

Abstract
Genealogical investigation of a large Norwegian family (F04) with autosomal dominant parkinsonism has identified 18 affected family members over four generations. Genetic studies have revealed a novel pathogenic LRRK2 mutation c.4309 C>A (p.Asn1437His) that co-segregates with disease manifestation (LOD=3.15, θ=0). Affected carriers have an early age at onset (48 ± 7.7 SD years) and are clinically asymmetric and levodopa-responsive. The variant was absent in 623 Norwegian control subjects. Further screening of patients from the same population identified one additional affected carrier (1/692) with familial parkinsonism who shares the same haplotype. The mutation is located within the Roc domain of the protein and enhances GTP-binding and kinase activity, further implicating these activities as the mechanisms that underlie LRRK2-linked parkinsonism.
Keywords: LRRK2, Parkinson’s disease, genetic, kinase
Introduction
Parkinson’s disease (PD) is a neurodegenerative movement disorder characterized by resting tremor, bradykinesia, rigidity and postural instability. The initial presentation is asymmetric and insidiously progressive, albeit responsive to levodopa replacement therapy. The overall prevalence is age associated and rare before 50 years, increasing up to 4% in the oldest populations 1. Most patients appear sporadic but 10–15% report a family history of parkinsonism and recent genetic studies have identified pathogenic mutations in several genes 2. This molecular etiology helps provide a rationale for novel therapeutic approaches and the models to develop them 2, 3.
Leucine-rich repeat kinase 2 (LRRK2) harbors six pathogenic substitutions (p.Arg1441Cys/Gly/His, p.Tyr1699Cys, p.Gly2019Ser and p.Ile2020Thr) and two susceptibility variants (p.Arg1628Pro and p.Gly2385Arg) implicated in PD (http:www.genetests.org). LRRK2 c.6055G>A (p.Gly2019Ser) confers the greatest population attributable risk and directly activates the kinase domain 4, 5. Observed in 1% of sporadic PD and 5% of familial parkinsonism in the US and Europe, the frequency of p.Gly2019Ser dramatically increases in patients of Ashkenazi Jewish and North-African Arab-Berber heritage 6–9. For LRRK2 mutations penetrance is age-related for example conservative estimates for p.G2019S from the larger studies suggest disease manifests in approximately 8% of carriers by 50 years to more than 80% at 75 years 8–10.
Our group has longitudinally followed a number of families with a history of parkinsonism from Central Norway 6, 11. Herein we describe the presentation of autosomal dominant parkinsonism in a large Norwegian family (F04) resulting from a novel LRRK2 mutation. The mutation segregates with disease in this multi-incident kindred providing proof of pathogenicity. In addition, the mutation is present in a second Norwegian family (F45) with parkinsonism. In vitro studies demonstrate the functional consequence of this amino acid substitution on Lrrk2 GTPase and kinase activities.
Methods
Genealogical research
Genealogical investigations of Families F04 and F45 included collection of historical material, family records and interviews with family members. Direct contact was initiated with other family members via the proband with prior informed consent. A standardized questionnaire was used to enquire about all 1–3° relatives (parents, siblings - first cousins) and whether there was a diagnosis of PD, symptoms of parkinsonism or other neurodegenerative disease. Confirmation of historic and demographic data employed the national/regional archives, the Norwegian censuses of 1801, 1865, 1875 and 1900, and local parish records.
Clinical exams
The medical records of all living and deceased patients were reviewed. Clinical examinations of affected family members, included the unified Parkinson’s disease rating scale (UPDRS) 12, Hoehn & Yahr scale 13 and mini-mental state examinations 14, and for the majority of patients were conducted longitudinally as part of a routine visit to a neurology clinic (by JOA). A clinical diagnosis of PD required the presence of at least two of three cardinal signs (resting tremor, bradykinesia, and rigidity), improvement through adequate dopaminergic therapy and the absence of atypical features or other causes of parkinsonism 15. Disease duration was calculated by subtracting the age at onset, obtained from the patient’s medical records, from the age at examination. When the age of onset was unknown it was estimated from the patient’s documented clinical history or by self report. Clinicogenetic studies were approved by the Institutional Review Boards at St. Olavs Hospital, Trondheim and Mayo Clinic, Jacksonville. Informed consent was obtained from all patients, by proxy if deceased, and from control subjects.
Population samples
The Norwegian series examined includes sequential new referrals to the Department of Neurology, St. Olav’s Hospital, Trondheim, Norway between May 1998 and January 2009. Eighty-percent are community-based and reside within Trondheim or within 50 miles. Most of the remainder derives from the surrounding district, within a 200 mile radius, and less than 5% from other parts of the country. All patients were examined and observed longitudinally by one movement disorders neurologist (JOA) and diagnosed with PD according to published criteria 15. Patients with PD have a mean age of 70 ± 11 years, n=692, with a mean age at disease onset of 58 ± 11 years; range 25–88 years. Unrelated Norwegian control subjects were volunteers recruited from two local blood banks (n=308), the Department of Ophthalmology (n=60) and from local senior citizen societies. Norwegian control subjects were matched by age (± 5 years) and gender (n=623).
SPECT imaging
At-risk clinically asymptomatic individuals older than 40 years were invited to participate in a neurological exam and single photon emission computed tomography (SPECT). These were selected from the offspring of affected individuals and were potential Lrrk2 mutation carriers. However, both neurologist and subjects were blinded to genetic data and SPECT results at the time of the clinical evaluation. SPECT imaging took place 3 to 4 hours after intravenous injection of 123I-Ioflupane (111–185 MBq; DaTSCAN GE Healthcare, Amersham, UK) as a ligand for the dopamine transporter (DAT). Image interpretation was based on visual assessment by an experienced physician. Normal images were considered to be those characterized by a largely symmetrical uptake of the tracer in both right and left putamen and caudate nuclei. Abnormal images included those with asymmetrical or reduced uptake.
Genetic Analysis
Blood samples were drawn and genomic DNA extracted using standard techniques. As parkinsonism within families F04 and F45 segregates in an autosomal dominant pattern, all affected subjects were screened for missense and multiplication mutations in α-synuclein 16. LRRK2 gene sequencing (51 exons) was performed as previously described 17. The LRRK2 sequence and amino acid notation were compared with the published sequence of LRRK2 (GenBank accession number AY792511). Genotyping of LRRK2 c.4309 C>A (p.Asn1437His) was subsequently performed on a Sequenom MassArray iPLEX platform (San Diego, CA); all primer sequences are available on request.
Protein biochemistry: Plasmids, cell culture and transfection
Mammalian expression constructs encoding human wild-type and mutant Lrrk2 (c.4321C>T p.Arg1441Cys; c.6055G>A p.Gly2019Ser) on the pcDNA3.1-myc/his backbone (Invitrogen) have been previously described 5. Additional variants including LRRK2 c.4309 C>A (p.Asn1437His) and c.4043A>C (p.Thr1348Asn) were generated using site specific mutagenesis (Stratagene). Primer sequences for the different variants are available on request. Individual maxi-scale plasmid preparations (Qiagen) were assessed for equivalent Lrrk2 expression by western blot (SDS-PAGE, described below) and resequenced in entirety prior to usage in experiments. Experiments were carried out in HEK-293FT cells cultured in Opti-MEM media supplemented with 10% fetal bovine serum (Invitrogen). One day prior to transfection, cells were passaged and transfection was carried out at ~ 80% confluence in Opti-MEM media supplemented with 2% fetal bovine serum using FugeneHD reagent at a 1:3 ratio (plasmid to FugeneHD, Roche), and cells were harvested 48 hours post.
Lrrk2 GTP-binding assays
Cells were harvested into PBS lysis buffer (PBS pH=7.4, 0.1% Triton X-100, protease and phosphatase inhibitors [Complete Mini Protease Inhibitor Cocktail, PhosStop, both Roche]), rotated for 1 hour at 4°C to obtain complete cell lysis and clarified by centrifugation at 20,000x g for 10min. The pre-clarified samples were adjusted for protein content by BCA assay (Pierce) and incubated with 60μl γ-aminohexyl-GTP-sepharose suspension (Jena Bioscience) by rotating at 16°C for 2 hours. After the incubation, beads were washed five times with 500μl lysis buffer supplemented to 500mM NaCl and three times with PBS alone. GTP-bound proteins were eluted in Laemmli sample buffer by heating for 10 min at 70°C, and protein resolved by SDS-PAGE and relative protein concentrations determined by western blot analysis with anti-myc-HRP (Roche) antibody as previously described 5.
Lrrk2 kinase assays
Cells were harvested, lysed and pre-clarified as described in the Lrrk2-GTP-binding assay above. After adjusting for protein content, cell lysates were combined with Protein-G Dynabeads (Invitrogen) precoupled with anti-Myc antibody (clone 9E10, Roche) and incubated at 4°C overnight on a rotor wheel. Dynabeads were subsequently washed five times with lysis buffer supplemented to 300mM NaCl and resuspended into kinase assay buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM EGTA, 20 mM β-glycerol phosphate). Individual kinase reactions were initiated with the addition of 10 μM ATP [0.5 μCi γ-32P-ATP (Perkin Elmer)] and 20 mM MgCl2. The kinase reactions were incubated at 30°C for 30 min with gentle shaking. The reaction was terminated by placing the samples on ice and supplementing with 2x Laemmli buffer. To achieve complete elution and to denature Lrrk2 protein complexes, samples were kept at 70°C for 10 min with gentle shaking After centrifugation and removal of the beads, kinase reactions were resolved using 3–12% gradient Bis-Tris acrylamide SDS gels (Invitrogen). Gels were subsequently stained with Biosafe G-250 dye (BioRad) and dried onto Whatman paper. Lrrk2 autophosphorylation signal was resolved with Kodak BioMax film and intensity of spots determined with NIH ImageJ.
Results
Historical and genealogical investigation
Probands of families F04 and F45 indicated a family history of parkinsonism within 1–3° of relationship. Detailed genealogic investigation of F04 revealed a multi-incident pedigree with 18 clinically affected family members spanning four generations. One patient was diagnosed with PD in the United States, subsequent to their emigration from Norway (F04-26). For F45 the genealogy is not as extensive and only the proband and his mother are known to be affected by parkinsonism. Segregation of disease is most consistent with autosomal dominant inheritance with reduced penetrance. Combining current and historical data from F04 and F45 kindreds the mean age at onset of disease was 47.8 ± 8 years (range 37–61 years, n=10), with a mean duration to death of 18 ± 8 years (range 2–26 years, n=7). To protect patient confidentiality the pedigrees illustrated are simplified and gender-disguised (Figure 1a).
Figure 1.


A) Pedigrees of F04 and F45
Pedigrees of F04 and F45. Individuals with DNA available are numbered beneath symbols, the proband in each family is arrowed, males are squares, circles are females, diamonds are gender disguised. A diagonal line indicates the subject is deceased, mt = carrier, wt = non-carrier. Asterisks refer to subjects with asymmetric SPECT images.
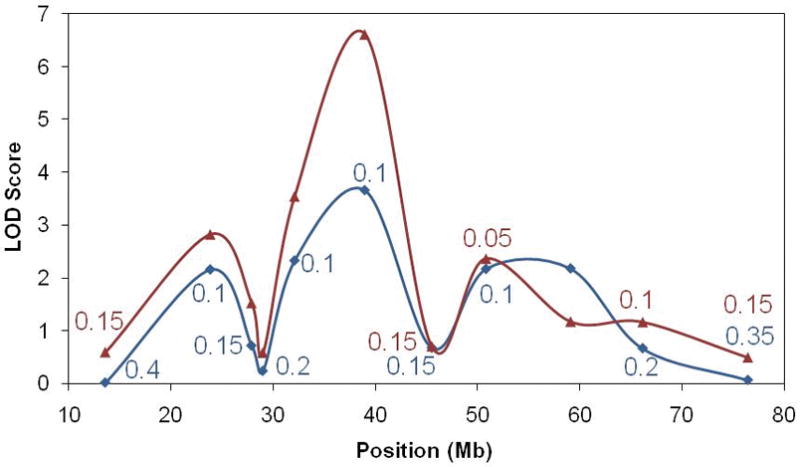
B) LOD scores
Model-based, maximum two-point LOD scores are illustrated across chromosome 12. The red line shows values obtained assuming F04-1 and F04-34 are disease phenocopies, whereas the blue line was calculated assuming they have the same cause of disease. The value of θ is only given for each marker when different from 0.0. The genomic location of LRRK2 is under the peak at 39Mb.
C) Disease-segregating chromosome 12q12 haplotype
The higher-resolution phase-known haplotype and genotypes of F04 and F45-3 are shown.
Genetic and clinical findings
Screening for α-synuclein missense and multiplication mutations was negative for all affected family members. None of the previously reported pathogenic mutations in the LRRK2 gene were identified. However, LRRK2 sequencing in F04-1 (the proband) and F04-2 revealed discrepant results in that only F04-2 had a LRRK2 c.4309 C>A transition leading to a novel Lrrk2 p.Asn1437His substitution in the Roc domain of the protein (Figure 2). Of the 33 subjects from Family F04, DNA was available for eleven individuals (denoted by a number below the symbol in Figure 1a; 6/14 female and 5/19 male) for screening of the LRRK2 c.4309 C>A transition. In addition to the five affected mutation carriers examined, three further affected carriers were inferred from the results of their spouse and offspring. Notably, both the affected proband F04-1 and a second affected cousin F04-34 (age at onset 50 and 70 years, respectively) do not have this LRRK2 mutation; in addition neither had an affected parent and therefore have a different etiology underlying disease. Chromosome 12 short-tandem repeat genotyping confirmed sample relationships within the pedigree. Two-point LOD scores were calculated under the assumption of an autosomal dominant model using MLINK 18. The frequency of the deleterious allele was set at 0.0001, using chromosome 12 marker-allele frequencies and intermarker distances, as previously described 6. Linkage analysis summarizes the genetic evidence for LRRK2 c.4309 C>A segregation with disease and remained significant under all models examined (minLOD = 3.66, θ=0.1; maxLOD = 6.02, θ=0) (Figure 1b).
Figure 2. LRRK2 idiogram of domains, chromatogram of LRRK2 c.4309 C>A, Roc domain protein structure and conservation across species.

Molecular model of the Lrrk2 Roc domain highlighting in yellow the p.Asn1437His and p.Arg1441Cys/Gly/His variants was obtained from the NCBI MMDB database [MMDB ID# 64897 (PDB# 3D6T)] 22. Species conservation for the p.Asn1437His and adjacent amino acids was obtained from the UCSC Genome browser and the pathogenic p.Arg.1441 and putative pathogenic p.Ala1442 mutated residues are indicated with asterisks.
Further screening of LRRK2 c.4309 C>A in a community-based sample of patients and control subjects from the same geographical region of Norway identified only one additional affected carrier (F45-3, the affected proband of family F45). This patient’s mother was reported to also have PD but was unavailable for genetic testing. Chromosome 12q12 short-tandem repeat genotyping was consistent with a shared haplotype between F04 and F45 suggesting both are part of one ancestral kindred (Figure 1c).
Combined the mean age of onset of obligate or genetically-confirmed Lrrk2 p.Asn1437His carriers in F04 and F45 was 47 ± 7.8 years (range 37–61 years, n=10), significantly younger than for idiopathic PD in Norway (58 ± 11 years). Otherwise, Lrrk2 p.Asn1437His carriers with parkinsonism were largely indistinguishable from idiopathic PD in terms of first presentation and predominant subtype and severity, based on Hoehn & Yahr and UPDRS scores. Although the number of affected carriers is relatively few, the duration of disease was also similar to idiopathic PD (18 ± 8 years, range 2–26 years, n=7). Both genetically-defined p.Asn1437His carriers and idiopathic patients have a favorable response to levodopa administration. In addition, one Lrrk2 p.Asn1437His carrier currently benefits from subthalamic deep brain stimulation (STN-DBS). SPECT imaging on two clinically asymptomatic carriers also revealed asymmetric deficits in DAT binding, mainly in the putamen. The average age at study was 49.5 ± 0.7 years and within the mean age of onset. Two years post-SPECT the UPDRS results, part III, were 10 (presenting bradykinetic finger-finger, finger flexion, and alternating movements, as well as walking and body bradykinesia) and 3 respectively, and both remain clinically asymptomatic.
Biochemical results
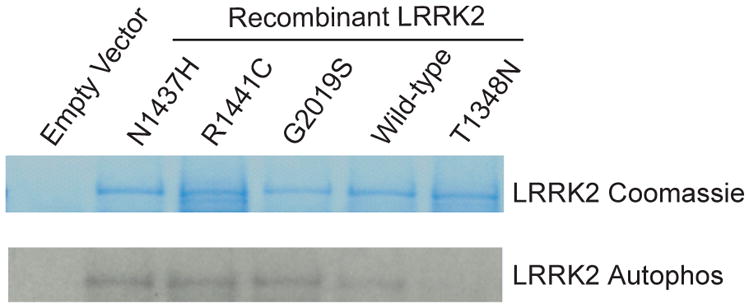
To assess the respective enzymatic properties of the newly identified variant, in vitro kinase and GTP-binding assays were performed. Compared to wild-type protein, Lrrk2 p.Asn1437His displays a significantly higher proportion of GTP-bound Lrrk2 in whole cell protein lysates relative to WT-Lrrk2 (p<0.01, Figure 3), consistent with reduced GTPase activity observed for the p.Arg1441Cys Lrrk2 pathogenic substitution. Also consistent with p.Arg1441Cys, p.Asn1437His enhances autophosphorylation activity comparable to the p.Gly2019Ser mutation in the described assay. An artificial p.Thr1348Asn in the Roc domain ablates both GTPase and kinase activity and further highlights the link between Roc and kinase activity in Lrrk2. Both GTP-bound Lrrk2 and autophosphorylation levels were normalized to protein inputs (Figure 3).
Figure 3. The p.Asn1437His mutation alters Lrrk2 GTP-binding and kinase activity.
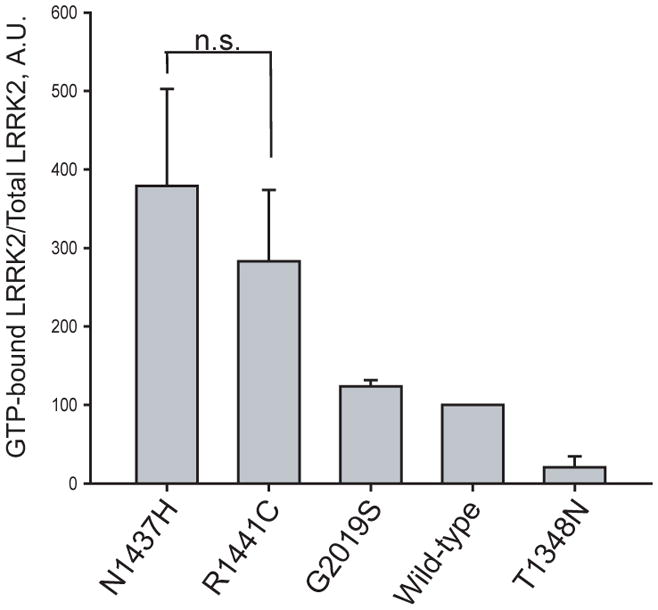
A) GTP-binding assay
GTP-binding assay, where the proportion of Lrrk2 bound to GTP-γ-sepharose beads normalized to total Lrrk2 in protein lysates shows that p.Asn1437His has a similar effect on activity compared with p.Arg1441Cys mutant Lrrk2 (n.s. is non-significant) and p.Asn1437His increases the proportion of GTP-bound Lrrk2 compared with wild-type protein. Data are derived from three independent experiments.
B) Lrrk2 kinase assay
Lrrk2 autophosphorylation kinase assay, with total Lrrk2 indicated by coomassie stain and kinase activity indicated by autoradiography. Normalized to input protein, p.Asn1437His demonstrates enhanced kinase activity compared with wild-type protein. The results are comparable to p.Gly2019Ser and p.Arg1441Cys Lrrk2 mutant proteins. Data are representative of three experiments.
Discussion
LRRK2 encodes a 2527 amino acid member of the ROCO protein family characterized by contiguous guanosine triphosphatase (GTPase Roc), C-terminal of Roc (COR) and kinase domains, and flanked by ankyrin/armadillo, leucine-rich repeat and WD40 (Trp-Asp 40) protein-protein interaction motifs 4. More than 75 coding substitutions have been described in the LRRK2 gene but the majority are rare and only six have been proven pathogenic 19. Herein we describe a novel LRRK2 c.4309 C>A transition resulting in a p.Asn1437His Roc domain substitution, with genetic and functional data providing an overwhelming burden-of-proof for pathogenicity. The identification of a second pathogenic variant in the Roc domain provides further opportunity for understanding the pathogenic mechanism associated with LRRK2-linked PD.
LRRK2 c.4309 C>A segregates with autosomal dominant parkinsonism in a multi-incident, multigenerational pedigree in which there is significant evidence for genetic linkage (LOD>3.0, θ=0), despite the presence of several affected phenocopies who are non-carriers. Genome-wide analysis did not identify any other regions suggestive for linkage (LOD score < 2.0). All carriers at or beyond the mean age-at-onset in the family manifest parkinsonism or have pathological uptake on DAT-scans. Genetic findings were confirmed in a seemingly unrelated kindred that suffers autosomal dominant parkinsonism, in which affected individuals share an ancestral LRRK2 c.4309 C>A haplotype. The frequency in patients with idiopathic PD from the same population is low (1/693 or 0.15%) and the mutation was not identified in matched control subjects. Nevertheless, given the dramatic population-specific differences in the frequency of other LRRK2 mutations genetic screening of other population series is warranted.
Clinically symptoms are comparable to idiopathic PD albeit with an earlier age at onset. Of note, asymptomatic carriers around the age-at-onset show suggestive asymmetric deficit in SPECT dopaminergic imaging, extending previous observations using positron emission tomography 20. Co-morbidities in autonomic, psychiatric and cognitive domains associated with idiopathic PD were similarly observed in affected Lrrk2 p.Asn1437His carriers, although numbers are too few for meaningful statistical comparison (data not shown). Post-mortem evaluations have yet to be performed but the majority of patients with Lrrk2-parkinsonism have Lewy body disease, consistent with a definite diagnosis of PD 21.
Biochemical studies of intrinsic enzymatic activities altered by the Lrrk2 p.Asn1437His mutation reveal enhanced Roc GTP-binding, with a consequent increase in kinase activity and autophosphorylation. Lrrk2 p.Asn1437His mutant effects on intramolecular and perhaps intermolecular Lrrk2 activation are consistent with past findings for other Roc domain mutants such as Lrrk2 p.Arg1441Cys, leading to a toxic gain-of-function 22. Identification of physiologically relevant Lrrk2 kinase substrates will help clarify the role of Lrrk2 enzymatic activation in neurodegeneration.
In summary, Lrrk2 p.Asn1437His is the seventh pathogenic mutation in Lrrk2 that leads to clinical parkinsonism consistent with idiopathic PD. Our observations highlight the issue of phenocopy rate in family-based studies of parkinsonism and the need for genetic screening in several affected members of multi-incident kindreds. Although the pathogenicity of Lrrk2 p.Asn1437His is unequivocal, the penetrance of disease within carriers, as with other Lrrk2 mutations, is variable. The range in age-at-onset clearly suggests the presence of genetic and environmental modifiers which may become therapeutic targets for neuroprotection. Given the frequency of Lrrk2-linked disease there is now the unprecedented opportunity to develop studies of the natural history of parkinsonism in terms of motor and non-motor features, in asymptomatic and affected carriers, and to develop clinical trials and drugs that may rectify the molecular etiology of disease.
Acknowledgments
The authors thank the patients and all family members who made this study possible. This study was supported by grants from the NIH Udall Parkinson’s Disease Research Center of Excellence (MJF), Michel J. Fox Foundation (OAR) and Research Council of Norway (JOA). Additional support from an NIH R00NS058111 award (ABW), NIH R01NS36960 (HP) and the American Parkinson’s Disease Association fellowship (PJW) is acknowledged.
Author Roles
1. Research project: A. Conception, B. Organization, C. Execution;
2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique;
3. Manuscript: A. Writing of the first draft, B. Review and Critique;
| Jan O. Aasly | 1A, 1B, 1C, 2C, 3A, 3B |
| Carles Vilariño-Güell | 1C, 2A, 2B, 2C, 3A, 3B |
| Justus C. Dachsel | 1A, 1B, 1C, 2C, 3A, 3B |
| Philip J. Webber | 1C, 2C, 3A, 3B |
| Andrew B. West | 1A, 1B, 1C, 2C, 3A, 3B |
| Kristoffer Haugarvoll | 1A, 1C, 2B, 2C, 3B |
| Krisztina K. Johansen | 1C, 2C, 3B |
| Mathias Toft | 1A, 1B, 1C, 2C, 3B |
| John G. Nutt | 1B, 2C, 3B |
| Haydeh Payami | 1B, 2C, 3B |
| Jennifer M. Kachergus | 1C, 2C, 3B |
| Sarah J. Lincoln | 1B, 1C, 2C, 3B |
| Amela Felic | 1C, 3A, 3B |
| Christian Wider | 1B, 1C, 2C, 3B |
| Alexandra I. Soto-Ortolaza | 1C, 2C, 3A |
| Stephanie A. Cobb | 1B, 2B, 2C, 3A |
| Linda R. White | 1A, 1B, 2C, 3B |
| Owen A. Ross | 1B, 1C, 2C, 3B |
| Matthew J. Farrer | 1A, 1B, 2A, 2C, 3B |
Full Financial Disclosures of all Authors for the Past Year
Stock Ownership in medically-related fields; Consultancies; Advisory Boards; Partnerships; Honoraria; Grants; Intellectual Property Rights; Expert Testimony; Contracts; Royalties; Other
Carles Vilariño-Güell, Justus C. Dachsel, Kristoffer Haugarvoll, Mathias Toft, Krisztina K. Johansen, Jennifer M. Kachergus, Sarah J. Lincoln, Amela Felic, Christian Wider, Alexandra I. Soto-Ortolaza, Stephanie A. Cobb, Linda R. White.
None
| Jan O. Aasly | Owen A. Ross | |
| Stock Ownership in medically-related fields | None | None |
| Consultancies | None | None |
| Advisory Boards | None | None |
| Partnerships | None | None |
| Honoraria | None | None |
| Grants | Research Council of Norway | Michael J. Fox Foundation Research Grant |
| Intellectual Property Rights | None | None |
| Expert Testimony | None | None |
| Contracts | None | None |
| Royalties | None | None |
| Other | None | None |
| Haydeh Payami | Andrew B. West | |
| Stock Ownership in medically-related fields | None | None |
| Consultancies | NIH ARRA Challenge Grant, NIH study section, HRI Inc. | None |
| Advisory Boards | None | None |
| Partnerships | None | None |
| Honoraria | None | None |
| Grants | NIH R01NS36960, NY Attorney General Office Research Grant | NIH R00NS058111 |
| Intellectual Property Rights | None | None |
| Expert Testimony | None | None |
| Contracts | None | None |
| Royalties | None | None |
| Other | None | None |
| Philip J. Webber | John G. Nutt | |
| Stock Ownership in medically-related fields | None | None |
| Consultancies | None | XenoPort, Impax Lab, Neurogen, Synosia |
| Advisory Boards | None | None |
| Partnerships | None | None |
| Honoraria | None | Novartis |
| Grants | APDA Fellowship | NIH |
| Intellectual Property Rights | None | None |
| Expert Testimony | None | None |
| Contracts | None | Schering-Plough |
| Royalties | None | None |
| Other | None | None |
| Matthew J. Farrer | ||
| Stock Ownership in medically-related fields | None | |
| Consultancies | Genzyme, H. Lundbeck A/S | |
| Advisory Boards | Michael J. Fox Foundation Scientific Advisory Board | |
| Partnerships | None | |
| Honoraria | None | |
| Grants | Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 #NS40256) | Michael J. Fox Foundation Research Grants |
| Intellectual Property Rights | None | |
| Expert Testimony | None | |
| Contracts | None | |
| Royalties | None | |
| Other | None |
| Mathias Toft |
| Stock Ownership in medically-related fields: None |
| Intellectual Property Rights: None |
| Consultancies: None |
| Expert Testimony: None |
| Advisory Boards: None |
| Employment: None |
| Partnerships: None |
| Contracts: None |
| Honoraria:GlaxoSmithKline, Lundbeck, Orion, Medtronic, Solvay |
| Royalties: None |
| Grants: South-Eastern Norway Regional Health Authority |
| Other: None |
Footnotes
Financial Disclosures: Mayo Clinic Jacksonville is a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 #NS40256). MJF and OAR are supported by the Michael J. Fox Foundation. JOA is supported by the Research Council of Norway. Additional support for ABW from an NIH R00NS058111 award, for HP from an NIH R01NS36960 and for PJW an American Parkinson’s Disease Association fellowship. CVG, JCD, KH, KKJ, MT, JGN, JMK, SJL, AF, CW, AIS, SAC, LRW have no financial disclosures to make.
References
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5(6):525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7(4):306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 3.Bagade S, Lill C, McQueen M, et al. The PDGene Database. Alzheimer Research Forum; 2009. [Google Scholar]
- 4.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 2006;29(5):286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 5.West AB, Moore DJ, Choi C, et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16(2):223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 6.Kachergus J, Mata IF, Hulihan M, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76(4):672–680. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozelius LJ, Senthil G, Saunders-Pullman R, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354(4):424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 8.Hulihan MM, Ishihara-Paul L, Kachergus J, et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 2008;7(7):591–594. doi: 10.1016/S1474-4422(08)70116-9. [DOI] [PubMed] [Google Scholar]
- 9.Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Latourelle JC, Sun M, Lew MF, et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med. 2008;6:32. doi: 10.1186/1741-7015-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aasly JO, Toft M, Fernandez-Mata I, et al. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol. 2005;57(5):762–765. doi: 10.1002/ana.20456. [DOI] [PubMed] [Google Scholar]
- 12.Fahn S, Elton RL. Unified Parkinson’s disease rating scale. Florham Park, NJ: Macmillan Healthcare Information; 1987. [Google Scholar]
- 13.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17(5):427–442. doi: 10.1212/wnl.17.5.427. [DOI] [PubMed] [Google Scholar]
- 14.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 15.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56(1):33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 16.Ross OA, Braithwaite AT, Skipper LM, et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63(6):743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mata IF, Kachergus JM, Taylor JP, et al. Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics. 2005;6(4):171–177. doi: 10.1007/s10048-005-0005-1. [DOI] [PubMed] [Google Scholar]
- 18.Terwilliger J, Ott J. Handbook of Human Genetic linkage. Baltimore: John Hopkins University Press; 1994. [Google Scholar]
- 19.Farrer M, Ross OA. LRRK2-related Parkinson’s disease. GeneReviews at GeneTests:: Medical Genetics Information Resource (database online) 2009 [Google Scholar]
- 20.Nandhagopal R, Mak E, Schulzer M, et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology. 2008;71(22):1790–1795. doi: 10.1212/01.wnl.0000335973.66333.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross OA, Toft M, Whittle AJ, et al. Lrrk2 and Lewy body disease. Ann Neurol. 2006;59(2):388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- 22.Deng J, Lewis PA, Greggio E, Sluch E, Beilina A, Cookson MR. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci U S A. 2008;105(5):1499–1504. doi: 10.1073/pnas.0709098105. [DOI] [PMC free article] [PubMed] [Google Scholar]