

Abstract
Mutations of the insulin-like 3 (INSL3) hormone or its receptor RXFP2 cause intraabdominal cryptorchidism in male mice. Specific RXFP2 expression in mouse gubernacula was detected at embryonic day 14.5 (E14.5) and markedly increased after birth in the developing cremaster muscle, as well as in the epididymis, testicular Leydig and germ cells. INSL3 treatment stimulated cell proliferation of embryonic gubernacular and Leydig cells implicating active INSL3-mediated signaling. The transcription factor SOX9, a known male sex determination factor, up-regulated the activity of the RXFP2 promoter. INSL3 is sufficient to direct the first transabdominal phase of testicular descent in the absence of hypothalamic-pituitary-gonadal axis signaling or Hoxa10 but their presence is important for inguinoscrotal testicular descent. Similarly, conditional ablation of the androgen receptor gene in gubernacular cells resulted in disruption of inguinoscrotal descent. We performed mutation screening of INSL3 and RXFP2 in human patients with cryptorchidism and control subjects from different populations in Europe and USA. Several missense mutations were described in both the INSL3 and RXFP2 genes. A novel V39G INSL3 mutation in a patient with cryptorchidism was identified, however the functional analysis of the mutant peptide did not reveal compromised function. In more than 2000 patients and controls analyzed to date the T222P RXFP2 mutation is the only one strongly associated with the mutant phenotype. The T222P mutant receptor transfected into 293T cells had severely decreased cell membrane expression, providing the basis for the functional deficiency of this mutation.
Keywords: testis, cryptorchidism, INSL3, RXFP2, hormones
Introduction
Most mammalian species manifest distinct sexual dimorphism in the position of the adult gonads. In females, the developing ovary remains in a high abdominal position. In contrast, male differentiation, triggered by the SRY gene on the Y-chromosome, leads to testis differentiation followed by its migration into the scrotum. The process is called testicular descent (TD). Despite notable differences in anatomy between mammals, a two stage model of TD was proposed - the transabdominal and inguinoscrotal descent (Fig. 1).1 Two mesentery ligaments attached to the developing gonad, the cranial suspensory (CSL) and gubernacular ligaments, are believed to play a major role in TD. During the transabdominal phase between 10 and 23 weeks of gestation in human embryos (in mouse - between 15.5E and 17.5E) the testes gradually move from their original position in the urogenital ridge to the inguinal region. The process of transabdominal descent occurs in parallel with the shortening of gubernacular ligament cord, an outgrowth of the gubernacular bulb, differentiation and eversion of the cremaster muscle. In the human fetus the second phase of TD occurs before birth at weeks 24-34 of gestation; in mice it is completed within 19 days after birth.2 This stage is characterized by the caudal extension of the gubernaculum, its involution and protrusion into the scrotal sac, development of the processus vaginalis, dilation of the inguinal canal by the gubernacular bulb, and intraabdominal pressure to force the testis through the canal. In mice, TD is mediated by the contractions of the cremasteric muscle. Human gubernacular histology evolves from a hydrated structure with a loose extracellular matrix and poorly differentiated fibroblasts into an essentially fibrous structure rich in collagen and elastic fibers. In rodents however, the adult gubernaculum is differentiated into cremaster muscle component of the spermatic cord.
FIGURE 1.
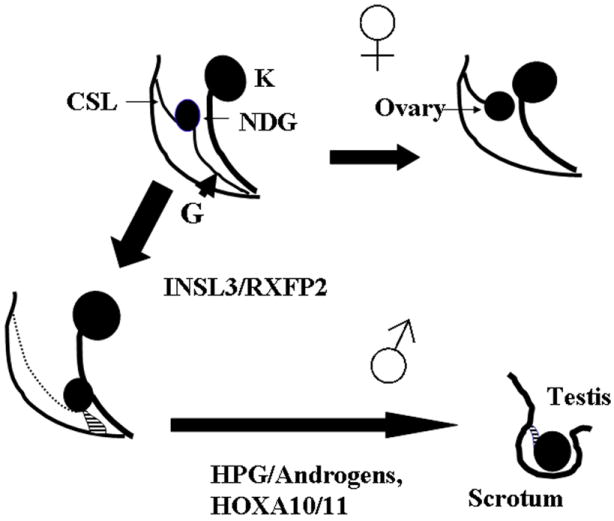
Two stage model of testicular descent (TD) in mammals. Non-differentiated gonad (ND G) is attached to the body walls by the cranial suspensory ligament (CSL) and gubernacular ligament (G). In females G regresses and the ovary is located in an abdominal position. During first stage of TD INSL3/RXFP2 signaling causes differentiation of G with TD to the low abdominal position. Androgens cause regression of CSL and in concert with other factors (as HOXA10/11) are responsible for inguinoscrotal TD.
Cryptorchidism is the most common congenital abnormality in newborn boys with a reported frequency of 2-4%. Cryptorchidism is an established risk factor for infertility and testicular cancer and thus requires surgical intervention.
INSL3/RXFP2 signaling controls the transabdominal stage of testicular descent in mice
Targeted ablation of the mouse Insl3 or INSL3 receptor gene Rxfp2, previously known as Great or Lgr8, caused cryptorchidism.3-6 Both loss-of-function alleles behaved as recessive mutations; the adult testes were located in high intraabdominal position just below the kidney. In fact, the similarity of these phenotypes led to the subsequent identification of the corresponding gene products as a cognate ligand-receptor pair. The gubernacular ligaments failed to differentiate due to an early arrest of proliferation and differentiation of the mesenchymal cells to myoblasts in outer layers of the gubernacular bulb in gene-deficient animals. Cell proliferation effects of INSL3 treatment on cultured primary gubernacular cells were established in vitro.7, 8
The expression of INSL3 primarily occurs in pre- and postnatal Leydig cells of the testis. Insl3 is first detected in murine testis at embryonic day 13.5 (E13.5), coinciding with Leydig cell differentiation and expression continues to be stable afterwards.9 No Insl3 transcript could be detected in prenatal ovary.
INSL3 is expressed in the population of fetal Leydig cells in hpg mutant mouse. These mice lack an active pituitary-gonadal axis, indicating that gonadotropins are not essential for the regulation of INSL3 expression during embryonic development. Prenatal exposure to 17β-estradiol and the nonsteroidal synthetic estrogen diethylstilbestrol (DES) causes de-masculinizing and feminizing effects in the male embryo, including impaired testicular descent (cryptorchidism) Maternal exposure to estrogens, as well as DES, downregulates Insl3 expression in embryonic Leydig cells, thereby suggesting a mechanism for cryptorchidism in male progeny.10, 11
In adult testis the expression of INSL3 is constitutive and appears to reflect the differentiation status of adult Leydig cells. What are the factors which regulate an increase of INSL3 expression during male development? It was shown that SF1 (NR5A1) up-regulated the activity of rodent, human and canine INSL3 promoters in vitro. 9,12 In mutant mice with Leydig cell-specific deficiency of Sf1, the testes are cryptorchid suggesting abnormal INSL3 expression. The recognized antagonist of SF1, DAX1 inhibited Insl3 promoter activity.13 NR4A1, another transcription factor, regulates INSL3 transcription in Leydig cells. Chromatin immunoprecipitation assays revealed that endogenous NR4A1 binds to the proximal Insl3 promoter in vivo. 14
At day E14.5 Rxfp2 expression is most prominent in the gubernacular ligaments.15 Remarkably, even after transabdominal TD there is a steady increase in the RXFP2 expression in gubernacula and cremaster muscle. Immunostaining for RXFP2 is first detected in the differentiating myoblasts in the gubernacular bulb, and in adults in cremaster muscle cells. The strong RXFP2 expression was demonstrated in adult Leydig cells, in pre- and postmeiotic spermatogenic cells, in epididymis epithelial cells and other reproductive and non-reproductive organs. Luciferase-reporters driven by human RXFP2 promoter fragments were activated by SOX9 transcription factor.16 Significantly, SF1, DAX1, and SOX9 are the crucial players in male differentiation and thus provide a link between TD and the sex determination process.
INSL3 overexpression in transgenic mice was sufficient to induce development of gubernacular ligaments in females.17 The ovaries descended into a position over the bladder and attached to the abdominal wall via the well developed CSL and the gubernaculum. Remarkably, the processus vaginalis was also apparent. All transgenic females developed age-progressing bilateral inguinal hernia. The results suggest that INSL3-mediated activity induces gubernaculum development and precludes a role of androgen in this process.
Strong INSL3/RXFP2 expression after transabdominal TD suggests an involvement in the second phase of TD descent. As mentioned above, abnormal signaling in hypothalamic-pituitary-gonadal axis as well as mutations in Hoxa10/11 homeobox genes cause disruption of inguinoscrotal TD.18 Combining mouse mutant alleles of Gnrhr receptor or Hoxa10 homeobox gene with transgenic Insl3 overexpression in pancreas, we demonstrated that while transabdominal TD could occur in such mice, the INSL3 expression does not rescue the cryptorchid phenotype.19
What is the target of androgen signaling in TD? Conditional CRE-loxP mediated deletion of the androgen receptor gene in different testicular cells did not cause cryptorchidism in mice, thus excluding the testis as the primary target. We used three different Cre-transgenic lines for androgen receptor (AR) ablation in gubernacular ligaments and cremaster muscle. The Rxfp2-Cre transgene previously generated in our laboratory drives the CRE recombinase expression in a fashion consistent with the endogenous RXFP2 expression including a prominent expression in gubernacular ligaments. The resulted phenotype of Ar-loxP/Y Tg(Rxfp2-Cre) was similar to the one observed in androgen-insensitive mice with testicular feminization (Tfm). Such males exhibit external female phenotype. This result was not however completely unexpected, as the expression of Rxfp2 was detected at the early stages of mouse development as well as in numerous reproductive and non-reproductive organs in late prenatal stages. The other Cre-transgenes used were Rarb2-Cre and tamoxifen-inducible HAS-Cre. Both transgenes are expressed in gubernacular ligaments. The Rarb2-Cre, Ar-loxP/Y males and postnatally tamoxifen-treated HAS-Cre, Ar-loxP/Y males both developed cryptorchidism due to an abnormal cremaster muscle differentiation. Thus expression of AR in differentiating gubernacular muscle cells is primarily responsible for the androgen effects on second stage of TD.
What is the INSL3 role in adult reproductive organs? Orchydopexy (the surgical correction of cryptorchidism) in Insl3-/- or Rxfp2-/- males restored spermatogenesis; moreover some Insl3-deficient males sired progeny. Thus, the presence of INSL3 is not critically important for testis function. Can INSL3 signaling play a supporting role in male reproduction? INSL3 treatment in the gonadotropin deprivation model decreased apoptosis of the germ cells,20 suggesting that this hormone may be a cell survival factor. The newly discovered non-reproductive function of INSL3 signaling in bone metabolism might demonstrate a broader role of this hormone in the adult organism.
Mutation analysis of INSL3 and RXFP2 in human patients
After discovery of the INSL3/RXFP2 role in TD in mice, a significant effort was made to uncover the gene mutations in patients with cryptorchidism. We and others performed screening of affected men and corresponding controls (see refs in 21, 22). Indeed a number of mutations in both genes were found. Perhaps due to the simple two-exon gene structure of INSL3 allowing direct sequencing, more mutations were found in this gene. RFXP2 has 18 exons and large population screens were performed using a less sensitive DHPLC analysis. Most of the mutant alleles were caused by a single nucleotide/amino acid substitution. They were found both in controls and in affected patients; some of them, however, were detected exclusively in the affected group (Fig. 2). Surprisingly, all the patients with the INSL3/RXFP2 mutations were heterozygous for the wild-type allele, implying that the potential mutation-dependent mechanism of testicular maldescent may be caused by reduced signaling.
FIGURE 2.

Identified sequence variants of INSL3 (A) and RXFP2 (B) genes in patients with cryptorchidism (shown above the protein structure) or in patients and control subjects (below protein structure). The T222P mutation is strongly associated with cryptorchidism, however some apparently normal individuals were described. SP, signal peptide; IC, C terminal intracellular domain. The amino acids are numbered from first translated methionine in non-processed protein. C-19G is a nucleotide substitution in the promoter region of INSL3.
INSL3 is translated as a pre-prohormone and contains signal sequence, B-chain, C-peptide, and A-chain. Upon hormone maturation, the signal sequence and C-peptide are removed, while A-and B-chains are assembled with two inter-chain and one intra-chain disulfide bonds. The mutations are found in all parts of the peptide, including C-19G variant of the promoter region.23 All but one of the mutations found are missense alleles. The only exception is the R73X mutation, resulting in the termination of translation and a putative expression of only the B peptide of INSL3. Recently, it was demonstrated that the B peptide of INSL3 retained binding but failed to induce signaling of RXFP2 receptor.24 Treatment of pregnant rats with truncated INSL3 peptide caused testicular retention in male progeny.25 The R73X mutation was found in a patient with unilateral cryptorchidism. B-chain mutation (P49S) in a patient with bilateral intra-abdominal testes and undermasculinized genitalia was reported. Functional analysis of the recombinant INSL3 peptides, containing the above-mentioned substitution, revealed that the P49S mutation affects the ability of INSL3 to activate its cognate receptor.26, 27 The mutations in the signal peptide (R4H, V18M) and A-chain (N110K) were identified in few patients. However, we did not find any compromised function of the recombinant peptides. The majority of mutations were identified in the C-peptide. Although this region is cleaved during peptide processing and not present in the mature hormone, it is possible that transcription, translation, or hormone processing are affected by these substitutions.
Analysis of familial cases of testicular maldescent provides an opportunity to follow the hereditary pattern of a mutant allele and the disease phenotype. Recently we analyzed one such family (Fig. 3). The affected child, two of his cousins and maternal grandfather all had unilateral undescended testis suggesting an inherited cause for the abnormality. We prepared genomic DNA samples from saliva of all members of the family. Following PCR amplification of the 2 exons of INSL3 and exons 8, 14, 17 of RFXP2, the PCR fragments were sequenced in both orientations. While no mutations were identified in the receptor gene and the second exon of INSL3 in proband or his parents, both mother and the affected child had previously unrecognized T to G substitution at position 116 beginning from the open reading frame. The substitution was predicted to cause valine-to-glycine substitution at position 39. The mutant exon also contained A60T substitution which became evident with the analysis of genomic DNA from his parents and grandparents. However, further sequencing analysis of INSL3 in the grandfather, affected cousins and mother’s sister did not reveal the mutant allele. Initial analysis of DNA isolated from grandmother’s saliva showed an absence of wild-type allele (Fig.3B), suggesting either the presence of the deletion or somatic mosaicism. To verify the results of the PCR we prepared DNA isolated from a peripheral blood sample obtained from the grandmother. Surprisingly, the sequence analysis revealed the presence of the wild-type allele. We suggest therefore that the grandmother was in fact mosaic with cells heterozygous and homozygous for V39G INSL3.
FIGURE 3.
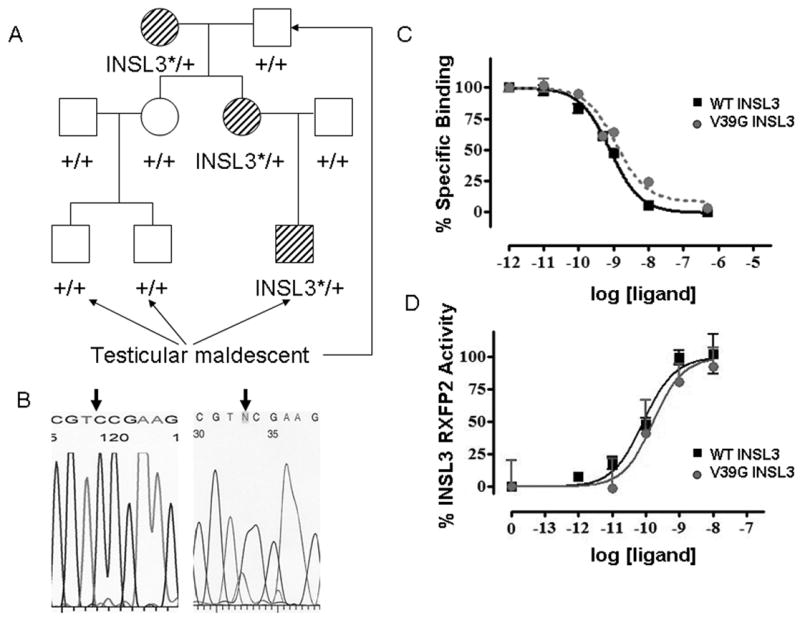
Novel V39G INSL3 mutation in a patient with cryptorchidism. A. Pedigree analysis of a family with cryptorchidism. The V39G mutation is marked as INSL3* and is detected in proband, his mother and grandmother. B. Sequencing of the PCR fragments obtained from the grandmothers genomic DNA isolated from saliva (left side) and peripheral blood (right side). The arrow shows the mutation site. No mutant allele was detected in DNA isolated from saliva. Reverse sequencing is presented. C. Competition binding curves of wild type and V39G INSL3 peptides using 0.5nM Eu-DTPA-INSL3 as labeled ligand in HEK293T cells stably transfected with RXFP2. Different concentrations of INSL3 were added to cells and incubated at room temperature for 60 min. GraphPad Prism software was used to fit the data from three experiments performed in triplicate using the nonlinear regression and one site competition model. D. Ligand-stimulated cAMP accumulation in RXFP2-expressing cells measured using a pCRE-β-galactosidase reporter gene system. Data are expressed as percentage of maximum INSL3 response and are pooled data from two experiments performed in triplicate.
Substitution of valine-to-alanine at position 39 affected binding but not signaling of the mutant peptide.28 To analyze the physiological properties of the V39G variant, the INSL3 peptide was synthesized. As shown on Fig. 3C and D, neither binding of the mutant peptide to the RXFP2 receptor nor cAMP induced signaling in cells transfected with RXFP2 was affected by the substitution as compared to the wild-type peptide.
Further functional, population, and, if possible, hereditary analysis of INSL3 variants is needed to ascertain the role of these mutations in the etiology of cryptorchidism.
The INSL3 receptor, RXFP2, is a G protein-coupled receptor with a large extracellular N-terminus, intracellular C-terminus and a central part composed of 7 transmembrane regions. The extracellular domain of RXFP2 includes ten leucine-rich repeats, believed to form a highly organized structure participating in the ligand binding. T222P represents the only mutation found so far that is present in several populations with higher frequency in cryptorchid patients than in control group (Fig. 2).5, 29 The severity of the phenotype varied with uni- and bilateral cryptorchidism; some patients had high intra-abdominal positioned testes, however others experienced spontaneous TD at puberty. All patients were heterozygotes. The fact that individuals with the same mutation presented with different clinical phenotypes suggests that other genetic and/or endocrine factors might affect the severity of INSL3-related deficiency. Indeed, the recent study by Nuti et al30 described several apparently normal subjects with the mutation. Although the misdiagnosis and/or non-recorded history of spontaneously corrected testicular maldescent in childhood is a possible explanation for contrasting results in this and other studies, the genetic or epigenetic influences on gene penetrance should be taken into account. Indeed, from an evolutionary perspective, the decreased male fertility associated with cryptorchidism might select those genetic factors suppressing manifestation of the abnormal phenotype in the population. It is important to compare the frequency of the mutation within specific populations. Whereas in Spain the mutation is present in a significant number of control subjects, in Italy the situation is quite different. Here the cumulative frequency of T222P mutation is 3.1% in affected males (41/1338) and 0.5% (8/1560) in controls, a value that is highly statistically significant (P < 0.0001, relative risk 5.97, 95% CI 2.86-12.51). Remarkably, in all cases where the parents of the affected patients were analyzed, the mutant allele was transmitted from the mother. At the amino acid level the mutation affects one of the leucine-rich repeats, and, according to functional analysis, renders the protein unable to be expressed on the cell surface membrane; thus suggesting possible haploinsufficiency as a mechanism of mutation effect in heterozygotes.29
In summary, despite overwhelming data supporting the role of INSL3/RXFP2 signaling in TD in mice, additional studies of its role in the etiology of human cryptorchidism are required. The gene-deficient phenotype in mice is viable, thus one can expect to find the homozygous mutant in human populations. Analysis of such patients combined with the studies of extended pedigrees might further the understanding of the role of INSL3 in testis descent.
Acknowledgments
The work in Dr. Agoulnik lab is supported by the National Institute of Health grants 5R01HD03706 and 1R21CA118362. Reproductive biology and cancer studies in Dr. Lamb laboratory are supported in part by the NIH grants 5P01HD36289, 1R01DK078121, 5T32DK00763, U54HD007495-31, and the Department Of Defense, U.S. Army Materiel Command grant PC061154.
References
- 1.Hutson JM. A biphasic model for the hormonal control of testicular descent. Lancet. 1985;2:419–421. doi: 10.1016/s0140-6736(85)92739-4. [DOI] [PubMed] [Google Scholar]
- 2.Hadziselimovic F, Herzog B. The development and descent of the epididymis. Eur J Pediatr. 1993;152(Suppl 2):S6–9. doi: 10.1007/BF02125424. [DOI] [PubMed] [Google Scholar]
- 3.Zimmermann S, et al. Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol Endocrinol. 1999;13:681–691. doi: 10.1210/mend.13.5.0272. [DOI] [PubMed] [Google Scholar]
- 4.Nef S, Parada LF. Cryptorchidism in mice mutant for Insl3. Nat Genet. 1999;22:295–299. doi: 10.1038/10364. [DOI] [PubMed] [Google Scholar]
- 5.Gorlov IP, et al. Mutations of the GREAT gene cause cryptorchidism. Hum Mol Genet. 2002;11:2309–2318. doi: 10.1093/hmg/11.19.2309. [DOI] [PubMed] [Google Scholar]
- 6.Overbeek PA, et al. A transgenic insertion causing cryptorchidism in mice. Genesis. 2001;30:26–35. doi: 10.1002/gene.1029. [DOI] [PubMed] [Google Scholar]
- 7.Kumagai J, et al. INSL3/Leydig insulin-like peptide activates the LGR8 receptor important in testis descent. J Biol Chem. 2002;277:31283–31286. doi: 10.1074/jbc.C200398200. [DOI] [PubMed] [Google Scholar]
- 8.Hsu SY, et al. Relaxin signaling in reproductive tissues. Mol Cell Endocrinol. 2003;202:165–170. doi: 10.1016/s0303-7207(03)00078-9. [DOI] [PubMed] [Google Scholar]
- 9.Sadeghian H, et al. Constitutive regulation of the Insl3 gene in rat Leydig cells. Mol Cell Endocrinol. 2005;241:10–20. doi: 10.1016/j.mce.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Emmen JM, et al. Involvement of insulin-like factor 3 (Insl3) in diethylstilbestrol-induced cryptorchidism. Endocrinology. 2000;141:846–849. doi: 10.1210/endo.141.2.7379. [DOI] [PubMed] [Google Scholar]
- 11.Nef S, Shipman T, Parada LF. A molecular basis for estrogen-induced cryptorchidism. Dev Biol. 2000;224:354–361. doi: 10.1006/dbio.2000.9785. [DOI] [PubMed] [Google Scholar]
- 12.Truong A, et al. Isolation and expression analysis of the canine insulin-like factor 3 gene. Biol Reprod. 2003;69:1658–1664. doi: 10.1095/biolreprod.103.019166. [DOI] [PubMed] [Google Scholar]
- 13.Koskimies P, et al. Murine relaxin-like factor promoter: functional characterization and regulation by transcription factors steroidogenic factor 1 and DAX-1. Endocrinology. 2002;143:909–919. doi: 10.1210/endo.143.3.8683. [DOI] [PubMed] [Google Scholar]
- 14.Robert NM, Martin LJ, Tremblay JJ. The orphan nuclear receptor NR4A1 regulates insulin-like 3 gene transcription in Leydig cells. Biol Reprod. 2006;74:322–330. doi: 10.1095/biolreprod.105.044560. [DOI] [PubMed] [Google Scholar]
- 15.Agoulnik AI. Mouse mutants of relaxin, insulin-like 3 peptide and their receptors. Curr Med Chem Immunol Endocr Metab Agents. 2005;5:411–419. [Google Scholar]
- 16.Feng S, et al. Developmental expression and gene regulation of insulin-like 3 receptor RXFP2 in mouse male reproductive organs. Biol Reprod. 2007;77:671–680. doi: 10.1095/biolreprod.107.060442. [DOI] [PubMed] [Google Scholar]
- 17.Adham IM, et al. The overexpression of the insl3 in female mice causes descent of the ovaries. Mol Endocrinol. 2002;16:244–252. doi: 10.1210/mend.16.2.0772. [DOI] [PubMed] [Google Scholar]
- 18.Klonisch T, Fowler PA, Hombach-Klonisch S. Molecular and genetic regulation of testis descent and external genitalia development. Dev Biol. 2004;270:1–18. doi: 10.1016/j.ydbio.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Feng S, et al. Over expression of insulin-like 3 does not prevent cryptorchidism in GNRHR or HOXA10 deficient mice. J Urol. 2006;176:399–404. doi: 10.1016/S0022-5347(06)00519-2. [DOI] [PubMed] [Google Scholar]
- 20.Kawamura K, et al. Paracrine regulation of mammalian oocyte maturation and male germ cell survival. Proc Natl Acad Sci U S A. 2004;101:7323–7328. doi: 10.1073/pnas.0307061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogatcheva NV, Agoulnik AI. INSL3/LGR8 role in testicular descent and cryptorchidism. Reprod Biomed Online. 2005;10:49–54. doi: 10.1016/s1472-6483(10)60803-6. [DOI] [PubMed] [Google Scholar]
- 22.Ferlin A, et al. Paracrine and endocrine roles of insulin-like factor 3. J Endocrinol Invest. 2006;29:657–664. doi: 10.1007/BF03344168. [DOI] [PubMed] [Google Scholar]
- 23.El Houate B, et al. Novel mutations involving the INSL3 gene associated with cryptorchidism. J Urol. 2007;177:1947–1951. doi: 10.1016/j.juro.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Del Borgo MP, et al. Analogs of insulin-like peptide 3 (INSL3) B-chain are LGR8 antagonists in vitro and in vivo. J Biol Chem. 2006;281:13068–13074. doi: 10.1074/jbc.M600472200. [DOI] [PubMed] [Google Scholar]
- 25.Bullesbach EE, et al. Cryptorchidism induced in normal rats by the relaxin-like factor inhibitor. Reproduction. 2008;135:351–355. doi: 10.1530/REP-07-0330. [DOI] [PubMed] [Google Scholar]
- 26.Bogatcheva NV, et al. GREAT/LGR8 is the only receptor for insulin-like 3 peptide. Mol Endocrinol. 2003;17:2639–2646. doi: 10.1210/me.2003-0096. [DOI] [PubMed] [Google Scholar]
- 27.Ferlin A, et al. Insulin-like factor 3 gene mutations in testicular dysgenesis syndrome: clinical and functional characterization. Mol Hum Reprod. 2006;12:401–406. doi: 10.1093/molehr/gal043. [DOI] [PubMed] [Google Scholar]
- 28.Bullesbach EE, Schwabe C. The mode of interaction of the relaxin-like factor (RLF) with the leucine-rich repeat G protein-activated receptor 8. J Biol Chem. 2006;281:26136–26143. doi: 10.1074/jbc.M601414200. [DOI] [PubMed] [Google Scholar]
- 29.Bogatcheva NV, et al. T222P mutation of the insulin-like 3 hormone receptor LGR8 is associated with testicular maldescent and hinders receptor expression on the cell surface membrane. Am J Physiol Endocrinol Metab. 2007;292:E138–144. doi: 10.1152/ajpendo.00228.2006. [DOI] [PubMed] [Google Scholar]
- 30.Nuti F, et al. The leucine-rich repeat-containing G protein-coupled receptor 8 gene T222P mutation does not cause cryptorchidism. J Clin Endocrinol Metab. 2008;93:1072–1076. doi: 10.1210/jc.2007-1993. [DOI] [PubMed] [Google Scholar]