

Abstract
Gastric cancer is the second most common cancer and a leading cause of cancer-related death worldwide. The KLF6 tumour suppressor gene has been previously shown to be inactivated in a number of human cancers through loss of heterozygosity (LOH), somatic mutation, decreased expression, and increased alternative splicing into a dominant negative oncogenic splice variant, KLF6-SV1. In this present study, thirty seven gastric cancer samples were analysed for the presence of loss of heterozygosity (LOH) of the KLF6 locus and somatic mutation. In total, 18 of 34 (53%) of the gastric cancer samples analysed demonstrated KLF6 locus specific loss. Four missense mutations, T179I, R198G, R71Q, and S180L were detected. Interestingly, two of these mutations R71Q and S180L have been identified independently by several groups in various malignancies including prostate, colorectal and gastric cancer. In addition, decreased wtKLF6 expression was associated with loss of the KLF6 locus and was present in 48% of primary gastric tumour samples analysed. Functional studies confirmed that wtKLF6 suppressed proliferation of gastric cancer cells via transcriptional regulation of the cyclin dependent kinase inhibitor p21 and the oncogene c-myc. Functional characterization of the common tumour-derived mutants demonstrated that the mutant proteins fail to suppress proliferation and function as dominant negative regulators of wtKLF6 function. Furthermore, stable overexpression of the R71Q and S180L tumour derived mutants in the gastric cancer cell line, Hs746T resulted in increased tumourigenicity in vivo. Combined, these findings suggest an important role for the KLF6 tumour suppressor gene in gastric cancer development and progression and identify several highly cancer-relevant signaling pathways regulated by the KLF6 tumour suppressor gene.
Keywords: Kruppel-like, tumour suppressor gene, gastric cancer, loss of heterozygosity, somatic mutation
Introduction
Gastric cancer is the second most common cancer in the world and one of the leading causes of cancer death worldwide (1). Many well characterized tumour suppressor and oncogenes have been analysed to better define their respective roles in gastric cancer development and progression but only a few consistent and frequent genetic alterations have been identified (2). Germline mutations in the E-cadherin tumour suppressor gene, loss and somatic mutation of the p53 tumour suppressor gene, oncogenic activation of B-catenin, and K-ras mutations have been found in a subset of gastric cancers (2).
Kruppel-like factor 6 (KLF6) belongs to the Kruppel-like family of transcription factors which have been shown to play roles in the regulation of diverse cellular processes including development, differentiation, proliferation, and apoptosis (3). Functional inactivation of the KLF6 gene has been implicated in a number of human cancers including prostate (4,5), colorectal (6,7), non-small cell lung (8,9), gastric (10), glioma (11,12), nasopharyngeal (13), hepatocellular (14-17), pancreatic (18) and ovarian carcinomas (19). In contrast to these studies, several groups have failed to identify somatic mutations in the KLF6 gene (20-22). These reported differences in mutational frequency highlight important differences in sample selection, numbers of samples, tissue isolation and the analytic techniques used. Further evidence supporting a role for KLF6 in tumour development is the reported associations between reduced KLF6 expression and decreased patient survival in prostate (23-25) and lung cancers (26). Depending on cell type and context, KLF6's growth suppressive properties have been associated with a number of highly relevant cancer pathways, including p53-independent upregulation of p21 (4), E-cadherin (27), disruption of cyclin D1 and CDK4 interaction (28), induction of apoptosis (8), and c-jun inhibition (29). Most recently, we have shown that a KLF6 single nucleotide polymorphism (SNP) is associated with increased prostate cancer risk (30). Previous studies in gastric cancer (10) have described LOH and mutation in the KLF6 gene in sporadic gastric cancer, mainly in the intestinal type. Interestingly, the KLF6 mutations were prevalent in advanced patient samples with evidence of lymph node metastases (10). In this present study, we sought to determine the KLF6 LOH, mutation status, and expression levels in a cohort of gastric cancer patient samples, and determine the biological role of the KLF6 tumour suppressor gene in gastric cancer cells.
Material and Methods
Tumour samples, preparation and DNA isolation
Tumour specimens were collected and analysed under IRB approval. Human gastric cancer and adjacent non-cancer tissues were obtained from gastric cancer patients in Prince of Wales Hospital of Hong Kong during endoscopy or surgery. Matched non-cancer gastric samples were obtained at least 2 cm distant from the tumour in which tumour cell infiltration was ruled out by histologic assessment. All specimens were snap frozen and stored at -80C. All patients gave informed written consent for obtaining the specimens and the study was approved by the Clinical Research Ethics Committee of the Chinese University of Hong Kong. Genomic DNA from frozen gastric tissues was extracted by using the High Pure PCR Template Preparation kit (Roche, Germany). Microdissection was performed on both tumour and surrounding normal tissue. The diagnosis was validated by pathology review. Additional clinical characteristics for the cohort analysed include average age 64 +/- 13 years of age; 70% of patients analyzed were male; the ethnicity of all of the patient analyzed was Chinese, and metastasis was present in 30% of patient samples analysed.
LOH and DNA mutation analysis
Fluorescent LOH analysis using genomic DNA from matched normal / tumour gastric tissue and markers has been previously described (4). Briefly, fluorescent LOH analysis using genomic DNA microdissected from matched normal / tumour gastric tissue was performed as previously described (see above references). Fluorescently labeled microsatellite markers flanking KLF6 and ordered according to the Marshfield map (http://research.marshfieldclinic.org/genetics/) were generated. PCR was performed according to manufacturer's suggestions (Perkin Elmer). The exponential range of the PCR was determined for each marker and each sample, and was between 30-38 cycles. The data were analysed by the ABI Genescan and Genotyper software packages (Perkin Elmer) and allelic loss was scored and by two independent observers. In our system, a relative allele ratio of less than 0.7, which correlates with an allele loss of approximately 40%, was defined as loss of heterozygosity. The XLOH was confirmed at least twice for each marker.
In order to finely map the region of chromosome 10p loss in gastric cancer to the KLF6 gene locus we used three microsatellite markers, KLF6M1, KLF6M2, and KLF6M4 that were designed specifically to tightly flank the KLF6 gene by 40 kb to the D10S591 and D10S594 markers that flank the gene by apprxoximately 1 Mb. The heterozygosity scores for these markers in the normal population were calculated by amplifying genomic DNA isolated from over 100 healthy Caucasian individuals. Samples that had either loss of at least one of the KLF6 specific markers flanking the KLF6 gene, M1, M2 & M4, or a flanking marker when the contiguous KLF6 specific marker was non-informative, were regarded as having LOH. All sample marker combinations were analysed at least twice.
PCR products were directly sequenced, following purification (Qiagen, QIAquick PCR purification kit), on an ABI Prism 3700 automated DNA Analyser and data analysed using the program Sequencher (Gene Codes Corporation). The following sets of intronic primers (sense and antisense, respectively) were used to amplify the coding region and intron/exon boundaries of KLF6 exon 2: Exon 2 Fwd: 5′- CGG GCA GCA ATG TTA TCT GTC CTT C - 3′ and Exon 2 Rev: 5′- CCC TCC AGG GCT GGT GCA - 3′. PCR cycling conditions were: 94°C (10 min) for 1 cycle, 94°C (30 sec), 55°C (30 sec), 72°C (1 min) for 45 cycles and a final extension of 72°C (5 min).
Real-time PCR analysis
For quantitating target gene expression, RNA isolation from cultured cells and tumour xenografts was performed using RNeasy Mini kits (Qiagen). All RNA was treated with DNAse (Qiagen). One ug of RNA was reverse transcribed for each reaction using first strand cDNA synthesis with random primers (Biorad). mRNA levels were quantified by qRT-PCR using the following PCR primers on an ABI PRISM 7900HT (Applied Biosystems): KLF6 Forward: 5′-CGG ACG CAC ACA GGA GAA AA-3′ and Reverse: 5′- CGG TGT GCT TTC GGA AGT G-3′; GAPDH Forward: 5′- CAA TGA CCC CTT CAT TGA CC-3′ and Reverse: 5′- GAT CTC GCT CCT GGA AGA TG-3′; c-myc Forward: 5′- CAG CTG CTT AGA CGC TGG ATT T and Reverse: 5′-ACC GAG TCG TAG TCG AGG TCA T. Quantitative real time PCR primers to p21 were described previously (23). All values were calculated by normalizing the levels of each target for each cDNA to GAPDH and b-actin and then using this normalized value to calculate fold change compared to control. All experiments were done in triplicate and repeated three independent times. Statistical significance was determined by one-way ANOVA using a Bonferroni correction.
Cell culture and transfection
AGS and HS746T gastric cancer cell lines were purchased from the American Tissue Culture Collection (ATCC). Stable cell lines were generated by retroviral infection of pBABE, pBABE-R71Q, or pBABE-S180L and selected with 2 μg/ml of puromycin, as previously described (23). Polyclonal pools of each infected cell line were collected and KLF6 expression was determined by qRT-PCR and western blotting. Genomic DNA was isolated from each cell line as previously described (4) and sequence analysis to confirm each point mutation was performed using the following sets of primers and PCR conditions Exon 2 Fwd: 5′- CGG GCA GCA ATG TTA TCT GTC CTT C - 3′ and Exon 2 Rev: 5′- CCC TCC AGG GCT GGT GCA - 3′. PCR cycling conditions were: 94°C (10 min) for 1 cycle, 94°C (30 sec), 55°C (30 sec), 72°C (1 min) for 45 cycles and a final extension of 72°C (5 min). Transient transfection and co-transfection of the pciNEO, pciNEO-KLF6, R71Q, and S180L and site directed mutagenesis was performed as previously described (4).
Western blot analysis and densitometric analysis
Cell extracts were harvested in RIPA buffer (Santa Cruz Biotechnology, standard protocol). Tumour tissue extracts were harvested and prepared in the T-PER reagent (Pierce). Equal amounts of protein (50 μg; determined by the BioRad DC Protein quantification assay (BioRad) were loaded and separated by polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Actin (SC-1616), KLF6 (SC-7158); p21 (H164) antibodies were obtained from Santa Cruz Biotechnology, C-myc (Calbiochem anti-cmyc (ab-1) mouse mab (9e10). Enhanced chemiluminescent immunoblot images were analysed by scanning densitometry and quantified with a BIOQUANT NOVA imaging system. Values were expressed as fold change relative to control and normalized to actin as a loading control.
Analysis of proliferation
Proliferation was determined by performing a tritiated thymidine incorporation assays as previously described (23). The stable cell lines containing pBABE, R71Q, and S180L were plated at a density of 100,000 cells / well in 12 well dishes.
Tumourigenicity assays
Stable HS746T cells (1 × 107) were injected into the left flank of female 6-8 week old BALB/c nu/nu mice as previously described (23). Tumour volume was assessed weekly and determined using the formula (length × width × width × 0.4). After 6 weeks, mice were sacrificed and tumours removed for RNA and protein analysis. All animal studies were approved by the MSSM IACUC.
Results
Functional Inactivation and decreased expression of KLF6 tumour suppressor gene in gastric cancer
A total of 37 patient samples with detailed clinical information were collected under Institution Review Board approval. Briefly, 65% were of the diffuse type, and 16% of the intestinal type of gastric cancer with a complete range of stages represented. The clinical-pathologic profile of these samples is representative of the varied clinical spectrum of gastric cancer. We determined the frequency of KLF6 loss using five microsatellite markers: KLF6M1, KLF6M2, and KLF6M4 which tightly flank the KLF6 gene locus (4) and D10S594 and D10S591 which are more distal to the gene. This marker set rendered 34 of the 37 samples analysed informative for at least one marker at the KLF6 locus (Fig. 1A). Fluorescent LOH analysis using genomic DNA from matched normal / tumour tissue and these specific microsatellite markers has been previously described (4). The exponential range of the PCR was determined for each microsatellite markers and a relative allele ratio of less than 0.7, correlating with allelic loss of approximately 40%, was defined as LOH (4). Samples that had either loss of at least one of the KLF6 specific markers flanking the KLF6 gene, M1, M2 & M4, or a flanking marker when the contiguous KLF6 specific marker was non-informative, were regarded as having LOH. All sample marker combinations were analysed at least twice. In addition, patients (18 and 22) with failed PCR reactions for both the M2 and M4 microsatellite markers that represent the smallest region of overlap for KLF6 loss in gastric cancer and patients with microsatellite instability (MSI) (patient 37) were removed from the analysis. Clinical information on the cohort of samples analysed is provided in Figure 1B. Overall, 53% (18/34) of the gastric cancer samples demonstrated LOH of the KLF6 locus. LOH was present in both the diffuse and intestinal types of gastric cancer at similar frequencies (data not shown). Interestingly, allelic loss of the KLF6 locus was significantly more frequent (p < 0.001) in advanced stage disease (III and IV) than when compared to stage I and II cancers (Fig. 1C). The frequency of KLF6 loss in this present study is consistent with the previous report by Cho et al. who reported allelic loss of the KLF6 gene in 43.2% of informative cases.
Figure 1.

A. LOH status of KLF6. LOH of the KLF6 locus was analysed using microsatellite markers (vertical axis) from the 10p15 region and KLF6-specific markers KLF6M1, M2 and M4, which have been previously described (4). Cases are arranged on the horizontal axis. Black filled square - LOH; gray - non-informative (NI); white square - no evidence of loss; “-“ - represents a PCR reaction that failed three or more times. Tumours were defined as having LOH if one or all of the markers flanking the KLF6 locus were lost. B. Clinical profile of gastric cancer patient samples. A summary of the relevant clinical information for the 37 gastric cancer patient samples analysed is presented. A complete range of stages and histological types are represented. C. Summary of KLF6 allelic loss by stage of disease. Statistical analysis was performed using a Student t-test with two-tailed distribution and two samples equal variance (p < 0.001). Allelic loss of the KLF6 locus was significantly more frequent in advanced disease (p < 0.001).
We next explored whether allelic loss was associated with decreased KLF6 mRNA expression. RNA was available from a total of 16 normal and 33 of the tumour samples from gastric tissue specimens. Quantitative real time PCR (qRT-PCR) using wtKLF6 specific primers (30) were used to determine KLF6 expression in all 33 RNA samples. One ug of RNA was reverse transcribed for each reaction using first strand cDNA synthesis with random primers and levels of wtKLF6 were determined as previously described (30). Both β-actin and GADPH were used as housekeeping genes for all analysis. On average, patients with KLF6 LOH had a 80% reduction in wtKLF6 expression when compared to tumours without LOH (p < 0.001) or when compared to normal gastric tissue (p < 0.001) (Fig. 2A). Interestingly, KLF6 expression levels in tumours without LOH was identical to normal gastric tissue (Figure 2A). There was a significant correlation (p < 0.01) between KLF6 loss and decreased KLF6 expression as demonstrated in Figure 2A (p < 0.01). KLF6 mutation status was examined by direct sequencing of exon 2 in all patient samples as previously described (4). This exon encodes three quarters of the wild-type protein and contains the majority of mutations previously identified in other human cancers (4-19). Consistent with its role as a tumour suppressor gene, mutations were identified in 4/37 cases (10%) (Figure 2b,c). All four of the identified mutations were somatic and resulted in non-conservative amino acid changes in the KLF6 protein (Fig 2c). Interestingly, three of the four mutations, S180L, R71Q, and T179I had been previously identified in prostate, colorectal, and hepatocellular carcinoma patient samples (4,6,7,14). Combined, these findings demonstrate that inactivation of the KLF6 tumour suppressor gene occurs through either LOH and/or somatic mutation with decreased expression in wtKLF6 expression occurring in a significant percentage of gastric cancer patients. Furthermore, loss of the KLF6 locus is associated with later stage disease suggesting a potential role for the KLF6 gene in gastric cancer progression.
Figure 2.
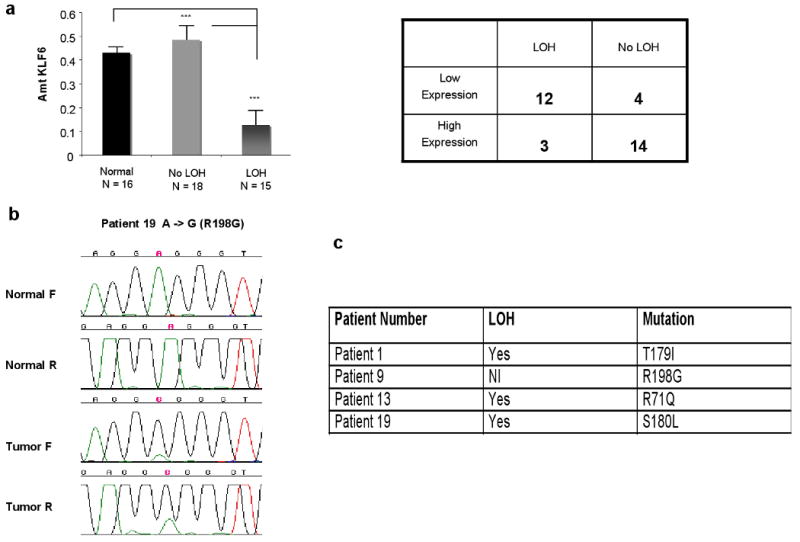
A. KLF6 expression in gastric cancer. Quantitative real time PCR using wtKLF6 specific primers as previously described (23) demonstrated a 80% reduction in wtKLF6 expression compared to both patient samples without LOH of the KLF6 locus and to normal gastric tissue (p < 0.0001). As the table demonstrates there was a significant correlation between KLF6 loss and decreased wtKLF6 expression. B. KLF6 mutations in gastric cancer. Microdissected tumour DNA was amplified using the following sets of intronic primers (sense and antisense, respectively) KLF6 exon 2: Exon 2 Fwd: 5′- CGG GCA GCA ATG TTA TCT GTC CTT C - 3′ and Exon 2 Rev: 5′- CCC TCC AGG GCT GGT GCA - 3′. PCR cycling conditions were: 94°C (10 min) for 1 cycle, 94°C (30 sec), 55°C (30 sec), 72°C (1 min) for 35 cycles and a final extension of 72°C (5 min). PCR products were directly sequenced, following purification (Qiagen, QIAquick PCR purification kit), on an ABI Prism 3700 automated DNA Analyser and data analysed using the program Sequencher (Gene Codes Corporation). All mutations were confirmed by an indendepent PCR reaction and all sequencing reactions were done in both the forward and reverse directions as previously described (4). C. Summary of KLF6 mutation and LOH status in gastric cancer patient samples.
Targeted reduction of KLF6 results in increased proliferation in gastric cancer cell lines
Having identified both genetic alterations in the KLF6 gene and correlations between KLF6 loss and clinical-pathological parameters in patient samples, we next directly explored the biological role of KLF6 in gastric cancer by using siRNA-mediated gene silencing to specifically downregulate its expression in gastric cancer cells. Both the AGS and Hs746T cell lines had no somatic mutations in the KLF6 gene and express approximately 50% less KLF6 than pooled normal gastric tissue (data not shown). Specific siRNA to wtKLF6 has been generated and validated for target specificity as previously described (19,30,31). Transient transfection of a siRNA specific to wtKLF6 in the AGS gastric cancer cell line resulted in 50% decrease in wtKLF6 expression at both the mRNA and protein level as measured by qRT-PCR analysis and western blotting when compared to a si-NTC non-targeting siRNA control (Fig 3A). No differences in KLF6 expression were seen at time point 0 and no effect on KLF6 expression was seen in control cells that had been not been transfected compare to cells that were transfected with either siRNAs (data not shown). Tritiated thymidine incorporation assays of transiently transfected cells demonstrated that targeted reduction of wtKLF6 resulted in a nearly two fold increase in cellular proliferation when compared to the si-NTC transfected control cells (Fig. 3B). Similar results were obtained with 2 additional siRNAs specifically targeting wtKLF6 (data not shown). This increase in cellular proliferation was associated with changes in the expression at of two well-characterized transcriptional targets of KLF6, the proto-onocgene c-myc and the cyclin dependent kinase inhibitor p21 (4). Targeted reduction of KLF6 resulted in increased expression of c-myc with concomitant decrease in p21 expression at both the mRNA and protein level (Fig 3c). Similar results were obtained in the HS746T gastric cancer cell line (data not shown). In addition, expression of KLF6 at the mRNA and protein level was not significantly different between the scrambled oligo control and si-KLF6 transfected cells at time zero and furthermore there were no major differences in KLF6 expression between transfected and untransfected cells. The incorporation of triatiated thymidine at time 0 was equal between the control and si-KLF6 transfected cells which is expected given that there was no difference in KLF6 expression at this time point (data not shown)
Figure 3.
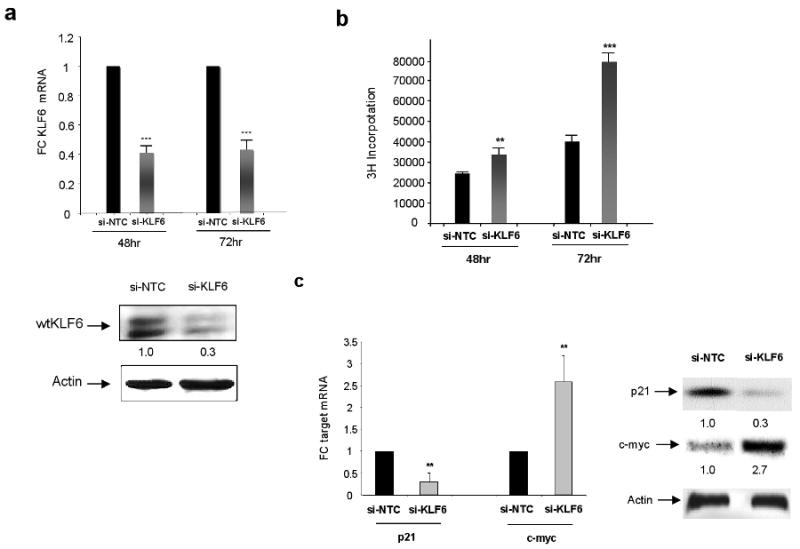
Targeted reduction of wtKLF6 in gastric cancer cell lines. A. Targeted reduction of wtKLF6 in gastric cancer cell lines results in decreased KLF6 expression at both the mRNA and protein level as measured by wtKLF6 specific quantitative real time PCR and western blotting respectively. AGS cells transfected with siRNA specific to wtKLF6 and a non targeting scrambled siRNA control were harvested 72 hrs after transfection. The experiment was repeated three independent times (*** p < 0.0001). B. Targeted reduction of wtKLF6 results in increased cellular proliferation. Triatiated thymydine incorporation was measured at 48 and 72hrs after siRNA transfection of AGS cells. At 72hrs there is a nearly 2 fold increase in cellular proliferation (*** p < 0.0001). The experiment was repeat three independent times. C. Targeted reduction of wtKLF6 results in dysregulated c-myc and p21 expression. Quantitative real time PCR and western blotting using c-myc and p21 specific primers and antibodies demonstrated a 70% reduction in p21 expression with a concomitant 2 fold increase in c-myc expression in gastric cancer cells transfected with KLF6 siRNAs (** p < 0.001).
Overexpression of KLF6 results in cell cycle arrest in gastric cancer cell lines
To further define the role of the wild type KLF6 tumour suppressor gene in gastric cancer cell lines, we overexpressed wtKLF6 in the AGS gastric cancer cell line. Transient transfection of wtKLF6 resulted in a greater than 40-fold increase in KLF6 expression at both the mRNA and protein level (Figure 4a). This was associated with a significant decrease in c-myc expression with a concomitant increase in p21 expression at both the mRNA and protein level (Figure 4a). Furthermore, overexpression of wtKLF6 resulted in a 80% decrease in cellular proliferation as measured by tritiated thymidine incorporation (Figure 4b). This decrease in cellular proliferation was associated with a G1/S arrest as demonstrated by FACS analysis (Fig 4c). Combined, this data demonstrates that wild type KLF6 is growth suppressive in gastric cancer cell lines through transcriptional regulation of p21 and c-myc.
Figure 4.
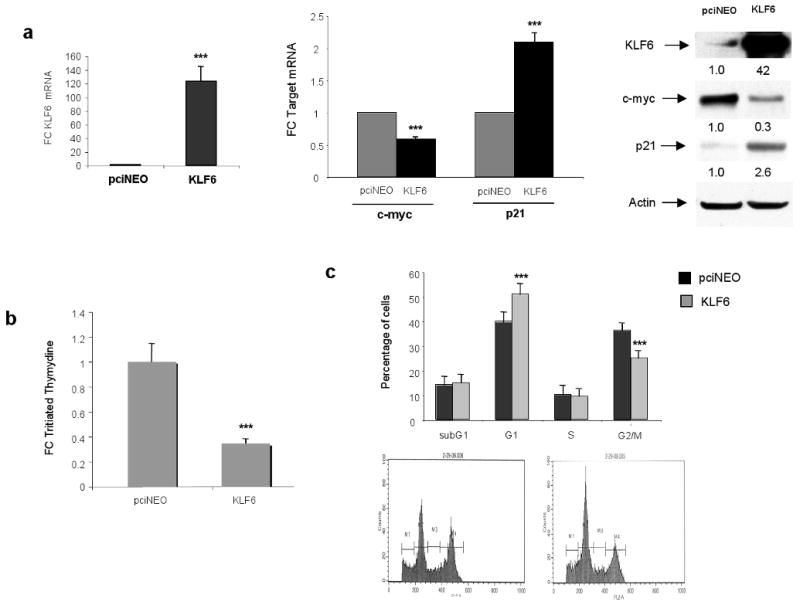
Functional role of wtKLF6 in gastric cancer cell lines. A. Overexpression of wtKLF6 Targeted reduction of wtKLF6 in gastric cancer cell lines results in decreased KLF6 expression at both the mRNA and protein level as measured by wtKLF6 specific quantitative real time PCR and western blotting respectively. AGS cells transfected with siRNA specific to wtKLF6 and a non targeting scrambled siRNA control were harvested 72 hrs after transfection. The experiment was repeated three independent times (*** p < 0.0001). B. Targeted reduction of wtKLF6 results in increased cellular proliferation. Triatiated thymydine incorporation was measured at 48 and 72hrs after siRNA transfection of AGS cells. At 72hrs there is a nearly 2 fold increase in cellular proliferation (*** p < 0.0001). The experiment was repeat three independent times. C. Targeted reduction of wtKLF6 results in dysregulated c-myc and p21 expression. Quantitative real time PCR and western blotting using c-myc and p21 specific primers and antibodies demonstrated a 70% reduction in p21 expression with a concomitant 2 fold increase in c-myc expression in gastric cancer cells transfected with KLF6 siRNAs (** p < 0.001).
Tumour derived KLF6 mutations fail to suppress cell growth
Having identified both genetic alterations in the KLF6 gene, correlations between KLF6 loss and clinical-pathological parameters in patient samples, and functionally characterized the biological function of wtKLF6 in gastric cancer cell lines, we next directly explored the biological role of KLF6 tumour-derived mutants in the gastric cancer cell lines, AGS and HS746T. Stable cell lines for each of these KLF6 mutants were generated as previously described (14,19) in each of the gastric cancer cell lines. We were unable to generate stable cell lines overexpressing the R198G and T179I tumour derived mutants for technical reasons. Previous reports, however have demonstrated that the T179I mutant fails to transactivate p21 and suppress cellular proliferation in colorectal cancer cell lines (6). In addition, the R198G mutation disrupts the predicted nuclear localization signal of the KLF6 tumour suppressor gene and would therefore most likely result in cytoplasmic accumulation of the mutant protein. Because of its growth suppressive effects on gastric cancer cell lines we were unable to successfully generate stable cell lines overexpressing wtKLF6 (data not shown). Sequence and quantitative real time PCR analysis of the R71Q and S180L retrovirally derived cell lines demonstrated overexpression of each KLF6 mutant in their respective cell lines compared to the control pBABE cell line. Overexpression of both the R71Q and S180L mutants resulted in a marked changes in the expression of both c-myc and p21 at both the mRNA and protein level (Fig 5a,b). These changes in c-myc and p21 expression were associated with changes in cellular morphology in the HS746T cell line and increased cellular proliferation when compared to control as shown in Figure 5c. Analysis of cellular proliferation using a assay of the R71Q and S180L cell lines demonstrated a greater than 3 fold increase in growth when compared to control cells (Fig 5c). These results were confirmed in the AGS cell line (data not shown).
Figure 5.
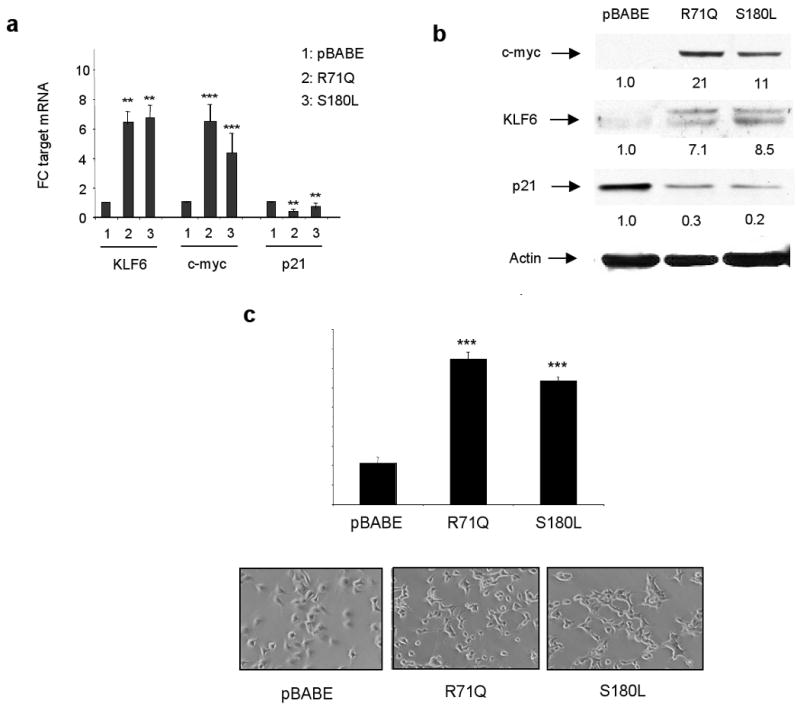
Functional characterization of the R71Q and S180L patient-derived mutations. A. Target gene expression was analysed by qRT-PCR and correlated to the control pBABE cell line. For quantitating target gene expression, one ug of RNA was reverse transcribed for each reaction using first strand cDNA synthesis with random primers. mRNA levels for each target was quantified by qRT-PCR and all values were calculated by normalizing the levels of each target for each cDNA to both GAPDH and B-actin to determine the relative amount of expression compared to control. All experiments were done in triplicate and repeated three independent times. KLF6 was overexpressed in the R71Q and S180L cell lines approximately eight fold (** p < 0.001). This was associated with a 7 fold increase in c-myc expression respectively with a concomitant reduction in expression of p21 in these cell lines compared to control. C. Western blot of the pBABE, R71Q, and S180L cells was performed using the following antibodies, c-myc, KLF6, p21, and actin as a control for protein loading. Consistent with qRT-PCR data, the R71Q and S180L mutants expressed increased levels of c-myc and decreased levels of p21 compared to control. D. The R71Q and S180L mutants resulted in increased cellular proliferation (*** p < 0.001). Photographs of retrovirally generated stable cell lines in HS746T gastric cancer cell line. The R71Q and S180L derived cells exhibit markedly different cellular morphology than the control pBABE cell line.
Dominant negative function of KLF6 tumour derived mutants
We next sought to determine if the R71Q and S180L had a dominant negative effect on wtKLF6 function. Transient cotransfection of either the R71Q or S180L mutant expression vectors with wtKLF6 resulted in equal levels of KLF6 ovexpression at both the mRNA and protein level (Fig 6a and data not shown). Consistent with previous results wtKLF6 overexpression resulted in 40% reduction in cellular proliferation in the HS746T cell line which was completely abrograted by cotransfection with either the R71Q or S180L KLF6 expression vectors. This dominant negative effect of the R71Q and S180L proteins was associated with suppression of p21 induction and failure to repress c-myc expression at both the mRNA and protein level (Fig 6c and data not shown). Combined, this data suggests that the R71Q and S180L KLF6 mutants function as dominant negative regulators of wild type KLF6 function through their ability to effect p21 and c-myc transcription.
Figure 6.

Dominant negative function of the KLF6 tumour derived mutants. A. KLF6 gene expression was analysed by qRT-PCR and compared to the control pBABE cell line. mRNA levels for wtKLF6 was quantified by qRT-PCR and all values were calculated by normalizing the levels of each target for each cDNA to both GAPDH and β-actin to determine the relative amount of expression compared to control. All experiments were done in triplicate and repeated three independent times. KLF6 was overexpressed in all three contransfected AGS cell lines to equal amounts. B. Triatiated thymydine incorporation was measured at 72hrs after transfection of AGS cells. WTKLF6 overexpression resulted in 40% reduction in cellular proliferation in the AGS cell line, which was completely abrograted by cotransfection with either the R71Q or S180L KLF6 expression vectors. C. KLF6 overexpression resulted in a 2 fold increase in p21 expression and a 50% reduction in C-myc expression as measured by qRT-PCR (*** p < 0.0001). These changes in target gene expression were completely abrogated by cotransfection with either the R71Q or S180L KLF6 tumour derived mutant constructs.
KLF6 tumour derived mutants increase in vivo tumourigenicity
To extend these findings to an in vivo tumourigenicity model, we injected the pBABE, R71Q, and S180L HS746T subcutaneously in nude mice. Consistent with our cell culture findings, tumours derived from the R71Q and S180L cell lines were significantly larger than the pBABE control derived tumours (Fig 7a). H&E staining of tumours revealed a marked difference in cellular differentiation between the R71Q, S180L and control tumours (Fig 7b). Specifically, tumours derived from the R71Q and S180L appeared less differentiated. Quantitative real time PCR (qRT-PCR) analysis of these tumours demonstrated a marked upregulation in c-myc expression and decreased p21 expression at both the mRNA and protein levels (Fig 7c) consistent with our previous findings in cell culture. Combined, these studies suggest that tumour-derived somatic mutations in the KLF6 gene result in increased in vivo tumourigenicity through regulation of c-myc and p21 expression.
Figure 7.
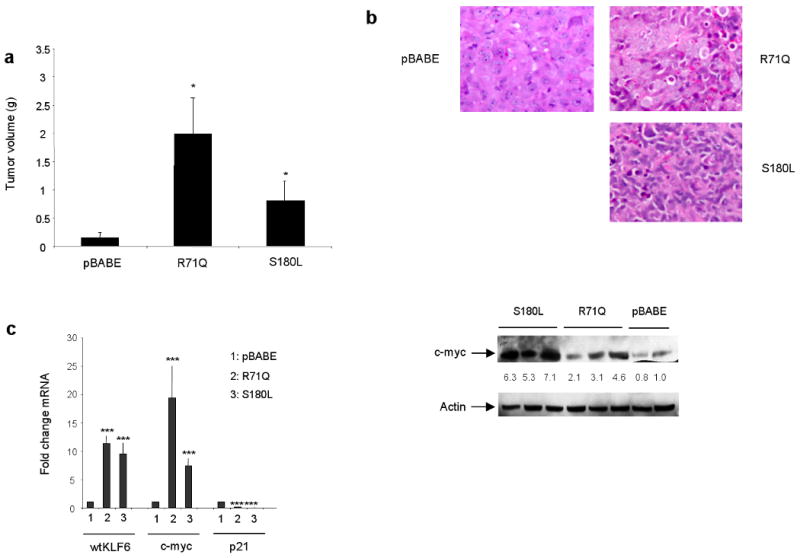
Tumour derived KLF6 mutations significantly increased in vivo tumourigenicity. Stable cell lines expressing the R71Q, S180L, or the control pBABE HS746T cell line were injected into nude mice and their volume assessed as described in Materials and Methods. A, Tumours derived from the R71Q and S180L cells were significantly larger (* p < 0.01) than control cell line derived tumours. For each group. five tumours were analysed. B, The R71Q and S180L derived tumours were markedly less differentiated as shown by H&E staining. C, mRNA levels for each target was quantified by qRT-PCR and all values were calculated by normalizing the levels of each target for each cDNA to both GAPDH and β-actin to determine the relative amount of expression compared to control. qRT-PCR of cDNA derived from each tumour revealed a marked increase in cmyc and decrease in p21 expression (*** p < 0.001). Western blotting confirmed increased c-myc expression in tumours expressing the either the R71Q or S180L patient-derived mutations. All experiments were repeated in triplicate and 5 tumours were analysed from each experimental group.
Discussion
Our findings in patient samples and biological model systems define a high frequency of KLF6 allelic loss and/ or somatic mutation suggesting that dysregulation of KLF6 may be an important event in the development and progression of gastric cancer. Mounting evidence in other tumours has highlighted a variety of KLF6 inactivating mechanisms relevant to tumour growth and spread. These mechanisms include: 1) LOH and somatic mutation in prostate (4,5), colorectal (6,7), malignant glioma (11,12), nasopharyngeal carcinomas (13), HCC (14-17) and gastric cancer (10), 2) transcriptional silencing through promoter hypermethylation in esophageal cancer cell lines (32) and; 3) dysregulated alternative splicing in prostate cancer and ovarian cancer (19,30,31). The current findings are consistent with a previous report in gastric cancer reporting a LOH frequency of 43.2% with somatic mutations occurring in approximately 10% of the 80 gastric cancer patient samples analysed. The results between these two studies are remarkably consistent and demonstrate that KLF6 loss is a frequent event in gastric cancer and that functional inactivation of the gene through somatic mutations occurs in a minority of cases.
Our findings demonstrate the importance and prevalence of KLF6 inactivation in gastric cancer. Consistent with previous reports, KLF6 LOH and/or somatic mutation occurs in a significant number of gastric cancer patient samples. In addition, we define the functional significance of commonly identified KLF6 mutations in multiple gastric cancer cell lines both in culture and in vivo. Specifically, we identify regulation of the the cyclin dependent kinase inhibitor p21 and the proto-oncogene c-myc by KLF6 tumour derived mutants resulting in increased tumourigenicity and decreased cellular differentiation. Combined, these studies highlight an important role for the KLF6 tumour suppressor gene in gastric cancer pathogenesis and demonstrate the diversity of mechanisms of functional inactivation and cellular pathways regulated by this gene in human cancer. Future studies of larger cohorts of patient samples will better define the exact prevalence and clinico-pathological associations with KLF6 loss and mutation. In addition, further studies of the KLF6 heterozygous mice which display significantly enlarged livers and reduced p21 expression (31), are warranted to identify if happloinsufficiency of the KLF6 gene plays a role in gastric cancer development and progression in vivo.
The association between loss of the KLF6 locus, decreased KLF6 expression and advanced stage disease is consistent with other previous reports of decreased KLF6 expression in advanced stages of disease in hepatocellular carcinoma and prostate cancer (31,33). These findings suggest not only the potential use of KLF6 as a prognostic marker in cancer, but also that additional epigenetic mechanisms of gene silencing such as promoter methylation may be responsible for this further decrease in KLF6 expression in advanced stage cancers.
In summary, these findings demonstrate that the tumour suppressor gene KLF6 plays an important role in gastric cancer development and progression. Ultimately, we believe these findings have relevance to both the identification of pathogenic associations between KLF6 loss, expression and mutation and clinically relevant parameters such as tumour stage, grade, and histological type.
Acknowledgments
Financial support: SLF: NIH Grants DK37340 and DK56621; Dept of Defense DAMD17-03-1-0100; The Bendheim Foundation, Samuel Waxman Foundation; JAM: DAMD17-02-1-0720 and DAMD17-03-10129; STK: NIH Training Grant T32 DK 07792-01; GN: Goutham Narla is a Howard Hughes Medical Institute Physician-Scientist Early Career Awardee.
Abbreviations
- KLF6
Kruppel like factor 6
- LOH
loss of heterozygosity
- wtKLF6
wild type KLF6
- qRT-PCR
quantitative real time polymerase chain reaction
Footnotes
Conflict of interest: There are no conflict of interests for any of the authors of this manuscript.
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Ushijima T, Sasako M. Focus on Gastric Cancer. Cancer Cell. 2004;2:121–25. doi: 10.1016/s1535-6108(04)00033-9. [DOI] [PubMed] [Google Scholar]
- 3.Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–60. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 4.Narla G, Heath KE, Reeves HL, et al. KLF6, a candidate tumour suppressor gene mutated in prostate cancer. Science. 2001;294:2563–6. doi: 10.1126/science.1066326. [DOI] [PubMed] [Google Scholar]
- 5.Chen C, Hyytinen ER, Sun X, et al. Deletion, mutation, and loss of expression of KLF6 in human prostate cancer. Am J Pathol. 2003;162:1349–54. doi: 10.1016/S0002-9440(10)63930-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reeves HL, Narla G, Ogunbiyi O, et al. Kruppel-like factor 6 (KLF6) is a tumour suppressor gene frequently inactivated in colorectal cancer. Gastroenterology. 2004;126:1090–103. doi: 10.1053/j.gastro.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Cho YG, Choi BJ, Kim CJ, et al. Genetic alterations of the KLF6 gene in colorectal cancers. APMIS. 2006;114:458–64. doi: 10.1111/j.1600-0463.2006.apm_431.x. [DOI] [PubMed] [Google Scholar]
- 8.Ito G, Uchiyama M, Kondo M, et al. Kruppel-like factor 6 is frequently down-regulated and induces apoptosis in non-small cell lung cancer cells. Cancer Res. 2004;64:3838–43. doi: 10.1158/0008-5472.CAN-04-0185. [DOI] [PubMed] [Google Scholar]
- 9.DiFeo A, Feld L, Rodriguez E, et al. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008;68:965–70. doi: 10.1158/0008-5472.CAN-07-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho YG, Kim CJ, Park CH, et al. Genetic alterations of the KLF6 gene in gastric cancer. Oncogene. 2005;24:4588–90. doi: 10.1038/sj.onc.1208670. [DOI] [PubMed] [Google Scholar]
- 11.Jeng YM, Hsu HC. KLF6, a putative tumour suppressor gene, is mutated in astrocytic gliomas. Int J Cancer. 2003;105:625–9. doi: 10.1002/ijc.11123. [DOI] [PubMed] [Google Scholar]
- 12.Camacho-Vanegas Olga, Narla G, Teixeira MS, et al. Functional inactivation of the KLF6 tumour suppressor gene by loss of heterozygosity and increased alternative splicing in glioblastoma. Int J Cancer. 2007;121:1390–5. doi: 10.1002/ijc.22809. [DOI] [PubMed] [Google Scholar]
- 13.Chen HK, Liu XQ, Lin J, Chen TY, Feng QS, Zeng YX. Mutation analysis of KLF6 gene in human nasopharyngeal carcinomas. Ai Zheng. 2002;21:1047–50. [PubMed] [Google Scholar]
- 14.Kremer-Tal S, Reeves HL, Narla G, et al. Frequent inactivation of the tumour suppressor Kruppel-like Factor 6 (KLF6) in hepatocellular carcinoma. Hepatology. 2004;40:1047–52. doi: 10.1002/hep.20460. [DOI] [PubMed] [Google Scholar]
- 15.Pan XC, Chen Z, Chen F, Chen XH, Jin HY, Xu XY. Inactivation of the tumour suppressor Krüppel-like factor 6 (KLF6) by mutation or decreased expression in hepatocellular carcinomas. J Zhejiang Univ Sci B. 2006;7:830–6. doi: 10.1631/jzus.2006.B0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song J, Kim CJ, Cho YG, et al. Genetic and epigenetic alterations of the KLF6 gene in hepatocellular carcinoma. J Gastroenterol Hepatol. 2006;21:1286–9. doi: 10.1111/j.1440-1746.2006.04445.x. [DOI] [PubMed] [Google Scholar]
- 17.Kremer-Tal S, Narla G, Chen Y, et al. Downregulation of KLF6 is an early event in hepatocarcinogenesis, and stimulates proliferation while reducing differentiation. J Hepatol. 2007 Apr;46:645–54. doi: 10.1016/j.jhep.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartel M, Narla G, Wente MN, et al. Increased alternative splicing of the KLF6 tumour suppressor gene correlates with prognosis and tumour grade in patients with pancreatic cancer. European Journal of Cancer. 2008 doi: 10.1016/j.ejca.2008.06.030. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiFeo A, Narla G, Hirshfeld J, et al. Role of KLF6 and KLF6-SV1 in ovarian cancer progression and intraperitoneal dissemination. Clin Cancer Res. 2006;12:3730–9. doi: 10.1158/1078-0432.CCR-06-0054. [DOI] [PubMed] [Google Scholar]
- 20.Muhlbauer KR, Grone HJ, Ernst T, et al. Analysis of human prostate cancers and cell lines for mutations in the TP53 and KLF6 tumour suppressor genes. Br J Cancer. 2003;89:687–90. doi: 10.1038/sj.bjc.6601164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lièvre A, Landi B, Côté JF, et al. Absence of mutation in the putative tumour-suppressor gene KLF6 in colorectal cancers. Oncogene. 2005;48:7253–6. doi: 10.1038/sj.onc.1208867. [DOI] [PubMed] [Google Scholar]
- 22.Boyault S, Herault A, Balabaud C, Zucman-Rossi J. Absence of KLF6 gene mutation in 71 hepatocellular carcinomas. Hepatology. 2005;41:681–2. doi: 10.1002/hep.20588. [DOI] [PubMed] [Google Scholar]
- 23.Glinsky GV, Glinskii AB, Stephenson AJ, Hoffman RM, Gerald WI. Gene expression profiling predicts clinical outcome of prostate cancer. J Clin Invest. 2004;113:913–23. doi: 10.1172/JCI20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh D, Febbo PG, Ross K, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;2:203–9. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 25.Narla G, Difeo A, Fernandez Y, et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J Clin Invest. 2008;118:2711–21. doi: 10.1172/JCI34780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kettunen E, Anttila S, Seppänen JK, et al. Differentially expressed genes in nonsmall cell lung cancer: expression profiling of cancer-related genes in squamous cell lung cancer. Cancer Genet Cytogenet. 2004;149:98–106. doi: 10.1016/S0165-4608(03)00300-5. [DOI] [PubMed] [Google Scholar]
- 27.DiFeo A, Narla G, Camacho-Vanegas O, et al. E-cadherin is a novel transcriptional target of the KLF6 tumour suppressor. Oncogene. 2006;44:6026–31. doi: 10.1038/sj.onc.1209611. [DOI] [PubMed] [Google Scholar]
- 28.Benzeno S, Narla G, Allina J, et al. Cyclin-dependent kinase inhibition by the KLF6 tumour suppressor protein through interaction with cyclin D1. Cancer Res. 2004;64:3885–91. doi: 10.1158/0008-5472.CAN-03-2818. [DOI] [PubMed] [Google Scholar]
- 29.Slavin DA, Koritschoner NP, Prieto CC, López-Díaz FJ, Chatton B, Bocco JL. A new role for the Kruppel-like transcription factor KLF6 as an inhibitor of c-jun proto-oncoprotein function. Oncogene. 2004;23:8196–205. doi: 10.1038/sj.onc.1208020. [DOI] [PubMed] [Google Scholar]
- 30.Narla G, DiFeo A, Reeves HL, et al. A germline DNA polymorphism associated with increased prostate cancer risk enhances alternative splicing of the KLF6 tumour suppressor gene. Cancer Res. 2005;65:1213–22. doi: 10.1158/0008-5472.CAN-04-4249. [DOI] [PubMed] [Google Scholar]
- 31.Narla G, Difeo A, Reeves HL, et al. Targeted Inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005;65:5761–8. doi: 10.1158/0008-5472.CAN-05-0217. [DOI] [PubMed] [Google Scholar]
- 32.Yamashita K, Upadhyay S, Osada M. Pharmacologic unmasking of epigenetically silenced tumour suppressor genes in esophageal squamous cell carcinoma. Cancer Cell. 2002;2:485–95. doi: 10.1016/s1535-6108(02)00215-5. [DOI] [PubMed] [Google Scholar]
- 33.Narla G, Kremer-Tal S, Matsumoto Nobuyuki, et al. In vivo Regulation of p21 by the KLF6 Tumour Suppressor Gene in Mouse Liver and Human Hepatocellular Carcinoma. Oncogene. 2007;26:4428–34. doi: 10.1038/sj.onc.1210223. [DOI] [PubMed] [Google Scholar]