

Abstract
Analgesic efficacy varies depending on the pain syndrome being treated. One reason for this may be a differential effect of individual pain syndromes on the function of the endogenous pain control circuits at which these drugs act to produce analgesia. To test this hypothesis, we examined the effects of diverse (i.e., ongoing inflammatory, neuropathic, or chronic widespread) pain syndromes on analgesia induced by activation of an opioid-mediated, noxious stimulus-induced endogenous pain control circuit. This circuit was activated by subdermal capsaicin injection at a site remote from the site of nociceptive testing. Analgesia was not affected by carrageenan-induced inflammatory pain or the early phase of oxaliplatin neuropathy (a complication of cancer chemotherapy). However, the duration of analgesia was markedly shorter in the late phase of oxaliplatin neuropathy and in alcoholic neuropathy. A model of fibromyalgia syndrome produced by chronic unpredictable stress and proinflammatory cytokines also shortened analgesia duration, but so did the same stress alone. Therefore, since chronic pain can activate neuroendocrine stress axes, we tested whether they are involved in the attenuation of analgesic duration induced by these pain syndromes. Rats in which the sympathoadrenal axis was ablated by adrenal medullectomy showed normal duration pain-induced analgesia in groups with either late-phase oxaliplatin neuropathy, alcoholic neuropathy, or exposure to sound stress. These results support the suggestion that pain syndromes can modulate activity in endogenous pain control circuits and that this effect is sympathoadrenal dependent.
Introduction
Variability in response to analgesics is recognized as a major cause of inadequate pain control. The contribution of genetic variation has been extensively investigated (Lee et al., 2006; Smith et al., 2008; Shabalina et al., 2009; Argoff, 2010). Another source of variability—one that has received less attention—is the differential effect of a given analgesic across diverse pain syndromes. Although it is a common clinical observation that analgesic efficacy varies for different pain syndromes, the reasons for this remain poorly understood. Many drugs are thought to produce analgesia by acting at endogenous pain control circuits [e.g., descending pain modulation (Fields et al., 2006), stimulation-induced analgesia (Mayer and Liebeskind, 1974), diffuse noxious inhibitory controls (Le Bars et al., 1979), ascending nociceptive control (ANC) (Gear et al., 1999), stress-induced analgesia (Lewis et al., 1980), and placebo analgesia (Levine et al., 1978). Therefore, if a pre-existing pain condition, particularly chronic pain, was to compromise the normal function of an endogenous pain circuit, the ability of that circuit to respond to analgesic therapies could be degraded.
In the current study we investigated the effect of diverse pain syndromes on the function of an endogenous analgesia circuit, ANC, which can be physiologically activated by a noxious stimulus, subdermal capsaicin (Gear et al., 1999). This endogenous analgesia circuit may mediate some counter-irritation therapies, many of which use capsaicin-containing substances (Mason et al., 2004). This form of endogenous analgesia is long lasting in that it remains undiminished after 1 h, independent of continuation of the eliciting stimulus, and can produce analgesia equivalent to that of high dose morphine (10 mg/kg) (Gear et al., 1999) Importantly, ANC is a heterosegmental system that allows nociceptive testing to be segmentally remote from the site of noxious stimulation (i.e., capsaicin administration), thereby eliminating the confound of testing within the same segment where the inducing noxious stimulation is administered. In this study we refer to this form of endogenous analgesia as capsaicin-induced analgesia.
Five rat models covering the three major classes of pain syndromes (i.e., inflammatory, neuropathic, and generalized) were tested. Inflammatory pain was produced by focal intradermal injection of carrageenan on the dorsum of the hindpaw (Guilbaud et al., 1989a,b; Dawson et al., 1991; Aley et al., 2000; Khasar et al., 2008). Three neuropathic pain models were tested; one of these was a model of alcoholic painful peripheral neuropathy (Diamond and Messing, 1994; Monforte et al., 1995; Kielhorn, 1996; Ortiz-Plata et al., 1998; Dina et al., 2000), and two models (early- and late-phase neuropathy) were induced by intravenous injection of the chemotherapeutic agent oxaliplatin (Joseph et al., 2008; Kowalski et al., 2008). Enhanced cytokine hyperalgesia, a model of chronic widespread pain syndromes such as fibromyalgia (Khasar et al., 2005), was induced by cytokine administration after chronic unpredictable stress.
Finally, because chronic unpredictable stress activates the sympathoadrenal stress axis (Khasar et al., 2005), we investigated whether the adverse effects produced by other pain models are similarly dependent on the sympathoadrenal stress axis.
Materials and Methods
Animals
Male Sprague Dawley rats (220–320 g, n = 66; Charles River) were used in all experiments. Animals were housed in the Laboratory Animal Resource Center of the University of California, San Francisco, CA under a 12 h light/dark cycle. All experimental protocols were approved by the University of California, San Francisco, Institutional Animal Care and Use Committee and conformed to National Institutes of Health Guidelines for the Care and Use of Experimental Animals. Effort was made to limit the number of animals used and their discomfort.
Drugs
Capsaicin (lot # 018K5051) was dissolved in a 1:1 mixture of Tween 80 and ethanol to an initial concentration of 50 μg/μl (stock solution); further dilutions were made in 0.9% saline to a final concentration of 2.5 μg/μl. Oxaliplatin and carrageenan were dissolved in 0.9% saline. Prostaglandin E2 (PGE2), a direct-acting hyperalgesic agent, was initially dissolved in absolute ethanol to a concentration of 1 μg/μl and then diluted with saline (0.9%) to a final concentration of 20 ng/μl for subdermal injection on the dorsum of the hindpaw using a 30 gauge hypodermic needle attached to a Hamilton syringe. The volume of administration was 5 μl (100 ng); the concentration of ethanol in the final solution was <2%. All drugs were purchased from Sigma.
Experimental protocols
In the models of the five pain syndromes and in a control group, endogenous analgesia was elicited by subdermal injection of capsaicin (125 μg in a volume of 50 μl) administered under brief isoflurane (3.5%) anesthesia on a forepaw (Gear et al., 1999).
Inflammatory hyperalgesia.
Inflammatory hyperalgesia was induced by intradermal injection of the inflammagen carrageenan (5 μl, 1% solution) on the dorsum of the hindpaw (Aley et al., 2000). This local route of administration was chosen to minimize systemic effects of inflammatory mediators. Nociceptive testing was performed 2 d later at this injection site.
Cancer chemotherapy neuropathy.
Cancer chemotherapy neuropathy was induced by a single intravenous injection of oxaliplatin (2 mg/kg) followed by a bolus injection of an equal volume of saline before removal of the injection needle (Joseph et al., 2008). Nociceptive testing was performed either 5 d (early phase) or 15 d (late phase) after administration of oxaliplatin in separate groups of rats, as the neuropathic pain at these two time points is thought to have different mechanisms (Joseph and Levine, 2009).
Alcoholic neuropathy.
Alcoholic neuropathy was produced by adding ethanol (6.5%) to a Lieber–DeCarli liquid diet (Dyets ) (Lieber and DeCarli, 1982, 1989; Lieber et al., 1989) as described previously (Dina et al., 2000). Briefly, rats were individually caged and fed a weekly regimen, 4 d with ethanol diet and 3 d with normal control diet, for a total of 3 weeks. Nociceptive testing was performed 2 weeks after the 3 week ethanol diet was completed.
Stress-enhanced cytokine hyperalgesia.
Chronic unpredictable sound stress was performed as described previously and used in our laboratory (Strausbaugh et al., 2003; Khasar et al., 2005). Groups of three animals were placed 25 cm from a speaker in a 12 × 15 × 9.5 inch wire mesh cage inside a 22 × 22 × 28 inch sound-insulated box. Sound pulses were emitted as pure tones at three frequencies (11, 15, and 19 kHz); amplitudes varied from 20 to110 dB independently for each frequency. The sound exposure protocol was initiated immediately after placing rats in the wire mesh cage and terminated 30 min later, when rats were returned to their home cages. Over the 30 min period, a 5 or 10 s tone was presented every minute at random times during the minute. This sound stress protocol was performed on days 1, 3, and 4, and nociceptive testing was performed 14 d after the last exposure to this stressor.
Adrenal medullectomy
In some experiments the adrenal medulla was surgically excised bilaterally 5 weeks before any further intervention (Wilkinson et al., 1981). This surgical procedure was performed under isoflurane anesthesia (2.5% isoflurane in O2). Each adrenal gland was located through a 2 cm lateral incision in the abdominal wall, the capsule of the gland was cut open, and the adrenal medulla was extirpated (Wilkinson et al., 1981; Khasar et al., 1998). Incisions were closed with silk sutures, and carprofen (4–5 mg/kg), an injectable nonsteroidal anti-inflammatory drug, was administered subcutaneously in accordance with the University of California, San Francisco policy on postsurgical care of laboratory animals.
Nociceptive testing
Mechanical nociceptive threshold was quantified using the Randall–Selitto paw pressure test (Randall and Selitto, 1957) in which a force that increases linearly over time is applied to the dorsum of the hindpaw (Taiwo et al., 1989) (Ugo Basile Algesymeter, Stoelting). Nociceptive threshold was defined as the force in grams at which the rat withdrew its paw.
Testing was performed with rats gently restrained in acrylic cylinders designed to allow extension of the hindleg for testing, minimize restraint stress, and provide unrestricted ventilation. Rats were prepared for testing by training them (i.e., placing them in the restrainers and performing the Randall–Selitto paw pressure test) for 1 h per day for 3 d before the day of the experiment. On the day of the experiment, rats were habituated to the restrainers for 30 min before testing.
The baseline paw-withdrawal threshold was defined as the mean of three readings. Each paw was treated as an independent measure, and each experiment was performed on a separate group of rats. All behavioral testing was conducted between the hours of 10:00 and 16:00 by one individual (L.F.), who was not blinded. Thresholds for paw withdrawal were measured for both hindpaws immediately before the subdermal administration of capsaicin (125 μg) into the right forepaw and then at 15 min intervals for 1 h postadministration.
Statistics
Because baseline paw-withdrawal thresholds for the control group differed from that in the neuropathic (hyperalgesic) pain groups, paw-withdrawal threshold data are presented as changes in force in grams from baseline to normalize the differences in all figures, except Figure 2. Data in Figure 2 are presented as absolute force in grams to evoke paw withdrawal. To determine whether there were significant differences between experimental groups, we employed repeated-measures ANOVA with one within-subjects factor (time) and one between-subjects factor (group). If there was a significant group × time interaction, indicating that the groups in the analysis demonstrated significantly different time courses, multivariate analyses (i.e., one-way ANOVAs with Scheffé post hoc analyses) were performed for all time points to determine which points accounted for the interaction. In these cases, a Bonferroni correction was applied to account for multiple comparisons. For within-subjects effects, the Mauchly criterion was used to determine whether the assumption of sphericity was met; if not, Greenhouse–Geiser p values are presented. If the group × time interaction was not significant (see Fig. 2) and the main effect of group was significant (p < 0.05), Scheffé post hoc tests were used to determine the basis of the significant difference.
Figure 2.

Paw-withdrawal threshold in pain models. Pain models were induced as follows: intradermal carrageenan injection (1% in a volume of 5 μl), intravenous oxaliplatin administration (2 mg/kg, early-phase and late-phase groups), and alcoholic diet; controls received no prior treatment. Responses were not significantly different among the pain models, but all models demonstrated significant mechanical hyperalgesia compared to control animals; n = 4 for all groups.
Results
Capsaicin-induced analgesia in naive controls
Control rats were administered capsaicin (125 μg) to confirm its ability to produce analgesia (Gear et al., 1999); a separate group of control rats was administered capsaicin vehicle. Compared to vehicle, capsaicin significantly elevated paw-withdrawal thresholds over the entire 1 h observation period (Fig. 1), demonstrating its analgesic effect as observed in previous studies (Gear et al., 1999). Two-way repeated-measures ANOVA showed a significant group × time interaction (F(4,40) = 15.968; p < 0.001), as well as a significant main effect of group (F(1,10) = 69.390; p < 0.001).
Figure 1.

Analgesic effect of capsaicin in naive control rats. The analgesic responses measured in the hindpaw in control rats produced by either capsaicin or vehicle injected subdermally in the forepaw were significantly different. Rats that received capsaicin showed significantly elevated paw-withdrawal thresholds (i.e., analgesia); n = 8 for the group that received capsaicin, and n = 4 for the vehicle group. In this and subsequent figures (except Fig. 2), data are presented as the difference in force in grams from baseline at which withdrawal occurred and are plotted as group mean ± SEM at each time point; PWT, Paw-withdrawal threshold.
Development of hyperalgesia
To test for the presence of hyperalgesia in the five models of inflammatory and neuropathic pain, paw-withdrawal thresholds of groups of rats, each with a different pain syndrome, as well as a control group, were compared (Fig. 2). Rats exposed to chronic unpredictable stress were not included in this experiment because this treatment does not by itself induce hyperalgesia (Khasar et al., 2005, 2009). Two-way repeated-measures ANOVA with one between-subjects factor, a group with five levels (carrageenan, alcohol, early-phase and late-phase oxaliplatin, and control groups), and one within-subjects factor, time with five levels (prevehicle baseline and 15, 30, 45, and 60 min), showed a significant main effect of group (F(4,21) = 21.031; p < 0.001) but not a significant time × group interaction. The Scheffé post hoc analysis showed that the control group was significantly different from each of the pain models (p < 0.002). However, the pain models were not significantly different from each other.
Capsaicin-induced analgesia in the presence of inflammatory pain
Capsaicin-induced analgesia in carrageenan-treated rats did not differ significantly in magnitude or duration from that in controls (Fig. 3). ANOVA included one between-subjects factor, a group with three levels (control rats, carrageenan-treated, and carrageenan-treated rats that received vehicle instead of capsaicin), and one within-subjects factor, time with five levels. This analysis revealed a significant main effect of group (F(2,17) = 22.890; p < 0.001); Scheffé post hoc analysis revealed that the control group and the carrageenan/capsaicin group both differed significantly from the carrageenan/vehicle group (p < 0.001), but not from each other (p = 0.841). There was also a significant group × time interaction (F(8,68) = 6.821; p < 0.001); however, this finding reflected the difference between the carrageenan/vehicle group and the other two groups, which were not different from each other. These results indicate that carrageenan does not affect capsaicin-induced analgesia.
Figure 3.

Effect of carrageenan-induced inflammatory pain on capsaicin-induced analgesia. The analgesic responses of the control and carrageenan (carrag)-pretreated groups to capsaicin (cap) were not significantly different, suggesting that local carrageenan-induced inflammatory hyperalgesia does not affect capsaicin-induced analgesia; n = 8 for the carrageenan/capsaicin group. Data for the control group are replotted from Figure 1, and data for the carrageenan/vehicle (veh) group are replotted from Figure 2 to allow comparison.
Capsaicin-induced analgesia in a model of chronic widespread pain
Two groups were tested 2 weeks after the last exposure to sound stress. These groups were injected with either PGE2 (100 ng), a model for fibromyalgia syndrome (Khasar et al., 2005, 2009), or its vehicle 2.5 h before the experiment. Rats exposed to the sound stress protocol used in this study exhibit markedly prolonged PGE2 hyperalgesia (Khasar et al., 2005).
Sound stress/PGE2
As observed previously (Khasar et al., 2005), the groups that received PGE2 demonstrated lowered paw-withdrawal thresholds than control rats (data not shown). In rats injected with PGE2, capsaicin induced analgesia at 15 min, which diminished to baseline by 45 min (Fig. 4A).
Figure 4.
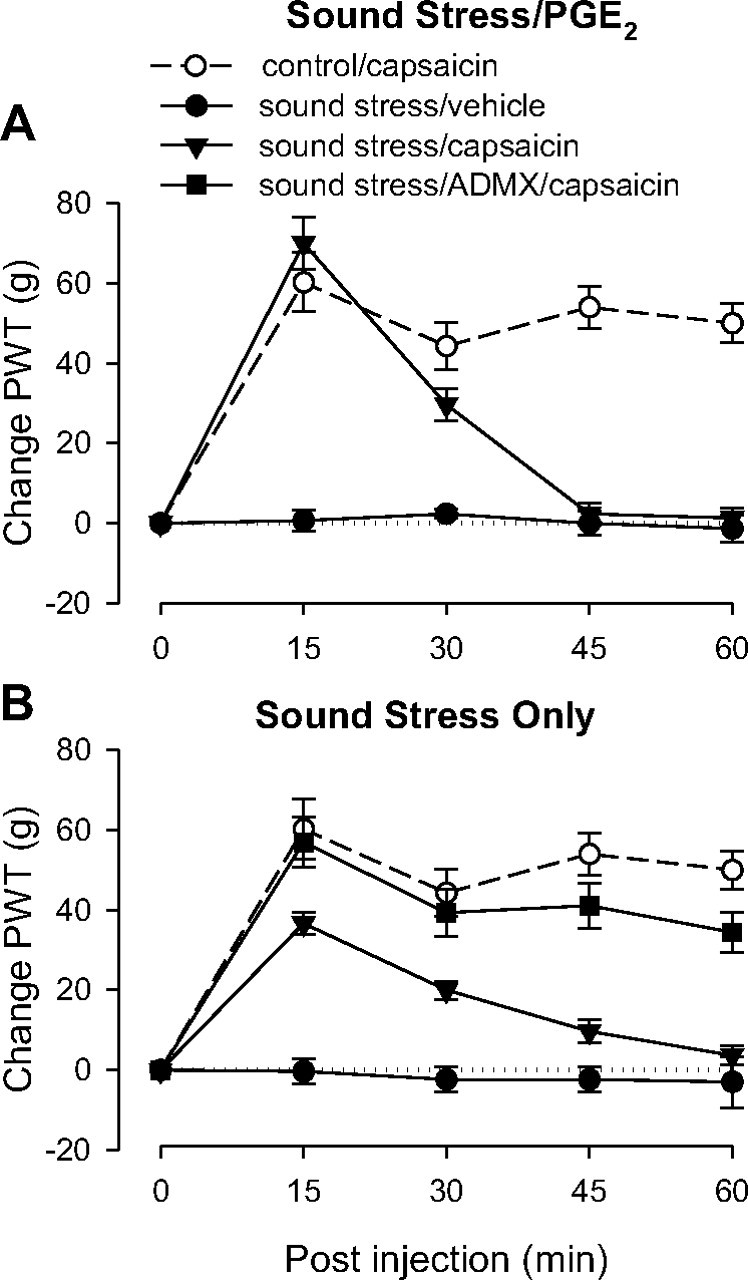
Effect of chronic widespread pain on capsaicin-induced analgesia. All groups of rats except controls experienced chronic unpredictable sound stress (see Materials and Methods) 14 d before testing. On the day of testing, 2.5 h before the experiment stressed rats received intradermal injections of either PGE2 or its vehicle on the dorsum of the hindpaw. A, PGE2. As expected, the pre-capsaicin baseline for both groups that received PGE2 was lower than the control group (data not shown). Paw-withdrawal thresholds (PWT) remained low for the group that received capsaicin vehicle, but the group that received capsaicin showed early antinociception that lasted only to the 30 min time point, indicating antagonism of capsaicin-induced analgesia duration; n = 6 for both sound stress groups. Data from control group are replotted from Figure 1 for comparison. B, No PGE2 pretreatment. Adrenal-intact rats showed short duration analgesia with a time course similar to that of the rats who received PGE2 (A), indicating that antagonism of capsaicin-induced analgesia was not due to PGE2 administration. This effect was abolished in ADMX rats, suggesting that the shortened analgesic duration is mediated by the sympathoadrenal axis.
ANOVA showed a significant group × time interaction (F(8,681) = 23.540; p < 0.001) and a significant main effect of group (F(2,17) = 51.924; p < 0.001). Scheffé post hoc analysis showed that both sound stress groups were significantly different from the control group (p < 0.001, p = 0.013) but also significantly different from each other (p < 0.001). Based on the significant interaction, multivariate analysis with Scheffé post hoc tests showed that the sound stress/capsaicin group differed significantly from the control group at 45 as well as 60 min (both p < 0.001).
Sound stress alone
The group that received vehicle instead of PGE2 allowed assessment of the effect of stress alone on the efficacy of capsaicin-induced analgesia (Fig. 4B). In this group capsaicin produced antinociception by 15 min, which, like the group that received PGE2, diminished to baseline nociceptive threshold by 45 min. Thus, the shorter duration of capsaicin-induced antinociception also occurred in the absence of PGE2. To determine whether this effect was dependent on the sympathoadrenal stress axis, a separate group of rats underwent bilateral removal of the adrenal medulla (ADMX). Five weeks after surgery, this group was exposed to the same sound stress protocol as above. Two weeks later their response to capsaicin was similar to that observed in control rats (Fig. 4B), suggesting that the attenuation of analgesic duration induced by sound stress is dependent on the sympathoadrenal axis.
A two-way repeated-measures ANOVA showed a significant group × time interaction (F(12,136) = 5.028; p < 0.001) and a significant main effect of group (F(3,34) = 23.386; p < 0.001). Based on the significant interaction, multivariate analysis with Scheffé post hoc tests showed that the sound stress/ADMX/capsaicin group did not differ significantly from the control group at any time point (p > 0.292), but the sound stress/capsaicin group differed significantly from the control group as well as the sound stress/ADMX/capsaicin group at 45 min (p = 0.014 and p = 0.002, respectively) and also at 60 min (p = 0.013 and p = 0.001, respectively).
Capsaicin-induced analgesia in the presence of neuropathic pain
Alcohol
Capsaicin elevated paw-withdrawal thresholds in rats with alcoholic neuropathy during the first 15 min, but the duration of the effect was shortened, with nociceptive thresholds virtually returning to baseline by 45 min (Fig. 5). To determine whether this effect was dependent on the sympathoadrenal stress axis, a separate ADMX group was tested. Alcoholic neuropathy was induced in this group in an identical manner to that of the non-ADMX group. In contrast to the intact rats, the magnitude of analgesia in the ADMX group remained elevated throughout the entire period of observation, suggesting that ADMX reconstituted capsaicin-induced analgesia in rats with alcoholic neuropathy.
Figure 5.

Effect of alcoholic neuropathy on capsaicin-induced analgesia. The responses of the control and the nonadrenal medullectomized alcohol (EtOH)-pretreated groups to capsaicin were significantly different at the 45 and 60 min time points, indicating that alcohol neuropathy shortens the duration of capsaicin-induced analgesia. This effect was abolished in adrenal medullectomized rats, suggesting that activation of the sympathoadrenal stress axis antagonizes capsaicin-induced analgesia; n = 8 for the EtOH/capsaicin group and the EtOH/capsaicin/ADMX group. Data for the control group are replotted from Figure 1, and data for the EtOH/vehicle group are replotted from Figure 2 to allow comparison. PWT, Paw-withdrawal threshold.
ANOVA showed a significant group × time interaction (F(12,96) = 9.224; p < 0.001) as well as a significant main effect of group (F(3,24) = 20.299; p < 0.001). Based on the significant interaction, multivariate analysis with Scheffé post hoc tests was performed to determine which time points were significantly different. This analysis showed that the control group did not differ significantly from the ethanol (EtOH)/ADMX/capsaicin group at any time point, but both of these groups differed significantly from intact EtOH/capsaicin group at 45 min (p < 0.001, p = 0.008, respectively) and at 60 min (p < 0.001, p = 0.023, respectively).
Oxaliplatin
Separate groups of oxaliplatin-treated rats were tested at one of two time points: those that received oxaliplatin 5 d before testing (early phase) and those that received oxaliplatin 15 d before testing (late phase) (Joseph and Levine, 2009). To assess involvement of the sympathoadrenal axis, these experiments were performed in separate groups of intact and ADMX rats.
Early-phase oxaliplatin
Capsaicin elevated paw-withdrawal thresholds in both early-phase oxaliplatin groups (i.e., ADMX and intact) similarly as that observed in control rats (Fig. 6A), and at no time point did these groups differ from controls, indicating that early-phase oxaliplatin neuropathy does not significantly affect capsaicin-induced analgesia.
Figure 6.
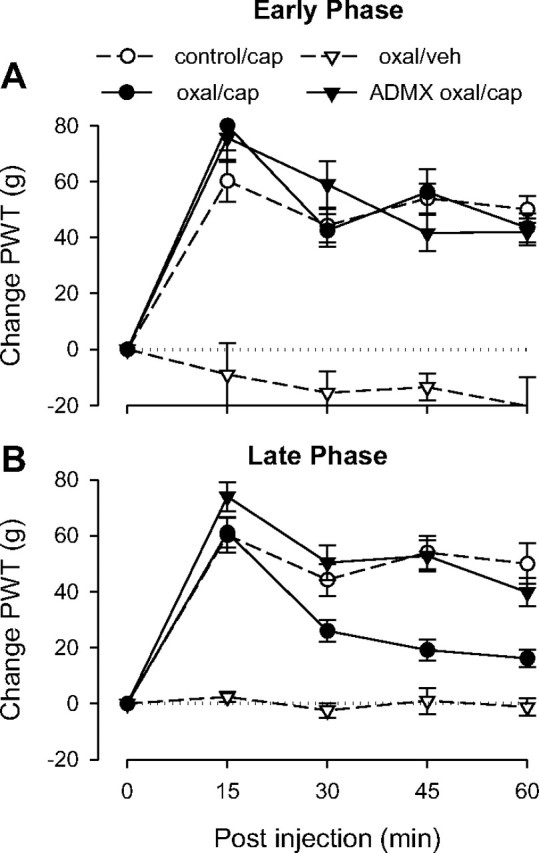
Effect of early- and late-phase oxaliplatin-induced neuropathy on capsaicin-induced analgesia. A single dose of oxaliplatin (oxal) was administered intravenously. Testing was performed in separate groups of rats either 2 d (“Early Phase”) or 15 d (“Late Phase”) after oxaliplatin administration. A, Early phase. The responses of the control and the intact and ADMX oxaliplatin-pretreated groups to capsaicin (cap) were not significantly different, indicating that early-phase oxaliplatin neuropathy does not affect capsaicin-induced analgesia; n = 8 for both the oxaliplatin/capsaicin and the ADMX/oxaliplatin/capsaicin groups. Data for the control group are replotted from Figure 2, and data for the oxaliplatin/vehicle (veh) group are replotted from Figure 1 to allow comparison. B, Capsaicin-induced analgesia was significantly shortened in the late-phase intact oxaliplatin group but not the adrenal medullectomized oxaliplatin group, suggesting that late-phase oxaliplatin neuropathy antagonizes capsaicin-induced analgesia and that this effect is mediated by the sympathoadrenal axis; n = 12 for the oxaliplatin/capsaicin group; n = 10 for the ADMX/oxaliplatin/capsaicin group. Data for the control group are replotted from Figure 1, and data for the oxaliplatin/vehicle group are replotted from Figure 2 to allow comparison.
ANOVA showed a significant time × group interaction (F(12,104) = 6.734; p < 0.001) as well as a significant main effect of group (F(3,26) = 23.723; p < 0.001). Based on the significant interaction, multivariate analysis was performed to determine which time points were significantly different. Early-phase oxaliplatin-treated rats that received vehicle instead of capsaicin differed significantly at all time points (p < 0.001) from the other three groups, which did not differ significantly from each other at any time point.
Late-phase oxaliplatin
Capsaicin elevated paw-withdrawal thresholds in both late-phase oxaliplatin groups (i.e., ADMX and intact) similarly as in control rats at 15 min (Fig. 6B). However, the intact late-phase oxaliplatin group, but not the ADMX group, showed significant shortening of capsaicin-induced analgesia compared to its duration in the control, suggesting that the adverse effect on analgesia observed in late-phase oxaliplatin neuropathy, as in alcoholic neuropathy, is sympathoadrenal dependent.
ANOVA showed a significant group × time interaction (F(12,144) = 13.121; p < 0.001) as well as a significant main effect of group (F(3,36) = 60.708; p < 0.001). Based on the significant interaction, multivariate analysis with Scheffé post hoc tests showed that the late-phase oxaliplatin ADMX group and the control group did not differ significantly at any time point. However, the ADMX group did differ significantly from the adrenal-intact group at 30 (p = 0.006), 45 (p < 0.001), and 60 min (p = 0.006).
Discussion
In these experiments we used models of the three major classes of clinical pain (i.e., inflammatory, neuropathic, and chronic widespread) to examine if and how they affect the function of an endogenous pain modulation system. Of the five tested pain models, two, inflammatory hyperalgesia and early-phase oxaliplatin neuropathy (5 d after oxaliplatin administration), had no effect on capsaicin-induced analgesia, suggesting that hyperalgesia alone is insufficient to adversely affect function of an endogenous analgesic circuit function. Three of the five tested pain models, alcoholic neuropathy, late-phase (15 d postadministration) oxaliplatin neuropathy, and chronic unpredictable stress, adversely affected capsaicin-induced analgesia by shortening its duration without significantly affecting its peak magnitude. These results support the underlying hypothesis that chronic pain syndromes can produce changes in endogenous analgesia circuits, effects that could degrade the efficacy of treatments whose analgesic effects are mediated by these circuits.
The differences in the effects of early- and late-phase oxaliplatin neuropathy may relate to recent clinical evidence that suggests two distinct oxaliplatin neuropathic pain syndromes (Gamelin et al., 2002; Lehky et al., 2004; Cersosimo, 2005). We have recently demonstrated these two syndromes in the rat and have shown that the mechanical hyperalgesia associated with these two forms of oxaliplatin-induced painful neuropathy are mediated by distinct mechanisms (Joseph and Levine, 2009). Early-phase oxaliplatin hyperalgesia is characterized by prominent cold allodynia and is not attenuated by inhibitors of caspase signaling (Joseph and Levine, 2009). In contrast, late-phase oxaliplatin hyperalgesia is characterized by no cold allodynia and attenuated by inhibitors of caspase signaling. The findings of the current study suggest that, in addition to these differences, late-phase but not early-phase oxaliplatin attenuates the duration of capsaicin-induced analgesia.
Chronic unpredictable stress produced a similar shortening of capsaicin-induced analgesia, both in the presence and absence of PGE2 administration. The lack of dependence on PGE2 hyperalgesia was unexpected, because our previous studies showed that the pronociceptive effect of stress could not be detected without PGE2 as an adjunctive pronociceptive treatment (Khasar et al., 2005, 2008). Those studies also demonstrated that the hypothalamic-pituitary-adrenal and sympathoadrenal stress axes both play a role in the ability of unpredictable sound stress to enhance cytokine hyperalgesia.
Adrenal medullectomy restored normal capsaicin-induced analgesia in late-phase oxaliplatin neuropathy, alcoholic neuropathy, and chronic unpredictable stress, suggesting a common sympathoadrenal-dependent mechanism. In contrast to these findings, we have observed this form of analgesia to be unchanged in otherwise normal adrenal medullectomized and hypophysectomized rats (R. Gear and J. Levine, unpublished observations), indicating that neuroendocrine stress axes do not play a role in producing this form of noxious stimulus-induced analgesia.
Previous studies from our laboratory found that the hyperalgesia associated with alcoholic neuropathy and chronic unpredictable stress (in combination with a proinflammatory cytokine) are sympathoadrenal dependent and are mediated by the effects of stress hormones at the level of the primary afferent nociceptor (Khasar et al., 2005; Dina et al., 2008; Khasar et al., 2008). Late-phase oxaliplatin hyperalgesia, however, is not sympathoadrenal dependent (see Table 1 for a summary of the role the sympathoadrenal axis in these models). But several lines of evidence point to a contribution of a CNS mechanism: first, the site of nociceptive testing (hindpaw) is segmentally remote from the site of capsaicin administration in the forepaw, implying a circuit-mediated effect; second, capsaicin-induced analgesia, once initiated, does not require continued peripheral input (Gear et al., 1999); and third, chronic unpredictable sound stress does not by itself produce hyperalgesia, arguing against a role for primary afferent nociceptors.
Table 1.
Effect of ADMX on hyperalgesia, preinduction and postinductiona
| Model | Preinductionb | Postinductionb | Intactc | ADMXc |
|---|---|---|---|---|
| Sound stress: intact | 102 ± 1.1 | 102 ± 0.9 | ||
| Alcohol: intact* | 110 ± 1.5 | 76 ± 1.8 | ||
| Alcohol: ADMX | 111 ± 1.7 | 112 ± 1.2 | ||
| Oxaliplatin late phase: intact* | 108 ± 1.6 | 81 ± 2.0 | ||
| Oxaliplatin late phase: ADMX* | 115 ± 2.7 | 90 ± 1.6 | ||
| Summary of above findings | ||||
| Sound stress | No | No | ||
| Alcohol | Yes | No | ||
| Oxaliplatin-late phase | Yes | Yes |
aAlthough adrenal medullectomy (ADMX) abolished the adverse effects of all three of these neuropathy models on capsaicin-induced analgesia, its effects on hyperalgesia were variable, suggesting first that hyperalgesia in these models has different underlying causes and second that hyperalgesia is not required for degrading the efficacy of capsaicin-induced analgesia.
bPaw-withdrawal thresholds were measured in groups of rats prior to induction of the pain model (“preinduction”) and again prior to capsaicin or vehicle administration (“postinduction”). Data are presented as mean ± SEM. Sound stress by itself did not induce hyperalgesia; the alcohol protocol induced hyperalgesia in intact rats, but not in ADMX rats, indicating the hyperalgesic effect in intact rats is sympathoadrenal dependent; both intact and ADMX late-phase oxaliplatin groups developed hyperalgesia, indicating that this form of hyperalgesia is not sympathoadrenal dependent. Asterisk (*) indicates significant difference (p < 0.05) between preinduction and postinduction.
cSummary of findings in the top portion of the table shows the presence of hyperalgesia in intact and ADMX rats for each of the three models. “Yes” indicates hyperalgesia was present; “No” indicates lack of hyperalgesia.
Although it is well established that nociceptive input can enhance pain (e.g., peripheral sensitization, wind-up, and other forms of central sensitization), these changes occur in what are generally regarded as pain transmission rather than pain modulation pathways. Pain modulation circuits can also be adversely affected: for example, neuropathy induced by spinal nerve ligation has been shown to produce descending nociceptive facilitation mediated in the rostral ventral medulla (RVM), a major component of the descending pain inhibitory system (Kovelowski et al., 2000); the RVM was also shown to play a facilitatory role in the persistent pain associated with pancreatitis (Vera-Portocarrero et al., 2006) as well as fentanyl hypersensitivity (Rivat et al., 2009) and by tolerance resulting from prolonged exposure to drugs like morphine, thereby degrading analgesic efficacy (Vanderah et al., 2001). The present results suggest that modulation of activity in endogenous pain control mechanisms by pain may be an important determinant of analgesic efficacy.
The ANC (Gear et al., 1999; Schmidt et al., 2002a,b) was chosen as the endogenous pain modulation mechanism to examine for the following reasons: (1) it is activated by physiologically relevant events [i.e., noxious stimulation (Gear et al., 1999); for example, capsaicin administration as used in the current study]; (2) much of the spinal circuitry (Tambeli et al., 2002, 2003a,b) and supraspinal circuitry (Gear et al., 1999; Schmidt et al., 2001; Schmidt et al., 2002a,b; Gear and Levine, 2009) is known; (3) it is of relatively long duration [>2 h relative to the duration of the initiating stimulus (M. Ku, R. Gear, and J. Levine, unpublished observations)]; and (4) it has been characterized in a variety of physiological conditions, including in naive animals (Gear et al., 1999; Schmidt et al., 2002a), animals made tolerant to morphine (Schmidt et al., 2002b), animals undergoing precipitated withdrawal from morphine (Schmidt et al., 2003), and animals made tolerant to nicotine (Schmidt et al., 2001). In each of these conditions, this endogenous analgesic circuit retains its ability to be activated by noxious stimuli to produce profound pain-induced analgesia, although it is altered in some cases in terms of the specific CNS receptors that mediate its effects (Schmidt et al., 2001, 2002b). It was therefore of interest to examine its interaction with chronic pain.
An important area for future research relates to how dysfunctional changes in analgesia produced by these diverse pain syndromes would compare to the effects of analgesic drugs across these same pain syndromes. For the method used to induce analgesia in the present study (i.e., subdermal capsaicin administration), we have previously shown an important role of μ- and δ-opioid receptors (Schmidt et al., 2002a) and other mechanisms [e.g., metabotropic glutamate receptors (Tambeli et al., 2002, 2003a), GABA receptors (Tambeli et al., 2003b), nicotinic receptors (Schmidt et al., 2001; Gear and Levine, 2009), and dopamine receptors (Gear et al., 1999; Schmidt et al., 2002b)] at spinal and supraspinal sites. As the response of neuropathic pain to opioids is especially variable [for review, see Backonja and Rowbotham (2006)], future experiments will compare the analgesic effect of opioids in different pain syndromes.
In conclusion, we have shown that diverse pain syndromes, including cancer chemotherapy, alcoholic neuropathy, and stress-induced chronic widespread pain, induce sympathoadrenal-dependent dysfunction of the ascending nociceptive control circuit, an endogenous form of pain control. These findings imply that the treatment of chronic pain—notoriously, the most difficult pain problem to manage—is made more difficult in part by the ability of the pain itself to degrade the responsiveness of endogenous pain controls. Although preliminary, these findings point to potentially fruitful research with clinical implications. For example, treatments designed to mitigate the adverse effects of sympathoadrenal activation might be useful adjuncts to other treatments meant to activate pain modulation circuits. Beyond this, capsaicin and many other counter-irritation interventions have long been used as therapies for chronic pain (Mason et al., 2004); therefore, these findings could help to explain the large variation in efficacy of counter-irritation-based therapies across pain syndromes. Other pain syndromes, including an acute effect of a cancer chemotherapy and focal inflammatory hyperalgesia, had no adverse effect on this pain-induced analgesia. Important questions that remain to be investigated are how the sympathoadrenal stress axis interacts with the ANC circuit to degrade its analgesic properties and whether this interaction is unique to this endogenous analgesia circuit or if other endogenous pain modulation circuits are similarly affected. Answering these questions could help explain the variable efficacy of analgesic treatments across diverse pain syndromes.
Footnotes
This work was funded by the National Institutes of Health Grant AR054635.
References
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argoff CE. Clinical implications of opioid pharmacogenetics. Clin J Pain. 2010;26(Suppl 10):S16–S20. doi: 10.1097/AJP.0b013e3181c49e11. [DOI] [PubMed] [Google Scholar]
- Backonja M, Rowbotham MC. Pharmacological therapy for neuropathic pain. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's Textbook of Pain. 5th Ed. Philadelphia: Elsevier/Churchill Livingstone; 2006. pp. 1075–1083. [Google Scholar]
- Cersosimo RJ. Oxaliplatin-associated neuropathy: a review. Ann Pharmacother. 2005;39:128–135. doi: 10.1345/aph.1E319. [DOI] [PubMed] [Google Scholar]
- Dawson J, Sedgwick AD, Edwards JC, Lees P. A comparative study of the cellular, exudative and histological responses to carrageenan, dextran and zymosan in the mouse. Int J Tissue React. 1991;13:171–185. [PubMed] [Google Scholar]
- Diamond I, Messing RO. Neurologic effects of alcoholism. West J Med. 1994;161:279–287. [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Barletta J, Chen X, Mutero A, Martin A, Messing RO, Levine JD. Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J Neurosci. 2000;20:8614–8619. doi: 10.1523/JNEUROSCI.20-22-08614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Alessandri-Haber N, Green PG, Messing RO, Levine JD. Alcohol-induced stress in painful alcoholic neuropathy. Eur J Neurosci. 2008;27:83–92. doi: 10.1111/j.1460-9568.2007.05987.x. [DOI] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's Textbook of Pain. 5th Ed. Philadelphia: Elsevier/Churchill Livingstone; 2006. pp. 125–142. [Google Scholar]
- Gamelin E, Gamelin L, Bossi L, Quasthoff S. Clinical aspects and molecular basis of oxaliplatin neurotoxicity: current management and development of preventive measures. Semin Oncol. 2002;29(5 Suppl 15):21–33. doi: 10.1053/sonc.2002.35525. [DOI] [PubMed] [Google Scholar]
- Gear RW, Levine JD. Rostral ventral medulla cholinergic mechanism in pain-induced analgesia. Neurosci Lett. 2009;464:170–172. doi: 10.1016/j.neulet.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gear RW, Aley KO, Levine JD. Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci. 1999;19:7175–7181. doi: 10.1523/JNEUROSCI.19-16-07175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Benoist JM, Eschalier A, Kayser V, Gautron M, Attal N. Evidence for central phenomena participating in the changes of responses of ventrobasal thalamic neurons in arthritic rats. Brain Res. 1989a;484:383–388. doi: 10.1016/0006-8993(89)90386-7. [DOI] [PubMed] [Google Scholar]
- Guilbaud G, Benoist JM, Eschalier A, Gautron M, Kayser V. Evidence for peripheral serotonergic mechanisms in the early sensitization after carrageenan-induced inflammation: electrophysiological studies in the ventrobasal complex of the rat thalamus using a potent specific antagonist of peripheral 5-HT receptors. Brain Res. 1989b;502:187–197. doi: 10.1016/0006-8993(89)90475-7. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Comparison of oxaliplatin- and cisplatin-induced painful peripheral neuropathy in the rat. J Pain. 2009;10:534–541. doi: 10.1016/j.jpain.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Bogen O, Levine JD. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain. 2008;9:463–472. doi: 10.1016/j.jpain.2008.01.335. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Miao FJ, Jänig W, Levine JD. Vagotomy-induced enhancement of mechanical hyperalgesia in the rat is sympathoadrenal-mediated. J Neurosci. 1998;18:3043–3049. doi: 10.1523/JNEUROSCI.18-08-03043.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Green PG, Levine JD. Repeated sound stress enhances inflammatory pain in the rat. Pain. 2005;116:79–86. doi: 10.1016/j.pain.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28:5721–5730. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Dina OA, Green PG, Levine JD. Sound stress-induced long-term enhancement of mechanical hyperalgesia in rats is maintained by sympathoadrenal catecholamines. J Pain. 2009;10:1073–1077. doi: 10.1016/j.jpain.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielhorn FW. The history of alcoholism: Brühl-Cramer's concepts and observations. Addiction. 1996;91:121–128. [PubMed] [Google Scholar]
- Kovelowski CJ, Ossipov MH, Sun H, Lai J, Malan TP, Porreca F. Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain. 2000;87:265–273. doi: 10.1016/S0304-3959(00)00290-6. [DOI] [PubMed] [Google Scholar]
- Kowalski T, Maier C, Reinacher-Schick A, Schlegel U. Painful hyperexcitability syndrome with oxaliplatin containing chemotherapy: clinical features, pathophysiology and therapeutic options. Schmerz. 2008;22:16–23. doi: 10.1007/s00482-007-0552-5. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Dickenson AH, Besson JM. Diffuse noxious inhibitory controls (DNIC). I. Effects on dorsal horn convergent neurones in the rat. Pain. 1979;6:283–304. doi: 10.1016/0304-3959(79)90049-6. [DOI] [PubMed] [Google Scholar]
- Lee YS, Kim H, Wu TX, Wang XM, Dionne RA. Genetically mediated interindividual variation in analgesic responses to cyclooxygenase inhibitory drugs. Clin Pharmacol Ther. 2006;79:407–418. doi: 10.1016/j.clpt.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Lehky TJ, Leonard GD, Wilson RH, Grem JL, Floeter MK. Oxaliplatin-induced neurotoxicity: acute hyperexcitability and chronic neuropathy. Muscle Nerve. 2004;29:387–392. doi: 10.1002/mus.10559. [DOI] [PubMed] [Google Scholar]
- Levine JD, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet. 1978;2:654–657. doi: 10.1016/s0140-6736(78)92762-9. [DOI] [PubMed] [Google Scholar]
- Lewis JW, Cannon JT, Stapleton JM, Liebeskind JC. Stress activates endogenous pain-inhibitory systems: opioid and nonopioid mechanisms. Proc West Pharmacol Soc. 1980;23:85–88. [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcohol Clin Exp Res. 1982;6:523–531. doi: 10.1111/j.1530-0277.1982.tb05017.x. [DOI] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989;24:197–211. [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology. 1989;10:501–510. doi: 10.1002/hep.1840100417. [DOI] [PubMed] [Google Scholar]
- Mason L, Moore RA, Derry S, Edwards JE, McQuay HJ. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991. doi: 10.1136/bmj.38042.506748.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer DJ, Liebeskind JC. Pain reduction by focal electrical stimulation of the brain: an anatomical and behavioral analysis. Brain Res. 1974;68:73–93. doi: 10.1016/0006-8993(74)90534-4. [DOI] [PubMed] [Google Scholar]
- Monforte R, Estruch R, Valls-Solé J, Nicolás J, Villalta J, Urbano-Marquez A. Autonomic and peripheral neuropathies in patients with chronic alcoholism: a dose-related toxic effect of alcohol. Arch Neurol. 1995;52:45–51. doi: 10.1001/archneur.1995.00540250049012. [DOI] [PubMed] [Google Scholar]
- Ortiz-Plata A, Palencia G, Garcia E, Perez R, Sotelo J. Ultrastructural changes in limb distal nerves of rats with alcoholism and/or malnutrition before and after dietary correction. J Appl Toxicol. 1998;18:89–92. doi: 10.1002/(sici)1099-1263(199803/04)18:2<89::aid-jat479>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–419. [PubMed] [Google Scholar]
- Rivat C, Vera-Portocarrero LP, Ibrahim MM, Mata HP, Stagg NJ, De Felice M, Porreca F, Malan TP. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci. 2009;29:727–737. doi: 10.1111/j.1460-9568.2009.06616.x. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Gear RW, Levine JD. Nicotine withdrawal hyperalgesia and opioid-mediated analgesia depend on nicotine receptors in nucleus accumbens. Neuroscience. 2001;106:129–136. doi: 10.1016/s0306-4522(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Levine JD, Gear RW. mu/delta Cooperativity and opposing kappa-opioid effects in nucleus accumbens-mediated antinociception in the rat. Eur J Neurosci. 2002a;15:861–868. doi: 10.1046/j.1460-9568.2002.01915.x. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Barletta J, Luo L, Green P, Levine JD, Gear RW. Altered nucleus accumbens circuitry mediates pain-induced antinociception in morphine-tolerant rats. J Neurosci. 2002b;22:6773–6780. doi: 10.1523/JNEUROSCI.22-15-06773.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Levine JD, Gear RW. Adaptations in nucleus accumbens circuitry during opioid withdrawal associated with persistence of noxious stimulus-induced antinociception in the rat. J Pain. 2003;4:141–147. doi: 10.1054/jpai.2003.12. [DOI] [PubMed] [Google Scholar]
- Shabalina SA, Zaykin DV, Gris P, Ogurtsov AY, Gauthier J, Shibata K, Tchivileva IE, Belfer I, Mishra B, Kiselycznyk C, Wallace MR, Staud R, Spiridonov NA, Max MB, Goldman D, Fillingim RB, Maixner W, Diatchenko L. Expansion of the human mu-opioid receptor gene architecture: novel functional variants. Hum Mol Genet. 2009;18:1037–1051. doi: 10.1093/hmg/ddn439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Marker CL, Perry C, Liao G, Sotocinal SG, Austin JS, Melmed K, Clark JD, Peltz G, Wickman K, Mogil JS. Quantitative trait locus and computational mapping identifies Kcnj9 (GIRK3) as a candidate gene affecting analgesia from multiple drug classes. Pharmacogenet Genomics. 2008;18:231–241. doi: 10.1097/FPC.0b013e3282f55ab2. [DOI] [PubMed] [Google Scholar]
- Strausbaugh HJ, Green PG, Dallman MF, Levine JD. Repeated, nonhabituating stress suppresses inflammatory plasma extravasation by a novel, sympathoadrenal dependent mechanism. Eur J Neurosci. 2003;17:805–812. doi: 10.1046/j.1460-9568.2003.02493.x. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Coderre TJ, Levine JD. The contribution of training to sensitivity in the nociceptive paw-withdrawal test. Brain Res. 1989;487:148–151. doi: 10.1016/0006-8993(89)90950-5. [DOI] [PubMed] [Google Scholar]
- Tambeli CH, Parada CA, Levine JD, Gear RW. Inhibition of tonic spinal glutamatergic activity induces antinociception in the rat. Eur J Neurosci. 2002;16:1547–1553. doi: 10.1046/j.1460-9568.2002.02204.x. [DOI] [PubMed] [Google Scholar]
- Tambeli CH, Young A, Levine JD, Gear RW. Contribution of spinal glutamatergic mechanisms in heterosegmental antinociception induced by noxious stimulation. Pain. 2003a;106:173–179. doi: 10.1016/s0304-3959(03)00332-4. [DOI] [PubMed] [Google Scholar]
- Tambeli CH, Quang P, Levine JD, Gear RW. Contribution of spinal inhibitory receptors in heterosegmental antinociception induced by noxious stimulation. Eur J Neurosci. 2003b;18:2999–3006. doi: 10.1111/j.1460-9568.2003.03031.x. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Xie JY, Yie JX, Kowal J, Ossipov MH, King T, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains visceral pain in rats with experimental pancreatitis. Gastroenterology. 2006;130:2155–2164. doi: 10.1053/j.gastro.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Wilkinson CW, Shinsako J, Dallman MF. Return of pituitary-adrenal function after adrenal enucleation or transplantation: diurnal rhythms and responses to ether. Endocrinology. 1981;109:162–169. doi: 10.1210/endo-109-1-162. [DOI] [PubMed] [Google Scholar]