

Abstract
In an effort to improve the bioavailability of the non-selective, cyclic enkephalin analogues H-Dmt-c[D-Cys-Gly-Phe-D(or L)-Cys]NH2 (Dmt = 2′,6′-dimethyltyrosine), analogues N-methylated at the Phe4 and/or Cys5 residue were synthesized. In comparison with the non-methylated parent peptides, all mono- and di-N-methylated analogues in general retained high binding affinities at all three opioid receptors and high opioid agonist potencies in functional opioid activity assays. The results indicate that the progressive conformational restriction in these compounds upon mono- and di-N-methylation did not significantly affect the in vitro opioid activity profile. A low-energy conformer identified for the conformationally most restricted analogue of the series, H-Dmt-c[D-Cys-Gly-Phe(NMe)-L-Cys(NMe)]NH2 (6), showed good spatial overlap of the essential pharmacophoric moieties with those in the proposed μ receptor-bound conformation of the μ-selective opioid peptide JOM-6 [H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]NH2] (Pen = penicillamine) [Mosberg M.I. and Fowler C.B. (2002) J Peptide Res; 60:329-335], in agreement with the moderate μ selectivity determined for this compound. An analogue of 6 containing (2S)-2-methyl-3-(2,6-dimethyl-4-hydroxyphenyl)propanoic acid [(2S)-Mdp] in place of Dmt1 was an opioid antagonist with quite high opioid receptor binding affinities and can be expected to show improved bioavailability due to its further increased lipophilicity and reduced hydrogen-bonding capacity.
Keywords: opioid peptide analogues, peptide synthesis, N-methylation of peptides, opioid activity profiles, theoretical conformational analysis of peptides, opioid peptide SAR
Cystine-containing cyclic opioid peptide analogues were first reported three decades ago. The two prototype cyclic enkephalin analogues of this type with a C-terminal carboxamide group, H-Tyr-c[D-Cys-Gly-Phe-D-(or L)-Cys]NH2 were independently synthesized by two groups (1,2). Both diasteroisomers showed high μ and δ opioid receptor binding affinities, high μ and δ opioid agonist potencies in vitro and no μ vs. δ receptor selectivity. Cyclic tetrapeptide analogues derived from these compounds by deletion of the Gly residue, H-Tyr-c[D-Cys-Phe-D(or L)-Cys]NH2 retained μ and δ opioid agonist activity, albeit with lower potency as compared to the parent cyclic pentapeptides, and the L-Cys4-analogue was μ-selective (3,4). Dicarba analogues of these cyclic penta- and tetrapeptide amides, containing a -CH=CH- (cis and trans) or a -CH2-CH2- bond in place of the disulfide linkage, were prepared (4,5). Both the olefinic and the saturated dicarba pentapeptide analogues retained high μ and δ receptor binding affinities and high μ and δ opioid agonist activity in vitro. In comparison with their respective disulfide-containing parent tetrapeptides, the dicarba tetrapeptides displayed comparable or reduced μ and δ agonist potencies. Another interesting structural modification of the tetrapeptide H-Tyr-c[D-Cys-Phe-D-Cys]NH2 resulted in the compound JOM-6 (H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]NH2), in which D-penicillamine (D-Pen) is substituted for D-Cys4,and the disulfide moiety is replaced by an ethylene dithioether (6). JOM-6 turned out to be a potent and selective μ opioid receptor ligand.
N-methylation of amino acid residues in biologically active peptides enhances their stability against enzymatic degradation and introduces conformational constraints in the peptide backbone, with the Φ angle at the N-methylated residue limited to positive values (energy minima at Φ = +60° and +150°). Importantly, N-methylated peptides have a decreased capacity to form hydrogen bonds with water molecules and, consequently, are better able to cross biological barriers. This is exemplified with the naturally occurring peptide cyclosporine which contains multiple N-methylated amino acid residues and is orally active. In the present paper we describe analogues of H-Tyr-c[D-Cys-Gly-Phe-D(or L)-Cys]NH2, in which the N-terminal tyrosine was replaced by 2′,6′-dimethyltyrosine (Dmt) and which are N-methylated at the Phe4 and/or Cys5 residue (Figure 1). N-methylation at the 4- and 5-position residues was carried out, because linear enkephalin analogues N-methylated at the 2- and 3-position residues are known to have in general weak opioid activity (7). Dmt was substituted for Tyr1 in these compounds because it has been shown that dimethylation at the 2′,6′-positions of Tyr1 in opioid peptides generally results in a significant increase in opioid agonist potency (8). These compounds have increased conformational integrity and can be expected to show improved blood-brain barrier (BBB) penetration. Replacement of the α-amino group of Dmt1 in opioid peptides with a methyl group, as achieved by substitution of (2S)-2-methyl-3-(2,6-dimethyl-4-hydroxyphenyl)propanoic acid [(2S)-Mdp], is a generally applicable structural modification for conversion of opioid peptide agonists to antagonists (9). In an effort to obtain an opioid antagonist with improved bioavailability, we also prepared an N-dimethylated analogue of H-Tyr-c[D-Cys-Gly-Phe-Cys]NH2 containing (2S)-Mdp in place of Tyr1 (Figure 1).
Figure 1.

Structural formulas of N-methylated cyclic enkephalin analogues.
The linear precursor peptides of the target compounds were prepared by solid-phase synthesis. In the case of compounds 1, 2, 7 and 8, peptides were assembled on a p-methylbenzhydrylamine resin with Nα-Boc or Fmoc- protection, 4-methylbenzyl protection of Cys and HF/anisole treatment for peptide cleavage. In the preparation of compounds 3-6 and 9, the linear precursor peptides were synthesized by using a Rink amide AM resin with Nα-Fmoc protection, S-tert-butyl protection of Cys or Cys(NMe) and peptide cleavage with 98% TFA/H2O. With all peptides disulfide bond-formation was carried out in solution with K3Fe(CN)6 as oxidation agent. Opioid activities of the compounds in vitro were determined using the guinea pig ileum (GPI) and mouse vas deferens (MVD) bioassays, and μ-, δ- and κ opioid receptor binding assays.
Methods and Materials
General Methods
Precoated plates (silica gel 60 F254, 250 μm, Merck, Darmstadt, Germany) were used for ascending TLC in the following systems (all v/v); (I) hexane/AcOEt (3:1); (II) CHCl3/MeOH (9:1); (III) n-BuOH/AcOH/H2O (4:1:1); (IV) n-BuOH/pyridine/AcOH/H2O (15:10:3:12). Preparative reversed-phase HPLC was performed on a Vydac 218-TP1022 column (22 × 250 mm) with a linear gradient of 20-40% MeOH in 0.1% TFA (peptides 1-8) or 30-70% MeOH in 0.1% TFA (peptide 9) over 30 min at a flow rate of 12 mL/min. Analytical reversed-phase HPLC was performed on a Vydac 218-TP54 column (5 × 250 mm) at a flow rate of 1.0 mL/min using the same linear gradients of MeOH in 0.1% TFA as in the preparative HPLC. The same column was also used for the determination of the capacity factors (K’ values) under the same conditions. Molecular masses of the compounds were determined by electrospray mass spectrometry on a Hybrid Q-Tof mass spectrometer interfaced to a MassLynx 4.0 data system.
Syntheses of Nα-methylcysteine derivatives
Fmoc-(NMe)-Cys(StBu)-OH was synthesized using the oxazolidinone procedure according to a literature procedure (10) and Fmoc-(NMe)-D-Cys(StBu)-OH was prepared in an analogous manner, as described in the following. Fmoc-D-Cys(StBu)OH was cyclized with formaldehyde and camphorsulfonic acid in benzene to afford (R)-Fmoc-4-((tert-butyldisulfanyl)methyl)-5-oxooxazolidine-3-carboxylate which was purified by flash chromatography on silica gel (hexane/AcOEt) and was obtained as an oil in 87% yield. TLC Rf 0.35 (I); [α]D20 -70.8 (c 1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, 2H, J = 7.0 Hz), 7.58, (d, 2H, J = 7.0 Hz), 7.42 (t, 2H, J = 7.0 Hz), 7.35 (m, 2H), 5.5-5.2 (br, m, 2H), 4.75-4.35 (br, m, 2H), 4.3 (br, s, 1H), 4.01 (br, s, 1H), 3.55 (br, s, 0.5H), 3.25 (br, s, 0.5H), 3.0 (br, s, 0.5H), 2.7 (br, s, 0.5H), 1.29 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.8, 152.2, 143.4, 141.4, 127.9, 127.2, 124.6, 120.0, 78.4, 73.9, 67.6, 55.3, 48.2, 47.2, 29.5; HRMS (ESI) m/e calcd for C23H26NO4S2 [M+H]+ 444.1303, obsd 444.1301.
Acid cleavage of the oxazolidinone with triethylsilane/TFA at room temperature for 16 h and purification by flash chromatography on silica gel (CHCl3/MeOH) afforded Fmoc-(NMe)-D-Cys(StBu)-OH as a white solid in 92% yield and in a 2.3:1.0 conformer ratio. TLC Rf 0.40 (II); [α]D20 +95 (c 1, CHCl3); 1H NMR (500 MHz, CDCl3) δ Major: 10.0 (br, s, 1H), 7.79 (m, 2H), 7.63 (m, 2H), 7.43 (m, 2H), 7.35 (m, 2H), 4.78 (d, 1H, J = 8.0 Hz), 4.58 (m, 2H), 4.32 (br, t, 1H), 3.38 (d, 1H, J =12.0 Hz), 3.20 (d, 1H, J =12.0 Hz), 3.07 (s, 3H), 1.37 (s, 9H); Minor: 10.0 (br, s, 1H), 7.75 (m, 2H), 7.60 (m, 2H), 7.40 (m, 2H), 7.31 (m, 2H), 4.72 (m, 1H), 4.53 (m, 1H), 4.26 (br, t, 1H), 3.10 (m, 0.5H), 2.97 (s, 3H), 2.73 (m, 0.5H), 1.33 (s, 9H); 13C NMR (125 MHz, CDCl3) δ Major: 175.6, 157.0, 144.0, 141.6, 127.5, 125.4, 68.4, 60.3, 47.4, 45.0, 39.2, 34.4, 30.2; Minor: 175.6, 157.0, 144.1, 141.6, 128.0, 125.4, 68.0, 59.1, 48.5, 45.0, 39.5, 33.5, 30.2; HRMS (ESI) m/e calcd for C23H28NO4S2 [M+H]+ 446.1460, obsd 446.1460.
Peptide Synthesis
The linear precursor peptides of compounds 1, 2, 7 and 8 were prepared by the manual solid-phase technique using Fmoc-protection for the α-amino group of Dmt, Gly and Phe(NMe), and Boc protection for the α-amino group of L- and D-Cys(4-MeBzl). Peptides were assembled on a methylbenzylhydrylamine resin (Bachem Americas, Torrance, CA) using 1,3-diisopropylcarbodiimide (DIC)/1-hydroxybenzotriazole (HOBt) as coupling agents according to a published protocol (9). Protected amino acids were purchased from Bachem or from RSP Amino Acids, Shirley, MA. Peptides were cleaved from the resin and completely deprotected by treatment with HF for 60 min at 0°C (10 mL of HF plus 1 mL of anisole/g resin). After evaporation of the HF, the resin was extracted three times with Et2O and, subsequently, three times with glacial AcOH. The peptides were obtained in solid form through lyophylization of the acetic acid extract. The linear precursor peptides of cyclic peptides 3, 4, 5 ,6 and 9 were assembled on a Rink amide AM resin (0.62 mmol/g) using Nα-Fmoc protection according to the standard Fmoc protocol. 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) in the presence of diisopropylethylamine (DIEA) was used as coupling agent and double couplings between Cys(NMe) and Phe (or Phe(NMe)) and between Phe(NMe) and Gly were performed. Fmoc deprotection was carried out with 30% piperidine in DMF and the StBu protecting group was removed by treatment with a mixture of 20% β-mercaptoethanol in DMF added to N-methylmorpholine (final concentration of β-mercaptoethanol = 0.1 M). Peptides were cleaved from the resin by treatment with 98% TFA/H2O in the usual manner. After evaporation, treatment with ethylether provided the peptides in solid form. For disulfide bond formation, a solution containing K3Fe(CN)6 in 0.05 M ammonium acetate was prepared with a 4-fold excess of K3Fe(CN)6 over the peptide to be oxidized. Peptides dissolved in MeOH were added to this solution at a rate of 8 mg/h/liter of oxidation solution. All cyclic peptides were purified by preparative reversed-phase HPLC and were found to be at least 98% pure, as assessed by HPLC and TLC. Molecular weights were confirmed by mass spectrometry. Analytical parameters are listed in Table 1.
Table 1.
Analytical parameters of N-methylated peptides
| Compound | Rf (III) | Rf (IV) | K’a | ES/ML (m/e) |
|---|---|---|---|---|
| 1 | 0.43 | 0.76 | 4.03 | 631 |
| 2 | 0.39 | 0.75 | 3.05 | 631 |
| 3 | 0.50 | 0.81 | 3.40 | 631 |
| 4 | 0.56 | 0.80 | 2.62 | 631 |
| 5 | 0.55 | 0.81 | 4.33 | 645 |
| 6 | 0.53 | 0.80 | 3.66 | 645 |
| 7 | 0.47 | 0.75 | 3.20 | 617 |
| 8 | 0.44 | 0.80 | 3.60 | 617 |
| 9 | 0.83 | 0.89 | 6.08b | 644 |
HPLC conditions: 20-40% MeOH/0.1% TFA-H2O, linear gradient over 30 min at a flow rate of 1 mL/min
30-70% MeOH/0.1% TFA-H2O, linear gradient over 30 min at a flow rate of 1 mL/min.
Opioid receptor binding assays and in vitro bioassays
Opioid receptor binding studies were performed as described in detail elsewhere (11). Binding affinities for μ and δ receptors were determined by displacing, respectively, [3H]DAMGO (Multiple Peptide Systems, San Diego, CA) and [3H]DSLET (Multiple Peptide Systems) from rat brain membrane binding sites, and κ opioid receptor binding affinities were measured by displacement of [3H]U69,593 (Amersham) from guinea pig brain membrane binding sites. Incubations were performed for 2h at 0°C with [3H]DAMGO, [3H]DSLET and [3H]U69,593 at respective concentrations of 0.72, 0.78 and 0.80 nM. IC50 values were determined from log-dose displacement curves, and Ki values were calculated from the obtained IC50 values by means of the equation of Cheng and Prusoff (12), using values of 1.3, 2.6 and 2.9 nM for the dissociation constants of [3H]DAMGO, [3H]DSLET, and [3H]U69,593, respectively. The GPI (13) and MVD (14) bioassays were carried out as reported in detail elsewhere (11,15). A dose-response curve was determined with [Leu5]enkephalin as standard for each ileum and vas preparation, and IC50 values of the compounds being tested were normalized according to a published procedure (16). Ke values for antagonists were determined from the ratio of IC50 values obtained with an agonist in the presence and absence of a fixed antagonist concentration (17). μ and κ antagonist Ke values of compounds were determined against the μ agonist TAPP (H-Tyr-D-Ala-Phe-Phe-NH2) (18) and the κ agonist U50,488, respectively, and δ antagonist Ke values were measured in the MVD assay against the δ agonist DPDPE.
Theoretical conformational analysis
All calculations were performed using the molecular modeling software SYBYL, version 7.0 (Tripos Associates, St. Louis, MO). The standard SYBYL force field was used for energy calculations, and a dielectric constant of 78 was chosen to simulate an aqueous environment. A stepwise approach was used to determine low-energy conformations of the cyclic peptides (19). For each peptide the “bare” ring structure consisting of only the atoms directly attached to the ring, along with associated hydrogen atoms, was first constructed. After minimization a systematic conformational grid search was carried out to identify low-energy ring structures. Each rotatable bound was rotated in 30° increments over all space. An allowed conformation was obtained if in a structure without unfavorable vdw contacts the ring could close within 0.4 Å of a normal bond. Each allowed ring structure was minimized and structures within 3.0 kca/mol of the lowest-energy ring structure were retained for further study. To each low-energy ring structure the exocyclic tyrosine residue and the phenylalanine side chain were attached and a second systematic grid search was performed on the exocyclic rotatable bonds. Energies were calculated, and the resulting conformations were ranked in order of increasing energy. Mu receptor-bound conformations were identified by spatial overlap with the proposed bioactive conformation of the cyclic μ opioid peptide agonist JOM-6 (H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]NH2) (20). The N-terminal amino group and the two aromatic rings of the peptide studied were superimposed on the corresponding pharmacophoric moieties in JOM-6.
Results
The two parent agonist peptides H-Dmt-c[D-Cys-Gly-Phe-D-Cys]NH2 (7) and H-Dmt-c[D-Cys-Gly-Phe-L-Cys]NH2 (8) showed subnanomolar μ-, δ- and κ-receptor binding affinities and essentially no selectivity for any of the three opioid receptor types (Table 2). Monomethylation at the Phe4 residue (compounds 1 and 2) or at the D- or L-Cys5 residue (compounds 3 and 4) resulted in compounds that retained subnanomolar μ receptor binding affinity and subnanomolar or low nanomolar δ and κ receptor binding affinities, with compounds 1 and 2 showing moderate preference for μ and κ receptors over δ receptors. The two N-dimethylated analogues (compounds 5 and 6) also displayed subnanomolar μ receptor binding affinities, very high κ receptor binding affinities and somewhat lower δ receptor binding affinities. Consequently, these two compounds showed modest μ vs. δ selectivity.
Table 2.
Opioid receptor binding data of N-methylated cyclic enkephalin analogues
| Ki (nM)a |
Ki ratio |
|||
|---|---|---|---|---|
| Compound | μ b | δ b | κ c | μ/δ/κ |
| 1 | 0.496 ± 0.037 | 2.29 ± 0.09 | 0.447 ± 0.070 | 1/5/1 |
| 2 | 0.354 ± 0.038 | 2.36 ± 0.48 | 0.855 ± 0.087 | 1/7/2 |
| 3 | 0.504 ± 0.039 | 0.525 ± 0.059 | 1.01 ± 0.06 | 1/1/2 |
| 4 | 0.586 ± 0.011 | 0.776 ± 0.050 | 0.894 ± 0.126 | 1/1/2 |
| 5 | 0.876 ± 0.059 | 6.07 ± 0.39 | 1.42 ± 0.16 | 1/7/2 |
| 6 | 0.641 ± 0.010 | 1.79 ± 0.03 | 0.875 ± 0.015 | 1/3/1 |
| 7 | 0.412 ± 0.035 | 0.202 ± 0.005 | 0.602 ± 0.152 | 1/1/1 |
| 8 | 0.282 ± 0.041 | 0.306 ± 0.011 | 0.677 ± 0.055 | 1/1/2 |
| 9 | 14.4 ± 1.0 | 35.9 ± 3.5 | 29.5 ± 1.4 | 1/2/2 |
Values represent means of 3-6 determinations ± SEM.
Displacement of [3H]DAMGO (μ-selective) and [3H]DSLET (δ-selective) from rat brain membrane binding sites.
Displacement of [3H]U69,593 (κ-selective) from guinea pig brain membrane binding sites.
In comparison with the two parent peptides (7 and 8), all N-mono- and N-dimethylated cyclic peptides also turned out to be full agonists in the GPI assay (μ receptor-representative) and in the MVD assay (δ receptor-representative) with subnanomolar or very low nanomolar potencies in both assays (Table 3). In general, there is good agreement between the receptor affinities measured in the binding assays and the agonist potencies determined in the functional GPI- and MVD assays, but some minor quantitative discrepancies are noticed. Such quantitative discrepancies have often been observed and could be due to possible differences in the structural requirements between central and peripheral receptors or to differences among the compounds studied with regard to their ability to access the receptors in the isolated tissue preparations.
Table 3.
GPI and MVD assay of N-methylated cyclic enkephalin analoguesa
| GPI |
MVD |
||||
|---|---|---|---|---|---|
| Compound | IC50 (nM) | Keμ (nM)b | Keκ (nM)c | IC50 (nM) | Keδ (nM)d |
| 1 | 1.07 ± 0.19 | 1.11 ± 0.13 | |||
| 2 | 0.457 ± 0.029 | 0.884 ± 0.110 | |||
| 3 | 1.81 ± 0.36 | 0.352 ± 0.020 | |||
| 4 | 1.36 ± 0.28 | 0.122 ± 0.016 | |||
| 5 | 1.34 ± 0.24 | 3.72 ± 1.35 | |||
| 6 | 0.394 ± 0.065 | 1.95 ± 0.20 | |||
| 7 | 0.586 ± 0.211 | 0.0530 ± 0.0153 | |||
| 8 | 0.812 ± 0.046 | 0.115 ± 0.005 | |||
| 9 | 71.0 ± 7.3 | 151 ± 16 | 277 ± 40 | ||
Values represent means of 3-6 determinations ± SEM.
Determined against TAPP (H-Tyr-D-Ala-Phe-Phe-NH2).
Determined against U50,488.
Determined against DPDPE.
Compound 9, the (2S)-Mdp1 analogue of cyclic peptide 6, showed quite high μ receptor binding affinity (Kiμ = 14.4 ± 10nM) and about 2-fold lower δ and κ receptor binding affinities (Table 2). As expected, peptide 9 showed μ opioid antagonist activity in the GPI assay with a Ke value of 71.0 ± 7.3 nM (Table 3). It also displayed κ and δ opioid antagonist properties with respective Ke values of 151 ±16 nM and 277 ± 40 nM.
The numbers of low-energy conformers within 3 kcal/mol of the lowest-energy conformation obtained for the “bare” ring structures of cyclic peptides 1-8 in the theoretical conformational analysis (systematic grid search and energy minimization) are listed in Table 4. The results indicate that the L-Cys(NMe)-containing rings are structurally more rigid than the corresponding D-Cys(NMe)-containing ones, as a consequence of a steric clash between the N-methyl group of L-Cys(NMe)5 and the C-terminal carboxamide group. The lowest-energy conformers of the ring structures in the eight compounds all contain all-trans peptide bonds. It is evident that N-mono- and dimethylation of the 14-membered ring structures produced a progressive decrease in conformational flexibility. The structurally most rigid ring structure is the one contained in cyclic peptide 6, for which only 4 low-energy conformers were obtained. As depicted in Figure 2, the lowest-energy conformer of the latter ring structure showed considerable similarity with the five lowest-energy conformers of the ring structure contained in compound 8 (H-c[D-Cys-Gly-Ala-Cys]NH2), indicating that N-methylation at the Ala and L-Cys residue did not significantly alter the overall low-energy ring conformation. Furthermore, the two N-methyl groups are oriented perpendicular to the peptide ring structure. After addition of the exocyclic Dmt1 residue and the Phe4 side chain to the bare ring structures and subsequent energy minimization, the resulting low-energy conformers of the moderately μ receptor-selective cyclic peptide 6 were superimposed on the proposed model of the μ receptor-bound conformation of the μ selective cyclic opioid peptide JOM-6 (H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]NH2 (20). Excellent spatial overlap was observed between the important pharmacophoric moieties (N-terminal amino group, Dmt/Tyr side chain, Phe side chain) in JOM-6 and in the 3rd-lowest energy conformer of 6, which is only 1.32 kcal/mol higher in energy than the lowest-energy conformer. The RMSD value for this overlap is 0.70 Å. Several conformers of 6 with somewhat higher energy showed a shorter intramolecular distance between the two aromatic rings, similar to the corresponding distance in the proposed δ receptor-bound conformation of the δ receptor-selective δ agonist JOM-13 (H-Tyr-c[D-Cys-Phe-D-Pen]OH (6,20)) (data not shown). These results may explain the modest μ vs. δ receptor selectivity of compound 6.
Table 4.
Number of low-energy conformers of the “bare” ring structures of compounds 1-8.
| Ring structure | Number of low energy ringsa |
|---|---|
| H-c[D-Cys-Gly-Ala(NMe)-D-Cys]NH2 | 28 |
| H-c[D-Cys-Gly-Ala(NMe)-L-Cys]NH2 | 28 |
| H-c[D-Cys-Gly-Ala-D-Cys(NMe)]NH2 | 28 |
| H-c[D-Cys-Gly-Ala-L-Cys(NMe)]NH2 | 16 |
| H-c[D-Cys-Gly-Ala(NMe)-D-Cys(NMe)]NH2 | 9 |
| H-c[D-Cys-Gly-Ala(NMe)-L-Cys(NMe)]NH2 | 4 |
| H-c[D-Cys-Gly-Ala-D-Cys]NH2 | 69 |
| H-c[D-Cys-Gly-Ala-L-Cys]NH2 | 109 |
Numbers of low-energy conformers within 3 kcal/mol of the lowest-energy conformation.
Figure 2.
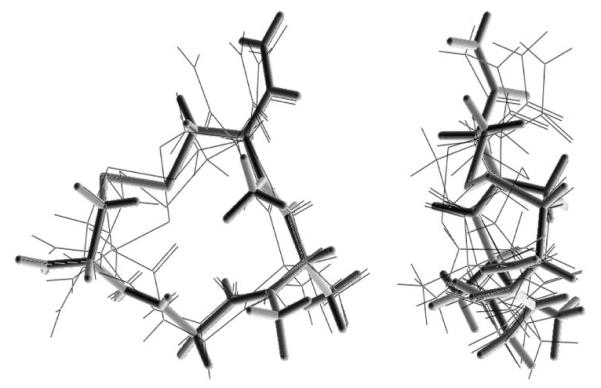
Spatial overlap of the lowest-energy conformation of H-c[D-Cys-Gly-Ala(NMe)-L-Cys(NMe)]NH2 (depicted in solid lines) with the 5 lowest-energy conformers of H-c[D-Cys-Gly-Ala-L-Cys]NH2 (depicted in light lines) (two views).
Discussion and Conclusions
In comparison with parent peptides 7 and 8 all mono- and di-N-methylated cyclic Dmt1-peptides retained similarly high μ and κ receptor binding affinities and in the case of the mono-N-methylated Cys(NMe)5-analogues (compounds 3 and 4) similarly high δ receptor binding affinity. Compounds that are N-methylated at the Phe4 residue (1,2) or at both the Phe4 and the D(or L)-Cys5 residue (5,6) showed somewhat lower δ receptor binding affinities and moderate μ vs. δ receptor selectivity. In agreement with the receptor binding data, the N-methylated Dmt1-analogues also showed high opioid agonist potencies in the GPI and MVD bioassays, comparable to the activities seen with the non-methylated parent peptides. These results indicate that the presence of the N-methyl groups per se at the 4- and 5-position residues and the progressive conformational restriction resulting from N-methylation at one or the other, or at both these residues do not have a major effect on the in vitro opioid activity profile. The conformationally most constrained peptide of this series is the moderately μ receptor-selective compound 6, a low-energy conformer of which showed good spatial overlap with the proposed μ receptor-bound conformation of the μ-selective cyclic opioid peptide JOM-6 (20). In contrast to the N-methylated cyclic enkephalin analogues described here, dimethylation of the β-carbons of the D-Cys2 and D-Cys5 residues in the cyclic enkephalin analogue H-Tyr-c[D-Cys-Gly-Phe-D-Cys]OH had a significant effect on opioid receptor binding affinity and selectivity (21). The resulting compound, H-Tyr-c[D-Pen-Gly-Phe-D-Pen]OH (DPDPE; Pen = penicillamine) showed somewhat lower δ receptor binding affinity but greatly increased δ receptor selectivity. In this case the altered opioid activity profile is not due to a significant change in the topography of the molecule but rather to steric interference caused by the β-methyl groups of the D-Pen2 residue (22). Replacement of the disulfide moiety in the cyclic opioid peptides H-Tyr-c[D-Cys-Gly-Phe-D(or L)-Cys]NH2 with a -CH=CH- (cis or trans) or a -CH2-CH2- linkage resulted in compounds that also retained high opioid activity but showed considerable differences in the low-energy conformations of their 14-membered ring structures among them and in comparison with the disulfide-containing parent peptide (5). Taken together, the results obtained with these various cyclic pentapeptide enkephalin analogues indicate that significant variation in the conformation and structural flexibility of the 14-membered ring structure is tolerated and that the ring component mainly served as a template for the proper spatial positioning of the exocyclic Tyr1 or Dmt1 residue and the Phe4 side chain.
N-methylation of three amino acid residues in the cyclic hexapeptide αIIbβ3 integrin receptor antagonist c[-Gly-Arg-Gly-Asp-D-Phe-Leu-] resulted in a compound which showed somewhat reduced receptor binding affinity but improved receptor selectivity (23). In this case, the selectivity enhancement was due to the reduced flexibility of the peptide. N-methylation at three amino acid residues of the somatostatin-derived hexapeptide c[-Pro-Phe-D-Trp-Lys-Thr-Phe-] somewhat reduced binding affinity for the hsst2 and hsst5 somatostatin receptors but, importantly, the resulting compound was found to be orally active (24). A linear dermorphin-derived tetrapeptide analogue containing two N-methylated residues, H-Tyr-D-Ala(NMe)-Phe-Sar-NH2, retained quite high opioid agonist activity in vitro with a μ receptor binding affinity 30-80-fold lower than those of the N-methylated cyclic peptides described here, and produced a centrally mediated analgesic effect after intravenous administration (25). The cyclic enkephalin analogues N-methylated at the 4- and 5-position residues described here (compounds 5 and 6) can be expected to have enhanced ability to cross the blood-brain barrier as compared to their non-methylated parents. The (2S)-Mdp1-containing antagonist 9 may show even further improved bioavailability because it contains a methyl group in place of the N-terminal amino group and, thus, has further enhanced lipophilicity and reduced hydrogen-bonding capacity.
Figure 3.
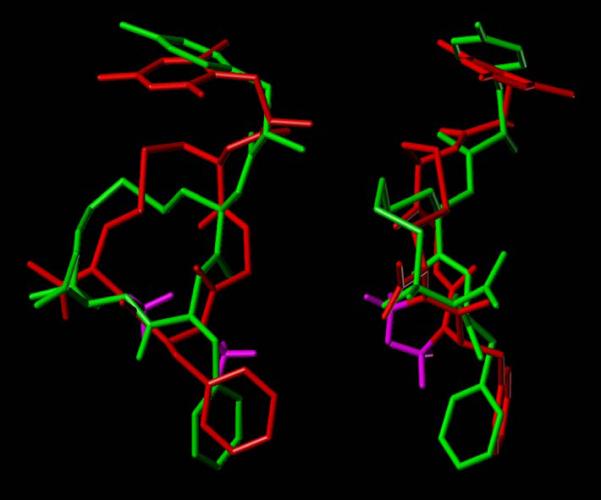
Spatial overlap of low-energy conformer of H-Dmt-c[D-Cys-Gly-Phe(NMe)-L-Cys(NMe)]NH2 (6, red, with N-methyl groups in magenta) with the proposed model of the μ-selective peptide JOM-6 (H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]NH2) in the μ receptor-bound conformation (green) (20) (two views).
Acknowledgements
This work was supported by grants from the National Institute on Drug Abuse, NIH (DA004443) and the Canadian Institutes of Health Research (MOP-89716).
Footnotes
- BBB
- blood-brain barrier
- DAMGO
- H-Tyr-D-Ala-Gly-Phe(NMe)-Gly-ol
- DIC
- 1,3-diisopropylcarbodiimide
- DIEA
- diisopropylethylamine
- Dmt
- 2′,6′-dimethyltyrosine
- DPDPE
- H-Tyr-c[D-Pen-Gly-Phe-D-Pen]OH
- DSLET
- H-Tyr-D-Ser-Gly-Phe-Leu-Thr-OH
- GPI
- guinea pig ileum
- HBTU
- 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HOBt
- 1-hydroxybenzotriazole
- HPLC
- high performance liquid chromatography
- JOM-6
- H-Tyr-c(S-Et-S)[D-Cys-Phe-D-Pen]NH2
- JOM-13
- H-Tyr-c[D-Cys-Phe-D-Pen]OH
- (2S)-Mdp
- (2S)-2-methyl-3-(2,6-dimethyl-4-hydroxyphenyl)propanoic acid
- MVD
- mouse vas deferens
- Pen
- penicillamine
- TFA
- trifluoroacetic acid
- U50,488
- trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]-benzeneacetamide
- U69,593
- (5α,7α,8β)-(—)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]benzeneacetamide.
References
- 1.Sarantakis D. Analgesic polypeptide. 4148786 U.S. Patent. 1979
- 2.Schiller PW, Eggimann B, DiMaio J, Lemieux C, Nguyen TM-D. Cyclic enkephalin analogs containing a cystine bridge. Biochem Boiophys Res Commun. 1981;101:337–343. doi: 10.1016/0006-291x(81)91265-1. [DOI] [PubMed] [Google Scholar]
- 3.Schiller PW, Nguyen TM-D, Maziak LA, Wilkes BC, Lemieux C. Structure-activity relationships of cyclic opioid peptide analogues containing a phenylalanine residue in the 3-position. J Med Chem. 1987;30:2094–2099. doi: 10.1021/jm00394a027. [DOI] [PubMed] [Google Scholar]
- 4.Berezowska I, Chung NN, Lemieux C, Wilkes BC, Schiller PW. Cyclic dermorphin tetrapeptide analogues obtained via ring-closing metathesis. Acta Biochim. Pol. 2006;53:73–76. [PubMed] [Google Scholar]
- 5.Berezowska I, Chung NN, Lemieux C, Wilkes BC, Schiller PW. Dicarba analogues of the cyclic enkephalin peptides H-Tyr-c[D-Cys-Gly-Phe-D(or L)-Cys]NH2 retain high opioid activity. J Med Chem. 2007;50:1414–1417. doi: 10.1021/jm061294n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosberg HI, Omnaas JR, Medzihradsky F, Smith CB. Cyclic disulfide- and dithioether-containing opioid tetrapeptides: development of a ligand with high delta opioid receptor selectivity and affinity. Life Sci. 1988;43:1013–1020. doi: 10.1016/0024-3205(88)90547-4. [DOI] [PubMed] [Google Scholar]
- 7.Hansen PE, Morgan BA. Structure-activity relationships in enkephalin peptides. In: Udenfriend S, Meienhofer J, editors. The Peptides: Analysis, Synthesis, Biology. vol. 6, “Opioid Peptides: Biology, Chemistry and Genetics”. Academic Press; Orlando, USA: 1984. pp. 269–321. [Google Scholar]
- 8.Hansen DW, Jr., Stapelfeld A, Savage MA, Reichman M, Hammond DL, Haaseth RC, Mosberg HI. Systemic analgesic activity and delta-opioid selectivity in [2,6-dimethyl-Tyr1,D-Pen2,D-Pen5]enkephalin. J Med Chem. 1992;35:684–687. doi: 10.1021/jm00082a008. [DOI] [PubMed] [Google Scholar]
- 9.Lu Y, Nguyen TM-D, Weltrowska G, Berezowska I, Lemieux C, Chung NN, Schiller PW. [2′,6′-Dimethyltyrosine]dynorphin A(1-11)-NH2 analogues lacking an N-terminal amino group: potent and selective κ opioid antagonists. J Med Chem. 2001;44:3048–3053. doi: 10.1021/jm0101186. [DOI] [PubMed] [Google Scholar]
- 10.Ruggles EL, Stevenson F, Jr., Hondal RJ. A viable synthesis of N-methyl cysteine. Biopolymers (Peptide Science) 2008;90:61–68. doi: 10.1002/bip.20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiller PW, Lipton A, Horrobin DF, Bodanszky M. Unsulfated C-terminal 7-peptide of cholecystokinin: a new ligand of the opiate receptor. Biochem Biophys Res Commun. 1978;85:1332–1338. doi: 10.1016/0006-291x(78)91149-x. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 13.Paton WD. The action of morphine and related substances on contraction and on acetylcholine output of coaxially stimulated guinea-pig ileum. Br. J. Pharmacol Chemother. 1957;12:119–127. doi: 10.1111/j.1476-5381.1957.tb01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson G, Hughes J, Kosterlitz HW. A new example of a morphine-sensitive neuro-effector junction: adrenergic transmission in the mouse vas deferens. Br J Pharmacol. 1972;46:764–766. doi: 10.1111/j.1476-5381.1972.tb06901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiMaio J, Nguyen TM-D, Lemieux C, Schiller PW. Synthesis and pharmacological characterization in vitro of cyclic enkephalin analogues: effect of conformational constraints on opiate receptor selectivity. J Med Chem. 1982;25:1432–1438. doi: 10.1021/jm00354a008. [DOI] [PubMed] [Google Scholar]
- 16.Waterfield AA, Leslie FM, Lord JA, Ling N, Kosterlitz HW. Opioid activities of fragments of β-endorphin and of its leucine65-analogue. Comparison of the binding properties of methionine- and leucine-enkephalin. Eur J Pharmacol. 1979;58:11–18. doi: 10.1016/0014-2999(79)90334-0. [DOI] [PubMed] [Google Scholar]
- 17.Kosterlitz HW, Watt A. Kinetic parameters of narcotic agonists and antagonists with particular reference to N-allylnormorphone (naloxone) Br J Pharmacol. 1968;33:261–276. doi: 10.1111/j.1476-5381.1968.tb00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiller PW, Nguyen TM-D, Chung NN, Lemieux C. Dermorphin analogues carrying an increased positive net charge in their “message domain” display extremely high μ opioid receptor selectivity. J Med Chem. 1989;32:698–703. doi: 10.1021/jm00123a035. 1989. [DOI] [PubMed] [Google Scholar]
- 19.Wilkes BC, Schiller PW. Conformation-activity relationships of cyclic dermorphin analogues. Biopolymers. 1990;29:89–95. doi: 10.1002/bip.360290113. [DOI] [PubMed] [Google Scholar]
- 20.Mosberg HI, Fowler CB. Development and validation of opioid ligand-receptor interaction models: the structural basis of mu vs delta selectivity. J Pept Res. 2002;60:329–335. doi: 10.1034/j.1399-3011.2002.21061.x. [DOI] [PubMed] [Google Scholar]
- 21.Mosberg HI, Hurst R, Hruby VJ, Gee K, Yamamura HI, Galligan JJ, Burks TF. Bis-penicillamine enkephalins possess highly improved specificity toward δ opioid receptors. Proc Natl Acad Sci USA. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosberg HI, Haaseth RC, Ramalingam K, Mansour A, Akil H, Woodard RW. Role of steric interactions in the delta opioid receptor selectivity of [D-Pen2,D-Pen5]enkephalin. Int J Peptide Protein Res. 1988;32:1–8. doi: 10.1111/j.1399-3011.1988.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee J, Ovadia O, Zahn G, Marinelli L, Hoffman A, Gilon C, Kessler H. Multiple N-methylation by a designed approach enhances receptor selectivity. J Med Chem. 2007;50:5878–5881. doi: 10.1021/jm701044r. [DOI] [PubMed] [Google Scholar]
- 24.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew Chem Int Ed. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 25.Ballet S, Misicka A, Kosson P, Lemieux C, Chung NN, Schiller PW, Lipkowski AW, Tourwé D. Blood-brain barrier penetration by two dermorphin tetrapeptide analogues: role of lipophilicity vs structural flexibility. J Med Chem. 2008;51:2571–2574. doi: 10.1021/jm701404s. [DOI] [PubMed] [Google Scholar]