF-α mRNA than do those from lean controls (1). We confirmed by ELISA that more TNF-α was released from an equal number of stromal-vascular cells from obese mice than from lean controls (data not shown). Since TNF-α is a potent activator of macrophages, it is reasonable to conclude that the macrophages accumulated in WAT in the obese state are functionally active.
Recently it was reported that preadipocytes could be converted to macrophages under certain conditions (26). Our study indeed showed that TNF-α could trigger preadipocytes to increase expression of some inflammation genes, although not the three macrophage markers we have studied (Supplemental Figure 1, http://www.jci.org/cgi/content/full/112/12/1821/DC1). This finding provides some support for converted preadipocytes as a source for the macrophage activity in obese WAT. It is possible that a combination of conversion of local preadipocytes to macrophages and activation and recruitment of resident macrophages and circulating monocytes has contributed to the increased macrophage activity observed in obese WAT. Future experiments are necessary to determine the significance of each mechanism during the physiological increase of adiposity.
This obesity-induced macrophage accumulation in WAT appears to be highly correlated with, if not causative of, insulin resistance in various mouse models. This is most strikingly demonstrated by tracking of the expression level of the macrophage markers over time in WAT of mice on a diet of high fat content. While body weight increases steadily over time on high-fat diet, fasting blood insulin levels remained relatively unchanged until 16 weeks on high-fat diet, when hyperinsulinemia became apparent. The expression levels of the selected macrophage and inflammation genes were upregulated before the increase in circulating-insulin levels. These data indicate that macrophage activities occur after the increase of adiposity but before insulin resistance.
The dramatic upregulation of macrophage-related genes is mostly restricted to WAT; the mRNA expression of these genes was barely detectable and essentially unchanged in muscle and liver of the obese mice. In addition, there was little change of expression of these genes in lung and spleen in the obese state, further demonstrating that the observed inflammation was adipose-specific. In the progression study, after 26 weeks on high-fat diet, a significant upregulation of CD68 was observed in the liver, although the absolute expression level was much lower than that in fat. These data suggest that obesity-induced inflammation is initiated in WAT but may become systemic with the steady increase of adiposity or insulin resistance.
Several recent studies have shown that TZDs can attenuate macrophage activation in vitro and improve atherosclerosis in vivo, suggesting direct involvement of these agonists in anti-inflammatory activities, most likely mediated by PPARγ in macrophages (30–33). Part of their inhibitory effect on macrophage activation may even be independent of PPARγ (34–36). Here we show that rosiglitazone suppressed the increased expression of the inflammation genes in WAT of ob/ob mice. We have also observed reduced macrophage numbers in ob/ob WAT upon rosiglitazone treatment (data not shown). In this scenario, reduced inflammation is correlated with increased insulin sensitivity. Our results are consistent with the known anti-inflammation function of TZDs and suggest the possibility that TZDs improve insulin sensitivity at least partially by suppressing the inflammatory events in obese WAT.
Macrophage accumulation is likely a direct response to the abnormal fat metabolism caused by the increasing adiposity. The molecular signals that trigger the macrophage activity in obese WAT are not yet known, but several good candidates exist. Adipocytes are known to secrete hormones, cytokines, and FFAs, most of which have been shown to play some role in inflammation and systemic insulin resistance. Adiponectin, known to improve insulin sensitivity, is downregulated in obese/diabetic states (37). Adiponectin can inhibit adhesion of macrophages to endothelial cells, an essential process in the pathogenesis of atherosclerosis (38, 39). The decrease in adiponectin therefore might have contributed to the increased macrophage activity in obese adipose tissue. Leptin, which increases in proportion to fat mass, promotes cholesterol ester synthesis in macrophages in a hyperglycemic environment, an important process in the formation of foam cells in atherosclerosis (25). This would suggest that lack of leptin signaling in the ob/ob and db/db models might have some protective effect against the inflammatory response. This is consistent with our observation that some inflammation genes are even more dramatically upregulated in DIO mice than in leptin-deficient mice, although more detailed studies are required to address this issue. Complement factor C3, the precursor of C3a, a potent activator and chemotaxin of macrophages (40, 41), could also play a role. Indeed C3 is highly expressed in adipocytes and has been related to obesity and insulin resistance previously (41–43). Another interesting candidate is MCP-1, an essential signal for macrophage activation and recruitment (44). In our own studies, increased MCP-1 expression in WAT both preceded (at 3 weeks on high-fat diet) and was concurrent with (by 16 weeks on high-fat diet) insulin resistance in obese mice, supporting a possible causative role of this chemokine in the observed macrophage infiltration.
Once macrophage activation and infiltration is initiated, it is reasonable to expect that this macrophage-mediated inflammatory response would lead to impaired insulin response in adipocytes. Macrophages, upon activation, secrete numerous cytokines and chemokines, such as TNF-α, IL-1, IL-6, and MCP-1, that are known to cause insulin resistance in adipocytes (2, 9, 45–47). These cytokines and chemokines further activate macrophages to increase lymphokine production and secretion. These feedback loops are schematically summarized in Figure 7. As a consequence, insulin signaling in adipocytes could become increasingly impaired, eventually leading to massive adipocyte lipolysis, necrosis, and systemic insulin resistance.
Figure 7.

Hypothetical model of chronic inflammation and adipocyte insulin resistance. When adiposity reaches a certain threshold, factors derived from adipocytes induce macrophage activation and infiltration. Activated macrophages secrete cytokines that can impair adipocyte insulin sensitivity and stimulate further activation and infiltration of peripheral monocytes and macrophages into fat. Preadipocytes can also secrete chemokines under the stimulation of TNF-α, which can contribute to macrophage infiltration. These amplifying signals increasingly impair adipocyte insulin signaling and eventually cause systemic insulin resistance.
It remains to be shown how the inflammatory response initiated in WAT ultimately causes systemic insulin resistance. Increased lipolysis is one possible piece of the puzzle. The observed increase of lipolysis in obese WAT could result in the release of a large amount of FFAs, and an increased FFA level in the circulation has been shown to result in resistance to insulin signaling in skeletal muscle and liver (48–52). Therefore, it is possible that FFAs are an important link between chronic adipose inflammation and systemic insulin resistance. Additional work will be required to explore the molecular mechanisms involved in the initial inflammation response and the subsequent insulin resistance in WAT, and how they might relate to defective response to insulin in other peripheral tissues.
Supplementary Material
Acknowledgments
We thank Effie Tozzo for providing rosiglitazone-treated materials; Rebecca Mosher, Alwin Schuller, Tracy Delaney, and Chris Groves for technical assistance in histopathology experiments; and Zhidan Wu and Frank An for useful discussions.
Footnotes
See the related Commentary beginning on page 1785.
Haiyan Xu and Glenn T. Barnes contributed equally to this work.
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: type 2 diabetes mellitus (T2DM); white adipose tissue (WAT); monocyte chemotactic protein-1 (MCP-1); diet-induced obesity (DIO); macrophage inflammatory protein-1α (MIP-1α); macrophage antigen-1 (MAC-1); thiazolidinedione (TZD).
References
- 1.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 2.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 3.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 4.Uysal KT, Wiesbrock SM, Hotamisligil GS. Functional analysis of tumor necrosis factor (TNF) receptors in TNF-alpha-mediated insulin resistance in genetic obesity. Endocrinology. 1998;139:4832–4838. doi: 10.1210/endo.139.12.6337. [DOI] [PubMed] [Google Scholar]
- 5.Ventre J, et al. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes. 1997;46:1526–1531. doi: 10.2337/diab.46.9.1526. [DOI] [PubMed] [Google Scholar]
- 6.Moller DE. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol. Metab. 2000;11:212–217. doi: 10.1016/s1043-2760(00)00272-1. [DOI] [PubMed] [Google Scholar]
- 7.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia. 1998;41:1241–1248. doi: 10.1007/s001250051058. [DOI] [PubMed] [Google Scholar]
- 9.Grimble RF. Inflammatory status and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care. 2002;5:551–559. doi: 10.1097/00075197-200209000-00015. [DOI] [PubMed] [Google Scholar]
- 10.Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–1292. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- 11.Festa A, et al. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS) Circulation. 2000;102:42–47. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- 12.Baron SH. Salicylates as hypoglycemic agents. Diabetes Care. 1982;5:64–71. doi: 10.2337/diacare.5.1.64. [DOI] [PubMed] [Google Scholar]
- 13.Yuan M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 14.Hundal RS, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J. Clin. Invest. 2002;109:1321–1326. doi:10.1172/JCI200214955. doi: 10.1172/JCI14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Hirosumi J, Uysal KT, Guler AD, Hotamisligil GS. Exclusive action of transmembrane TNF alpha in adipose tissue leads to reduced adipose mass and local but not systemic insulin resistance. Endocrinology. 2002;143:1502–1511. doi: 10.1210/endo.143.4.8715. [DOI] [PubMed] [Google Scholar]
- 17.Stahl A, et al. Identification of the major intestinal fatty acid transport protein. Mol. Cell. 1999;4:299–308. doi: 10.1016/s1097-2765(00)80332-9. [DOI] [PubMed] [Google Scholar]
- 18.Soukas A, Cohen P, Socci ND, Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev. 2000;14:963–980. [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida S, Setoguchi M, Higuchi Y, Akizuki S, Yamamoto S. Molecular cloning of cDNA encoding MS2 antigen, a novel cell surface antigen strongly expressed in murine monocytic lineage. Int. Immunol. 1990;2:585–591. doi: 10.1093/intimm/2.6.585. [DOI] [PubMed] [Google Scholar]
- 20.Wolpe SD, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J. Exp. Med. 1988;167:570–581. doi: 10.1084/jem.167.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshimura T, et al. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 1989;244:487–493. doi: 10.1016/0014-5793(89)80590-3. [DOI] [PubMed] [Google Scholar]
- 22.Pytela R. Amino acid sequence of the murine Mac-1 alpha chain reveals homology with the integrin family and an additional domain related to von Willebrand factor. EMBO J. 1988;7:1371–1378. doi: 10.1002/j.1460-2075.1988.tb02953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKnight AJ, et al. Molecular cloning of F4/80, a murine macrophage-restricted cell surface glycoprotein with homology to the G-protein-linked transmembrane 7 hormone receptor family. J. Biol. Chem. 1996;271:486–489. doi: 10.1074/jbc.271.1.486. [DOI] [PubMed] [Google Scholar]
- 24.Holness CL, da Silva RP, Fawcett J, Gordon S, Simmons DL. Macrosialin, a mouse macrophage-restricted glycoprotein, is a member of the lamp/lgp family. J. Biol. Chem. 1993;268:9661–9666. [PubMed] [Google Scholar]
- 25.O’Rourke L, Gronning LM, Yeaman SJ, Shepherd PR. Glucose-dependent regulation of cholesterol ester metabolism in macrophages by insulin and leptin. J. Biol. Chem. 2002;277:42557–42562. doi: 10.1074/jbc.M202151200. [DOI] [PubMed] [Google Scholar]
- 26.Charriere G, et al. Preadipocyte conversion to macrophage. Evidence of plasticity. J. Biol. Chem. 2003;278:9850–9855. doi: 10.1074/jbc.M210811200. [DOI] [PubMed] [Google Scholar]
- 27.Goldsby, R.A., Kindt, T.J., and Osborne, B.A. 2000. Kuby immunology. 4th Edition. W.H. Freeman and Co. New York, New York, USA. 371–393.
- 28.Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 29.Lowell BB. PPARgamma: an essential regulator of adipogenesis and modulator of fat cell function. Cell. 1999;99:239–242. doi: 10.1016/s0092-8674(00)81654-2. [DOI] [PubMed] [Google Scholar]
- 30.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 31.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 32.Marx N, Libby P, Plutzky J. Peroxisome proliferator-activated receptors (PPARs) and their role in the vessel wall: possible mediators of cardiovascular risk? J. Cardiovasc. Risk. 2001;8:203–210. doi: 10.1177/174182670100800404. [DOI] [PubMed] [Google Scholar]
- 33.Li AC, et al. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor–deficient mice. J. Clin. Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chawla A, et al. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 35.Moore KJ, et al. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 36.Klappacher GW, Glass CK. Roles of peroxisome proliferator-activated receptor gamma in lipid homeostasis and inflammatory responses of macrophages. Curr. Opin. Lipidol. 2002;13:305–312. doi: 10.1097/00041433-200206000-00011. [DOI] [PubMed] [Google Scholar]
- 37.Hotta K, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 38.Ouchi N, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 39.Ouchi N, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001;103:1057–1063. doi: 10.1161/01.cir.103.8.1057. [DOI] [PubMed] [Google Scholar]
- 40.Damerau B. Biological activities of complement-derived peptides. Rev. Physiol. Biochem. Pharmacol. 1987;108:151–206. doi: 10.1007/BFb0034073. [DOI] [PubMed] [Google Scholar]
- 41.Koistinen HA, et al. Plasma acylation stimulating protein concentration and subcutaneous adipose tissue C3 mRNA expression in nondiabetic and type 2 diabetic men. Arterioscler. Thromb. Vasc. Biol. 2001;21:1034–1039. doi: 10.1161/01.atv.21.6.1034. [DOI] [PubMed] [Google Scholar]
- 42.Choy LN, Rosen BS, Spiegelman BM. Adipsin and an endogenous pathway of complement from adipose cells. J. Biol. Chem. 1992;267:12736–12741. [PubMed] [Google Scholar]
- 43.Boggs RD, et al. Increased expression of complement component C3 in the plasma of obese Zucker fa and LA/N fa(f) rats compared with their lean counterparts. Obes. Res. 1998;6:361–367. doi: 10.1002/j.1550-8528.1998.tb00364.x. [DOI] [PubMed] [Google Scholar]
- 44.Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hrnciar J, et al. Relation between cytokines (TNF-alpha, IL-1 and 6) and homocysteine in android obesity and the phenomenon of insulin resistance syndromes. Vnitr. Lek. 1999;45:11–16. [PubMed] [Google Scholar]
- 46.Amrani A, et al. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. J. Endocrinol. 1996;148:139–148. doi: 10.1677/joe.0.1480139. [DOI] [PubMed] [Google Scholar]
- 47.Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 1998;83:847–850. doi: 10.1210/jcem.83.3.4660. [DOI] [PubMed] [Google Scholar]
- 48.McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992;258:766–770. doi: 10.1126/science.1439783. [DOI] [PubMed] [Google Scholar]
- 49.Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am. J. Physiol. 1999;276:E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- 50.Krssak M, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 51.Yu C, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 52.Shulman GI. Cellular mechanisms of insulin resistance. J. Clin. Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}