

Abstract
Vascular Ehlers-Danlos syndrome is a rare genetic disorder resulting from mutations in the α-1 chain of type III collagen (COL3A1) and manifesting as tissue fragility with spontaneous rupture of the bowel, gravid uterus, or large or medium arteries. The heterozygous Col3a1 knockout mouse was investigated as a model for this disease. The collagen content in the abdominal aorta of heterozygotes was reduced, and functional testing revealed diminishing wall strength of the aorta in these mice. Colons were grossly and histologically normal, but reduced strength and increased compliance of the wall were found in heterozygotes via pressure testing. Although mice demonstrated no life-threatening clinical signs or gross lesions of vascular subtype Ehlers-Danlos syndrome type IV, thorough histological examination of the aorta of heterozygous mice revealed the presence of a spectrum of lesions similar to those observed in human patients. Lesions increased in number and severity with age (0/5 [0%] in 2-month-old males vs 9/9 [100%] in 14-month-old males, P < .05) and were more common in male than female mice (23/26 [88.5%] vs 14/30 [46.7%] in 9- to 21-month-old animals, P < .05). Haploinsufficiency for Col3a1 in mice recapitulates features of vascular Ehlers-Danlos syndrome in humans and can be used as an experimental model.
Keywords: Col3a1, mouse model, vascular Ehlers-Danlos, haploinsufficient
The Ehlers-Danlos syndromes are a group of related genetic connective tissue disorders, named for the dermatologists Edvard Lauritz Ehlers and Henri-Alexander Danlos, who independently described the features of hyperextensibility of the skin, hyper-mobility of the joints, and tissue fragility with easy bruising, delayed wound healing, and atrophic scars in the early 20th century.23 The disease is subdivided into 6 clinical types according to the Villefranche nosology.4 Ehlers-Danlos syndrome type IV, vascular subtype (vEDS, aka Sack-Barabas syndrome, OMIM 130050), is a rare autosomal dominant disorder resulting from mutations in the α-1 chain of type III collagen (COL3A1).10,11,24 Type III collagen is a homotrimeric fibrillar collagen found abundantly in the wall of arteries, the gastrointestinal tract, the uterus, and the skin. Within arteries, type III collagen is integrated into the elastin lamellae of the media and the collagenous network of the adventitia.3 Major clinical disease in vEDS manifests as tissue fragility with spontaneous rupture of the bowel (usually sigmoid colon), gravid uterus, or large or medium artery.11 Vascular rupture often occurs without preceding vascular dilatation or aneurysm formation1,2 and commonly affects the large elastic arteries of the thorax and abdomen.24 Initial presentation typically occurs in the third or fourth decade. Most described human vEDS cases result from missense mutations within the canonical Gly-X-Y repeat or from exon skipping mutations, with a net reduction in structurally normal extracellular type III collagen.24,32 From genetic and ultra-structural investigations, a dominant negative mechanism of pathogenesis has been inferred.1,3,37 In this paradigm, 50% of procollagen chains are structurally abnormal, meaning that only 1 in 8 mature collagen homotrimers is structurally sound. However, there are a number of cases, both published32 and unpublished (N.B McDonnell, personal communication, 2007), of phenotypically indistinguishable disease arising from mutations that result in haploinsufficiency for COL3A1.
There are no described spontaneous animal models for this disease, but a Col3a1 knockout mouse (Col3a1tm1Jae) has been previously developed.18 In this strain, a targeted mutation replaces the promoter and first exon of the Col3a1 gene with a PGKneo cassette, resulting in a total absence of collagen III product from the mutated allele. This mutation was reported to produce a severe phenotype in homozygous knockouts, with 90% perinatal mortality and a reduced life span in surviving homozygotes, most often due to spontaneous vascular rupture. Heterozygotes were reported to have no phenotype.
Although vEDS is rare, with an estimated incidence of 1 in 100,000 live births, the disease typically has an aggressive course with high morbidity and mortality. Therapeutic interventions in vEDS are limited to symptomatic or palliative measures.39 Therefore an appropriate animal model is vital for understanding the pathophysiology of disease as well as for the development and testing of adequate treatment strategies. The previously reported homozygous knockout mouse is an insufficient model for 2 reasons: first, only a single human homozygous COL3A1 null individual has ever been reported, and the individual did not present as a typical vEDS phenotype.26 Second, the homozygous mice have an extremely high preweaning mortality, in comparison with a disease that typically manifests clinically in the third or fourth decade in humans. Therefore, this study was designed to evaluate the heterozygous knockout mouse as a potential animal model for vEDS. Because large-vessel vascular complications are the most serious cause of morbidity and mortality in this disease, this phenotyping strategy included a detailed histologic assessment of pathology in the aorta across the life span and in both sexes, including characterization of lesions observed. Complementary functional and biomechanical studies of the heart and large elastic (aorta) and medium muscular (gracilis) arteries were undertaken to assist in the evaluation of the vascular phenotype as well. Finally, anatomic, biomechanical, and transcriptional studies of the colon, a second major site of complications, were performed.
Methods
Animals
Strain C.129S4(B6)-Col3a1tm1Jae/J mice were obtained from Jackson Laboratories (stock no. 002907, Bar Harbor, ME) and maintained on a BALB/c background. Mice were housed in individually ventilated cages in a barrier facility. Routine sentinel monitoring was positive for mouse norovirus-1 only. All mice were genotyped by polymerase chain reaction (PCR).36 Animal care and use were in accordance with the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication 85-23, revised 1996), and all mouse experimental procedures were approved by the Institutional Animal Care and Use Committee.
Histology
Age- and sex-matched male and female heterozygous and wild-type mice were sacrificed at 2, 5, 9, 14, and 21 months with carbon dioxide. Immediately following sacrifice, the abdomen and thorax were opened. The right ventricular free wall of the heart was incised, and a 21-g × ¾-in. needle on a 12-in. butterfly catheter was inserted into the left ventricle. The vasculature was flushed with phosphate-buffered saline (PBS) until clear and then infused with a 50:50 mixture of distilled water and latex for injection (9 and 14 months, Ward’s Natural Science, Rochester, NY) or 3% low-melting-temperature agarose (wt/vol; SeaPlaque GTG Agarose, low-melt [Lonza, Allendale, NJ], diluted in saline, and colored with Evans blue [Sigma-Aldrich, St. Louis, MO] at 37°C, 2, 5, and 21 months) until the vessels were filled to prevent collapse during tissue processing. The infused latex was allowed to harden for 2 hours at 4°C, and mice were immersion fixed in 10% neutral buffered formalin (NBF) for 5–7 days. After the infused agarose had solidified, the heart and the aorta were dissected free from the surrounding connective tissue and pinned onto a wax block before fixation in 10% NBF. Cross-sections of the aorta, 2 mm in length, were taken starting at the root and ordered in 8% bacto-agar (Fisher Scientific, Pittsburgh, PA). Aortas were processed and embedded in paraffin, cut at 5 μm, and stained with hematoxylin and eosin (HE) and Masson’s tri-chrome. All slides were examined by a single pathologist blinded to genotype. Lesion number, location, and severity (on a subjective 1–4 scale) were recorded.
The colon from 9-month-old male and female mice was flushed with physiological saline solution and fixed in 10% NBF for 2 days. Cross-sections of the colon, 5 mm in length, were ordered in 8% bacto-agar, processed and embedded in paraffin, cut at 5 μm, and stained with HE and Masson’s trichrome.
Smoothelin Immunofluorescence
For smoothelin immunofluorescence, 5-μm aorta sections were mounted on plus slides, rehydrated in a descending concentration of ethanols, and washed in distilled water. Antigen retrieval was performed by steaming in 10 mM sodium citrate buffer, pH 6.0, for 30 minutes. Slides were washed in PBS followed by a 10-minute incubation in 3% hydrogen peroxide and then additional PBS washes. Slides were blocked in 5%bovine serum albumin in PBS for 1 hour at room temperature and incubated overnight at 4°C in 1:200 rabbit polyclonal anti-smoothelin (cat. no. 28562, Santa Cruz Biotechnology, Santa Cruz, CA). Slides were washed in PBS and incubated for 2 hours at room temperature in Alexa Fluor 594 donkey anti-rabbit IgG (cat. no. A21207, Invitrogen, Carlsbad, CA). Slides were coverslipped with Vectashield Dapi mounting media (Vector Laboratories, Burlingame, CA) and stored in the dark at 4°C.
Collagen Detection by Picro-Sirius Red Staining
To examine collagen content in the vascular wall, 5-μm sections of aorta from 5-month-old male mice were cut and stained with picro-sirius red. Digital images of stained sections of thoracic, transverse, descending, and abdominal aorta (wild-type = 13, heterozygous = 9) were obtained from light microscopy using polarized filters and analyzed using a digital imaging analysis system (MCID, InterFocus Imaging Ltd., Cambridge, UK). The collagen content was calculated as a percentage of the total area of tunica media.33 Assessment was performed by a single individual (HJT) blinded to genotype of the animal.
Western Blotting of Aorta
Four age-matched sibling pairs (wild-type and heterozygous) of 9-month-old male mice were sacrificed with carbon dioxide. Immediately following sacrifice, animals were perfused transcardially with ice-cold PBS until clear and then with PBS containing both phosphatase (Millipore, Billerica, MA) and protease inhibitors (Roche, Indianapolis, IN). The aorta was dissected free, and the adventitia was stripped. The descending thoracic and abdominal aortas were individually snap frozen in liquid nitrogen, ground in Tissue Protein Extraction Reagent (TPER; Pierce, Rockford, IL) containing protease and phosphatase inhibitors using a tissue grinder (Kimble-Kontes, Vineland, NJ), and then sonicated in a cup horn (Fisher Scientific). Protein was quantitated using a Bicinchoninic Acid assay (BCA; Pierce), and 10 μg of total protein was loaded per well for gel electrophoresis and Western blotting. Blots were probed with antibodies to type III collagen (SC-28888, Santa Cruz Biotechnology, Santa Cruz, CA), type I collagen (SC-28784-R, Santa Cruz Biotechnology), and actin (A2066, Sigma-Aldrich).
Urinary Type I Collagen Carboxy-Terminal Peptide Quantitation
The carboxy-terminal peptide of type I collagen (PICP) was analyzed with the RatLaps enzyme-linked immunosorbent assay (ELISA) (Nordic Bioscience Diagnostics A/S, Herlev, Denmark) in 24-hour urine collected in mouse metabolic cages (Nalgene) from 9-month-old male and female mice. The result of the carboxy-terminal peptide of type I collagen was normalized to creatinine, which was detected in a 1:40 dilution with the Metra creatinine assay (Quidel, San Diego, CA).
The carboxy-terminal peptide of type I collagen was analyzed in 24-hour urine from 9-month-old male and female mice by ELISA and normalized to urinary creatinine.
In Vivo Heart and Artery Function
Echocardiography (Vevo 770 with a 30-MHz transducer 704; VisualSonics Inc., Toronto, Canada) was conducted under light anesthesia of 2% of isoflurane in oxygen. Mice were examined at 9 months (female wild-type n = 8, female heterozygous n = 4, male wild-type n = 5, male heterozygous n = 6), 14 months (female wild-type n = 7, female heterozygous n = 4, male wild-type n = 8, male heterozygous n = 8), and 20 months (female wild-type n = 2, female heterozygous n = 3, male wild-type n = 4, male heterozygous n = 4). Parasternal long-axis views were obtained and recorded to ensure that the mitral and aortic valves, as well as the apex, were visualized. Short-axis views were recorded at the midpapillary muscle level. End-diastolic volume (EDV) and end-systolic volume (ESV) were calculated by a modified Simpson’s method using measurements taken from M-mode of short axis at. Ejection fraction (EF) was then derived as EF = (ED – ESV)/ED × 100. All measurements were made by a single observer (MK) blind to the identity of the tracings. Posterior wall thickness was calculated from 2-dimensional mode. The abdominal aorta and carotid arteries were measured from M-mode of longitudinal views.12 The carotid was viewed approximately 1 mm below the bifurcation, and the abdominal aorta was measured in 3 locations: under the liver, at the renal arteries, and approximately 1 mm above the bifurcation. All measurements were averaged over 35 consecutive cardiac cycles. The reproducibility of measurements was assessed in 2 sets of baseline measurements in 10 randomly selected mice, and the repeated measure variability did not exceed 5%.
Biomechanical Properties of the Arteries
The maximum pressure in the abdominal aorta was measured from 14-month-old males (wild-type n = 7, heterozygous n = 8) and females (wild-type n = 8, heterozygous n = 5) and from 21-month-old males (wild-type n = 4, heterozygous n = 8) and females (wild-type n = 5, heterozygous n = 12). The abdominal aorta was dissected and cleaned of connective tissue. A catheter was introduced in the abdominal aorta above the bifurcation, connected to a syringe and a 72.5-PSI pressure transducer (Sper Scientific, Scottsdale, AZ). The aorta was closed below the caudal mesenteric artery and physiological saline injected until the vessel was ruptured. The pressure was measured with the wide-range pressure meter (Sper Scientific) and recorded by DataLab graphical software (Sper Scientific).
In another set of 10-month-old males (wild-type n = 7, heterozygous n = 7), the thoracic aorta was dissected and cleaned of connective tissue. A 1.8- to 2.0-mm-long piece of the aorta was threaded on two 40-μm diameter stainless steel wires and mounted isometrically on a wire-myograph (Danish Myo Technology, Aarhus, Denmark) containing physiological salt solution (PSS) consisting of (in mmol/L) 140 NaCl, 4.5 KCl, 1.2 NaH2PO4, 1.0 MgSO4, 1.6 CaCl2, 0.025 EDTA, 5.5 glucose, and 5 HEPES at pH 7.4, which was continuously bubbled with carbogen. Isometric tension was recorded with the program Myodaq (Danish Myo Technology). After the temperature of the myograph had reached 37.0 ± 0.5°C, the vessels were stretched radially to their optimal lumen diameter corresponding to 90% of the passive diameter of the vessel at 100 mm Hg and were allowed to stabilize for 15 minutes. Thereafter, the reactivity of the vessels was tested by stimulation with 10 μmol/L serotonin in PSS. All drugs were applied directly into the experimental chamber. Distensibility was tested in PSS where calcium was omitted, and 1 mM EGTA was added to ensure complete passive behavior of the vessel.
Small gracilis arteries were dissected and mounted on 2 cannulas in an experimental chamber containing PSS. The microscope image of the vessel was viewed with a CCD camera and digitized by a frame-grabber board, and diameter reactions were analyzed online. The vessels were pressurized to 80 mm Hg. Leaking vessels were discarded at any stage of the experiment to ensure complete nonflow conditions. The temperature was set to 37.0 ± 0.5°C, and the pH was set to 7.40 ± 0.05. After a spontaneous myogenic tone had developed (constriction to a diameter between 60% and 80% of the fully relaxed diameter at 80 mm Hg; the fully relaxed diameter was in the range of 170–300 μm), vessel viability was tested with 1 μmol/L acetylcholine (viability criterion: dilation to diameter the vessel had before the development of the myogenic tone) and 0.1 μmol/L serotonin (viability criterion: at least 25% constriction).
Biomechanical Properties of the Colon
An approximately 13-mm-long section of the transverse colon from 9-month-old females (wild-type n = 7, heterozygous n = 8), 14-month-old females (wild-type n = 9, heterozygous n = 6) and males (wild-type n = 7, heterozygous n = 8), and 21-month-old females (wild-type n = 5, heterozygous n = 12) and males (wild-type n = 6, heterozygous n = 8) was injected with incremented volumes of PSS by a Hamilton syringe, and the luminal pressure was measured with a pressure transducer (Millar Instruments Inc., Houston, US-TX) until the burst pressure was reached. The pressure was amplified and recorded by the PowerLab system (ADInstruments, Colorado Springs, CO).
RNA Isolation and Quantitative Real-Time PCR
The colon of 9-month-old male and female mice of both genotypes was excised after sacrificing the animal. Feces were removed and the colon was washed in PSS, frozen in liquid nitrogen, and stored at −80°C. Total RNA was extracted with the RNeasy kit (Qiagen, Valencia, CA), and the complementary DNA was synthesized with a random primer with the MultiScribe reverse transcriptase kit (Applied Biosystems, Carlsbad, CA). The SYBR green QuantiTect mix (Qiagen) was used for real-time PCR detection with the primers described in Table 1. The reaction was run in 4 technical replicates of a messenger RNA (mRNA) pool of the colon tissue from 6 female and 6 male of each genotype mice in an ABI PRISM 7900HT with an annealing temperature of 60°C. The result was analyzed by the 2−Δ ΔCT method normalized to glyceraldehyde 3-phosphate dehydrogenase.
Table 1.
Primer Sequences for Polymerase Chain Reaction
| Gene | Also Known As | Forward Primer/Reverse Primer | Product Size, Base Pairs |
|---|---|---|---|
| Col1a1 | TCCGGCTCCTGCTCCTCTTA4 GTATGCAGCTGACTTCAGGGATGT27 |
77 | |
| Col3a1 | GCCCACAGCCTTCTACAC9 CCAGGGTCACCATTTCTC9 |
108 | |
| Mmp2 | GelA | AACTACGATGATGACCGGAAGTG34 TGGCATGGCCGAACTCA34 |
87 |
| Mmp9 | GelB | CGAACTTCGACACTGACAAGAAGT34 GCACGCTGGAATGATCTAAGC34 |
113 |
| Mmp14 | MT1-MMP | AGGAGACAGAGGTGATCATCATTG34 GTCCCATGGCGTCTGAAGA34 |
141 |
| Mmp15 | MT2-MMP | ATCCCCTATGACCGCATTGAC34 CCCCTGCCAGACACTGATG34 |
146 |
| Mmp16 | MT3-MMP | GGCTACCTTCCACCGACTGA34 CTTCATCCAGTCGATTGTGTTTCT34 |
140 |
| Mmp17 | MT4-MMP | GGCAGTATGTTCCTGCACTTCA34 GCTAGCACTGCCCTCAGGAT34 |
114 |
| Mmp24 | MT5-MMP | TATCATGGCTCCCTTCTACCAATAC34 CTGCGGACCGGGAGTGT34 |
143 |
| Timp1 | TCCTCTTGTTGCTATCACTGATAGCTT27 CGCTGGTATAAGGTGGTCTCGTT27 |
147 | |
| Timp2 | CCAGAAGAAGAGCCTGAACCA27 GTCCATCCAGAGGCACTCATC27 |
111 | |
| Timp3 | GGCCTCAATTACCGCTACCA34 CTGATAGCCAGGGTACCCAAAA34 |
134 | |
| Timp4 | TGCAGAGGGAGAGCCTGAA34 GGTACATGGCACTGCATAGCA34 |
79 | |
| Tgfb1 | AGAGGTCACCCGCGTGCTAA27 TCCCGAATGTCTGACGTATTGA27 |
107 | |
| Tgfb2 | AGCTAAAGTCCTTGGGAAAGC22 ATCATGGTCGTCATCGTTGTC22 |
77 | |
| Tgfb3 | CCACAATCAGCCTCTCTCTGT22 AATGGCTTCCACCCTCTTC22 |
76 | |
| Tgfbr1 | ATCTATGCAATGGGCTTAGTGTT22 TAGTCTTCATGGATTCCACCAAT22 |
70 | |
| Tgfbr2 | TGTCTACTCCATGGCTCTGGTA22 GGCTCGTAATCCTTCACTTCTC22 |
77 | |
| Gapdh | TGCACCACCAACTGCTTAG8 GGATGCAGGGATGATGTTC8 |
176 |
Statistical Analysis
Numerical data were analyzed and expressed as mean ± SEM. A multiple-sample comparison (analysis of variance [ANOVA] and the multiple range test as post hoc test using the criterion of the least significant differences) was applied to test the differences between the groups. A value of P < .05 was considered to be significant. Cumulative survival was analyzed with Kaplan-Meier method using Prism software. Fisher’s exact test and logistic regression models were used to analyze vessel pathology. Aortic collagen content was analyzed using 1-way ANOVA followed by Duncan’s post hoc comparison.
Results
Appearance and Lifespan
No observed differences in morbidity or mortality between heterozygous and wild-type mice were observed, nor were there any overt clinical signs related to cardiovascular or gastrointestinal function in mice observed up to 2 years of age (data not shown). There were no obvious differences between heterozygous and wild-type animals in appearance or posture, except that the dorsal skin of heterozygous mice seemed subjectively slightly more loose when the mice were scruffed for handling.
Histopathology of the Aorta
A spectrum of lesions were present in the aorta in which fragmentation of the internal elastic lamina (IEL) was a consistent feature. Grade 1 lesions appeared as small break in the IEL with no significant spindle cell proliferation (not shown). The broken ends of the laminae frequently curled under into the media. In grade 2 lesions, the distance between the fragmented ends of the IEL was wider and the intervening space was filled with a moderate proliferation of spindle cells and the accumulation of an abundant collagen-rich extracellular matrix (Fig. 1). Grade 3 lesions were larger, with more florid medial spindle cell proliferation and often fragmentation of 1–2 medial elastic laminae (Fig. 2). In grade 4 lesions, there was marked and abrupt attenuation of the wall thickness with abundant fibrosis and more severe fragmentation of several elastic laminae (Fig. 3). These florid lesions rarely formed a plaque-like eccentric thickening of the wall with a narrowing of the vascular lumen (Fig. 4). A lack of infiltrating leukocytes as well as a relative sparing of the aortic root clearly distinguished the lesions in these mice from the recently described aortitis of BALB/c mice.29 Grade 1 lesions were seen in 55% of analyzed aortas (93 of 170) with almost no discrimination between wild-type and heterozygous mice. Because of their mild nature, as well as a high frequency in both sexes and genotypes, grade 1 lesions were excluded from statistical analysis.
Figure 1.

Aorta. Grade 2 lesion with a large defect in the internal elastic lamina (IEL) and significant subintimal spindle cell proliferation with deposition of collagen. Masson’s trichrome.
Figure 2.
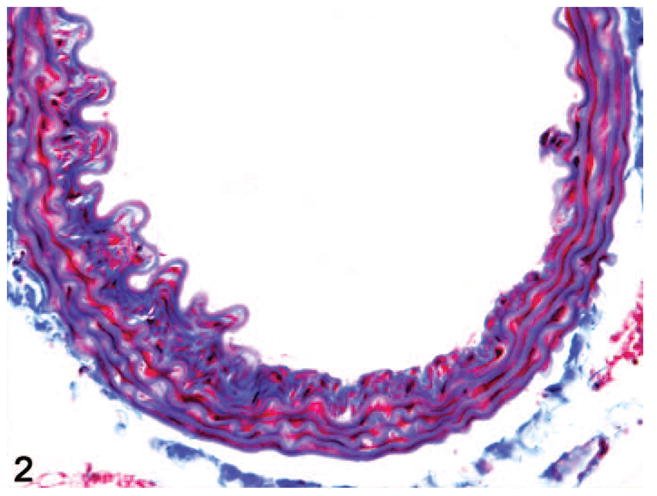
Aorta. Grade 3 lesion with a larger IEL defect, more florid spindle cell proliferation, and extensive deposition of collagenous matrix. Masson’s trichrome.
Figure 3.

Aorta. Grade 4 lesion with fragmentation of IEL as well as multiple medial elastic laminae, extensive medial interlamellar deposition of collagen, and jumbled irregular elastic fibers at the edge of a focally extensive attenuation in the thickness of the aortic wall. Masson’s trichrome.
Figure 4.

Aorta. A thick eccentric subintimal plaque of spindle cells and collagenous matrix segmentally expands the inner aortic wall and narrows the lumen. Outer media and adventitia are unremarkable. Masson’s trichrome.
Lesions in the aorta from 9- to 21-month-old animals were observed in 14 of the 30 (47%) female and 23 of the 26 (88%) male heterozygotes (Fig. 5). This sex distribution is similar to that reported in humans.2 Lesions were observed in 2 of the 24 (8%) female wild-type mice and 7 of the 21 (33%) male wild-type mice. Lesions occurred throughout the aorta but were most common in the distal half of the descending thoracic aorta and the proximal two-thirds of the abdominal aorta. No lesions were observed in any 2-month-old mice, whereas among 5-month-old mice lesions were only observed in 2 of the 16 (12.5%) male heterozygotes (Fig. 6). Lesions became more significant in older heterozygotes, with a higher cumulative lesion score with age (Fig. 7). A similar age-dependent increase in lesions was noted in male and female wild-type mice, although the cumulative severity of lesions is significantly lower (Table 2).
Figure 5.
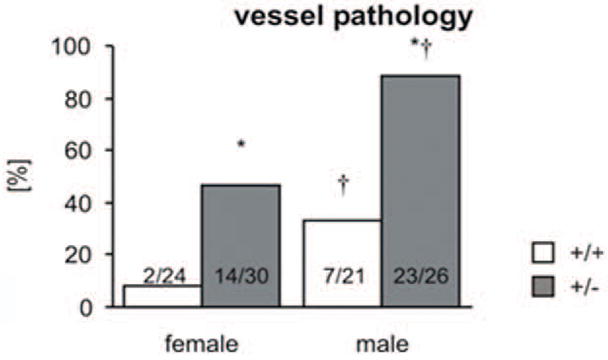
Comparison of the presence or absence of aortic pathology (lesions > grade 2) in 9- to 21-month-old animals demonstrates a significant association with genotype and sex (*P < .05 vs wild-type, †P < .05 vs female).
Figure 6.

Evaluation of the presence or absence of aortic pathology (lesions > grade 2) in each age group demonstrates a significant association with age, sex, and genotype (*P < .05 vs wild-type, †P < .05 vs female).
Figure 7.

Cumulative lesion score (lesions > grade 2) in each age group demonstrates a significant association with age, sex, and genotype (*P < .05 vs all female or male wild-type (respectively), †P < .05 vs female, #P < .05 vs 5 months).
Table 2.
Estimated Probability of the Development of Vessel Pathology in 5- to 21-Month-Old Mice
| Age Group, Months |
||||
|---|---|---|---|---|
| 5 | 9 | 14 | 21 | |
| Female WT | 0.0000 | 0.0147 | 0.0909 | 0.1185 |
| Female HT | 0.0151 | 0.5799 | 0.8617 | 0.2653 |
| Male WT | 0.0000 | 0.1103 | 0.4546 | 0.5284 |
| Male HT | 0.1136 | 0.9201 | 0.9811 | 0.7506 |
HT, heterozygous; WT, wild-type.
The probability (P) of developing vessel pathology was estimated by a logistic regression model using combinations of the 3 variables age, genotype, and sex. Sex was highly significant—P(pathology | female) < P(pathology | male)—and the genotype–age group interaction was significant. The estimates probabilities show that P(pathology | HT) > P(pathology | WT) within each sex by age group.
Smoothelin Immunofluorescence
Smoothelin immunostaining was performed to identify the types of spindle cells present within the aortic lesions. Unlike α-smooth muscle actin, which also stains myofibroblasts as well as other types of smooth muscle cells, smoothelin is considered to be a specific marker for vascular smooth muscle.38 Smoothelin immunofluorescence staining of a subset of these lesions confirmed that the spindle cells included both smoothelin-positive vascular smooth muscle cells and smoothelin-negative cells interpreted as fibroblasts and myofibroblasts (Fig. 8).
Figure 8.
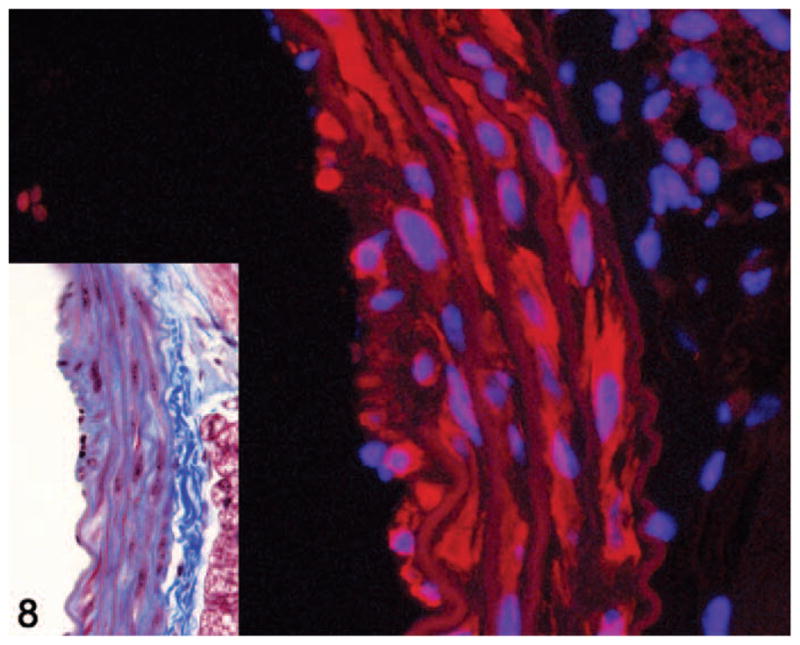
Aorta. Smoothelin immunofluorescence of a grade 2 lesion showing smoothelin-positive (red staining) and smoothelin-negative spindle cells within the media. Alexa Fluor 594 conjugate with DAPI counterstain. Inset shows histology of the same lesion. Masson’s trichrome.
Picro-Sirius Red Staining
There was no significant difference in total collagen content of the ascending or transverse aorta between genotypes as determined by evaluation of picro-sirius red–stained sections. There was a nonsignificant decrease in total collagen in the descending thoracic aorta of heterozygotes. Heterozygotes had significantly less total collagen present in the abdominal aorta (Figs. 8–11, P < .05).
Figure 11.

By picro-sirius red staining, wild-type (WT) mice had significantly higher collagen per unit area than heterozygotes (HT) in the abdominal aorta. AC, ascending aorta; Trn, transverse aorta; Tho, thoracic aorta; Abd, abdominal aorta (*P < .05 vs WT).
Western Blotting of Aorta
Levels of type III collagen were significantly reduced in the abdominal aorta of heterozygous mice relative to wild-type controls (Fig. 12). There was no significant difference in type III collagen levels noted in the descending thoracic aorta. Although the decreased collagen in the abdominal aorta as measured by picro-sirius red staining could not be specifically attributed to a reduction of type III collagen, but might result from reduced amounts of type I collagen (the most abundant collagen in the vessel wall), by Western blotting there was no detectable difference in type I collagen levels between genotypes in either the descending thoracic or abdominal aorta (data not shown).
Figure 12.

Western blotting of isolated abdominal aorta shows significant reduction of type III collagen levels in HT compared with age- and sex-matched WT littermates.
Determination of Type I Collagen Turnover
In 9-month-old male heterozygotes, there was significantly elevated urinary excretion of the PICP relative to wild-type, consistent with increased type I collagen formation (Fig. 13). There was no difference between heterozygous and wild-type females at this age.
Figure 13.

Evaluation of type I collagen carboxy-terminal peptide from 24-hour urine (normalized to creatinine) in 9-month-old animals demonstrates significantly higher urinary PICP in male HT mice relative to WT males and to females (P < .05). There is no significant difference between WT and HT females.
In Vivo Heart and Vessel Function
There were no detectable differences by echocardiography in the heart, aorta, or carotid arteries at any age in either sex.
Aorta and Small Artery Biomechanics
The distensibilities of the aorta of wild-type and heterozygous mice were not different (n = 7; P = .85) at physiological pressures (data not shown). The maximum pressure (rupture pressure) in the abdominal aorta was similar in wild-type and heterozygous mice at 14 months but significantly lower in heterozygotes at 21 months (Fig. 14, P < .05). The contractile behavior of the aorta of wild-type and heterozygous mice was not different (contractions to phenylephrine [n = 7; P = .57]; relaxations to acetylcholine [n = 7; P =.58] and nitric oxide [NO] (n = 7; P = .67), data not shown).
Figure 14.

The maximum pressure in the abdominal aorta was measured in 14- and 21-month-old wild-type (+/+) and heterozygous (+/−) mice. Number of experiments shown in parentheses.
Higher compliance was detected in small arteries from heterozygous mice compared with wild-type (Fig. 15, n = 7; P < .05). Specifically, a higher compliance of the arteria gracilis of heterozygous mice, measured as larger diameter changes in response to incremental increases of pressure, was observed in the pressure range between 20 and 60 mm Hg but not at higher pressures. The contractile behavior of the arteria gracilis of wild-type and heterozygous mice was not different (contractions to pressure changes [n = 6; P = .91] and phenylephrine [n = 7; P = .96]; relaxations to acetylcholine [n = 6; P = .75] and NO [n = 6; P = .69], data not shown).
Figure 15.

The compliance of arteria gracilis was measured using isobaric myography in 10-month-old wild-type (+/+) and heterozygous (+/−) mice (n = 7). *P <.05 vs wild-type.
Colon Strength Testing
The colon of heterozygous mice was significantly less stiff (more compliant) than in wild-type mice at all ages tested (Fig. 16). The maximum pressure (Fig. 17) was also significantly lower in heterozygotes. Furthermore, in both genotypes, maximum bursting pressure increased with age and was significantly higher in 14- and 21-month-old animals than in 9-month-old animals. No pathology was observed in any of the colon segments by routine HE examination (data not shown).5
Figure 16.
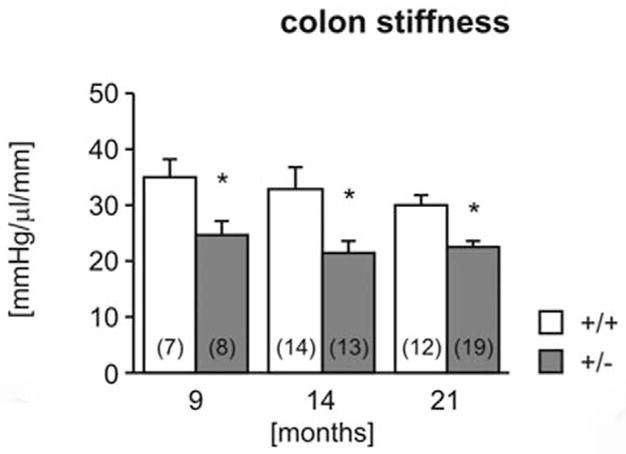
Biomechanical properties of the colon. Colon stiffness (Fig. 16) and maximal holding pressure (Fig. 17) were measured in 9-, 14-, and 21-month-old wild-type (+/+) and heterozygous (+/−) mice. Number of experiments shown in parentheses. *P < .05 vs wild-type, †P <.05 vs 9 months.
Figure 17.
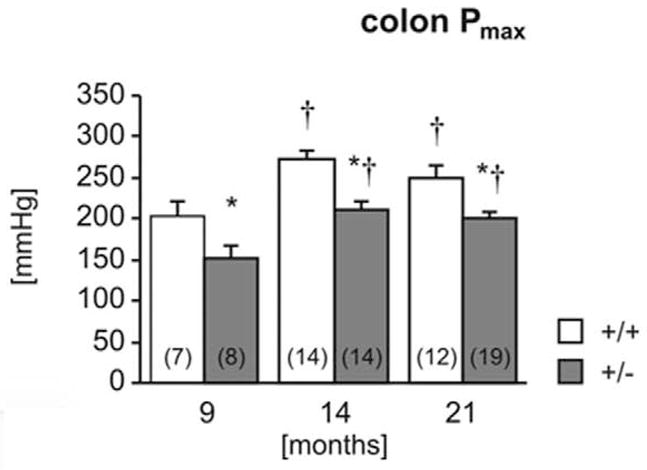
Biomechanical properties of the colon. Colon stiffness (Fig. 16) and maximal holding pressure (Fig. 17) were measured in 9-, 14-, and 21-month-old wild-type (+/+) and heterozygous (+/−) mice. Number of experiments shown in parentheses. *P < .05 vs wild-type, †P <.05 vs 9 months.
Real-Time PCR of Colon Tissue
Transcript levels for Col3a1 in heterozygotes were slightly less than 50% of those in wild-type mice, consistent with genotype and Western blotting. There was no apparent difference in Col1a1 transcript levels. The tissue inhibitors of metalloproteinases, matrix metalloproteinases, and the tumor growth factor-β (TGF-β) signaling family play a critical role in the homeostasis and remodeling of the extracellular matrix. Transcript levels for Timp4, Mmp15, and Tgfbr2 were significantly lower in heterozygotes, although differences in expression for the latter 2 may not be biologically significant (Table 3).
Table 3.
Expression of Extracellular Matrix–Related Transcripts
| Wild-Type | Heterozygous | P | |
|---|---|---|---|
| Col1a1 | 100 ± 13.2 | 99.3 ± 3.2 | .967 |
| Col3a1 | 100 ± 5.9 | 42.4 ± 4.6 | .000 |
| Mmp2 | 100 ± 9.7 | 73.3 ± 8.9 | .088 |
| Mmp9 | 100 ± 8.9 | 81.5 ± 10.2 | .222 |
| Mmp14 | 100 ± 14.6 | 68.7 ± 15.2 | .188 |
| Mmp15 | 100 ± 1.1 | 79.6 ± 4.3a | .011 |
| Mmp16 | 100 ± 15.3 | 65.1 ± 1.8 | .153 |
| Mmp17 | 100 ± 15.3 | 91.0 ± 5.7 | .298 |
| Mmp24 | 100 ± 32.0 | 69.9 ± 13.4 | .420 |
| Timp1 | 100 ± 27.1 | 88.6 ± 35.1 | .804 |
| Timp2 | 100 ± 9.8 | 85.9 ± 2.9 | .219 |
| Timp3 | 100 ± 5.3 | 99.6 ± 7.1 | .967 |
| Timp4 | 100 ± 12.1 | 54.9 ± 4.7a | .011 |
| Tgfb1 | 100 ± 12.9 | 83.3 ± 6.5 | .291 |
| Tgfb2 | 100 ± 12.9 | 68.6 ± 8.3 | .087 |
| Tgfb3 | 100 ± 7.7 | 81.0 ± 8.5 | .149 |
| Tgfbr1 | 100 ± 7.6 | 95.1 ± 5.4 | .616 |
| Tgfbr2 | 100 ± 4.8 | 81.8 ± 4.5a | .033 |
The amounts of Col1a1, Col3a1, and other transcripts involved in remodeling of the extracellular matrix were analyzed by real-time polymerase chain reaction. The average ± standard error of the mean of 4 technical replicates of a pool of the colon tissue from 6 female and 6 male mice is shown. The result was analyzed by the 2−ΔΔCT method normalized to Gapdh. The result of wild-type pool was set 100%.
P < .05 vs wild-type.
Discussion
At both the mRNA and protein level, Col3a1tmJae1 heterozygotes show reduced expression of type III collagen. Thorough analysis of heterozygous mice compared with their wild-type littermates demonstrates a subclinical phenotype of vascular fragility, evidenced by both the histologic and biomechanical analysis, which is age-dependent in its expression. A similar biomechanical phenotype was observed in the gastrointestinal tract, despite the absence of gross or histologic lesions.
Lesions observed by histology in the aortas of Col3a1tmJae1 heterozygous mice are similar to those reported in humans with Ehlers-Danlos type IV.1,2,7,35 Consistent involvement of the intima with progressive involvement of the media and sparing of the adventitia suggests an “inside-out” pathogenesis, possibly associated with circulatory flow. The microscopic nature of the lesions, with fragmentation of the IEL and variable rupture of additional laminae, is consistent with focal injury (intimal tearing), leading to proliferation of smoothelin-positive medial vascular smooth muscle cells as well as fibroblasts with production of a collagenous scar. These lesions are focal to multi-focal and apparently stochastic in distribution. Additionally, the nature of the lesions in haploinsufficient Col3a1 mice overlaps with the morphologically similar but milder lesions noted in older wild-type mice, indicating that the observed lesions are themselves nonspecific. Rather, heterozygotes are frequently identified by both the number and the cumulative severity of lesions, suggesting that the differences between genotypes are quantitative rather than qualitative. Similar quantitative rather than qualitative differences in diseased and “normal” control aortas have been noted previously in humans with vEDS as well as for other genetic and acquired aortic diseases.15,21,30,31 Picro-sirius red staining of the aorta sections shows diminished total collagen in heterozygotes relative to wild-type. These changes are observed only in the abdominal aorta, where there are relatively more collagen and less elastin,14,30 and occur despite the increased urinary excretion of PICP suggestive of increased type I collagen deposition consistent with wound repair. These observations are confirmed by Western blotting and are consistent with the current paradigm of a vascular wall that is structurally weak while still apparently normal at the gross and microscopic levels of resolution. Functional testing of the strength of the aortic wall supports this conclusion, as does demonstration of reduced type III collagen in the extra-vascular tissues at the mRNA and protein level.18 Results of analysis of colons with normal histology and reduced functional strength testing further reinforce this interpretation.
It is notable that the lesions seen in haploinsufficient Col3a1 mice do not culminate in vascular rupture. It is possible that there is a different pathogenetic sequence with Col3a1 mutations that cause haploinsufficiency, as opposed to those with dominant-negative potential. Although Schwarze et al32 reported a series of patients with null mutations phenotypically indistinguishable from those with missense mutations, a recently published case report details a single case of homozygosity for a null allele of COL3A1 producing a severe phenotype with a nontypical presentation in a young girl.26 Both parents, who were consanguineous and in the fifth decade, were heterozygous for the mutation and reported to be “healthy” and “phenotypically normal,” demonstrating the probability of modifier genes affecting the phenotypic presentation of the disease. However, analysis of the parents was limited and the possibility of nonclinical vascular lesions cannot be excluded. Regardless, presence of disease in haploinsufficient humans, and of lesions in mice, suggests that a dominant negative mechanism of pathogenesis is not necessary to cause vascular lesions. The ability of homozygous null mice and humans to survive, albeit for a limited time, demonstrates that type III collagen is not a requisite for viability. Rather, there may be 2 distinct but interrelated mechanisms at play, one involving a primary structural deficiency stemming from a qualitative or quantitative deficiency of structurally normal type III collagen and the second arising from perturbations of the TGF-β family signaling pathway mediated by the conserved amino-terminal peptide of type III collagen.6 In particular, dysregulated TGF-β signaling has been shown to be associated with a maladaptive vascular extracellular matrix remodeling phenotype,16 and altered TGF-β signaling has been show to play a central and defining role in the genetic vascular diseases Marfan syndrome and Loeys-Dietz syndrome.13,19,20 In this paradigm, a structural deficiency would result in some degree of tissue fragility sufficient to result in histologic lesions, whereas defective modulation of TGF-β signaling would be necessary for clinical manifestation of the disease due to unbalanced extracellular matrix degradation/synthesis at the site of injury. The second mechanism may be reduced or absent in haploin-sufficient mice compared with those with dominant negative-type mutations. The presence of a spectrum of clinically silent lesions in an aorta with a structurally weakened wall in these haploinsufficient mice supports this hypothesis, as does the fact that transcript levels for matrix metalloproteinases, tissue inhibitors of matrix metalloproteinases, and TGF-β ligands and receptors in the colon show limited differences between the 2 genotypes. A more detailed interrogation of the TGF-β family signaling pathways in this model may be warranted. Finally, further support can be inferred from the observation that vEDS usually manifests clinically during the third or fourth decade of life, rather than the first or second, hinting that significant wear and tear must accumulate in the tissues.
Although the lack of clinical signs or end points (eg, death due to vascular or intestinal rupture) is a limitation of the Col3a1tm1Jae haploinsufficient mouse model, this limitation is not unique to this particular model. The failure of progression to clinically apparent dissection in heterozygotes, with severe disease in homozygotes, is observed in several other mouse models of human autosomal dominant vascular disease, including knock-in mutations (FbnC1039G),17 hypomorphs (Fbntm2Rmz),25 and knockouts (Col1a1tm1Jae).28 This pattern may reflect the underlying limits of the mouse model, a small and short-lived animal with lower aortic medial tensions at physiologic pressures,40 for genetic vascular dissection syndromes rather than any particular deficiency of the Col3a1tm1Jae model. Development of a new vEDS mouse model arising from a glycine substitution mutation is presently underway. It remains to be seen whether dissection in heterozygotes will be a feature of this model. In the interim, application of cardiovascular stress, such as through the use of angiotensin II or aortic banding, may exacerbate the pathology in the Col3a1tm1Jae haploinsufficient mouse and result in a useful clinical end point.
Figure 9.

Abdominal aorta of wild-type (Fig. 9) and heterozygote (Fig. 10) mice stained with picro-sirius red and viewed under polarized light demonstrates reduced adventitial and, to a lesser extent medial, total collagen in the heterozygote, consistent with genotype.
Figure 10.
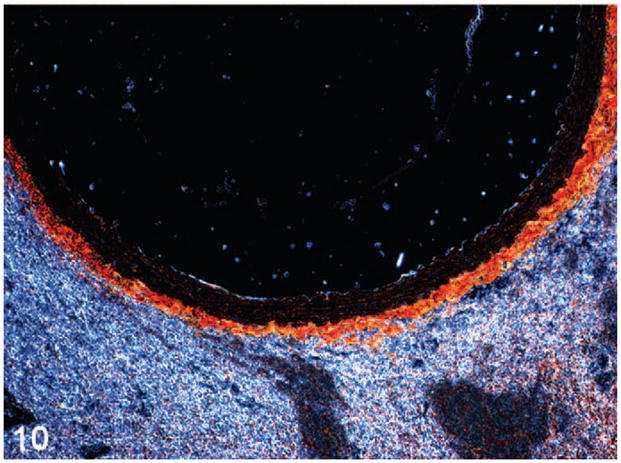
Abdominal aorta of wild-type (Fig. 9) and heterozygote (Fig. 10) mice stained with picro-sirius red and viewed under polarized light demonstrates reduced adventitial and, to a lesser extent medial, total collagen in the heterozygote, consistent with genotype.
Acknowledgments
Thanks to Pat Wilcox (JHU) and Ellen Mullady, Weifang Lin, and Trey Bruggeman (Penn State University, Milton S. Hershey Medical Center) for histology assistance. Thanks to Tia Turner (National Institute on Aging [NIA]) and Shannon Marshall (NIA) for technical assistance and Chris Morrell (NIA) for the statistical calculation of the vessel pathology. Thanks to Dr. NB McDonnell (NIA) for guidance.
Footnotes
Declaration of Conflict of Interest
The authors declared that they had no conflicts of interests with respect to their authorship or the publication of this article.
Financial Disclosure/Funding
Work was funded in part by the Intramural Research Program of the National Institutes of Health National Institute on Aging, the Howard Hughes Medical Institute, the William S. Smilow Center for Marfan Syndrome Research (JHU), and the National Institutes of Health grant R01AR41135.
References
- 1.Arteaga-Solis E, Gayraud B, Ramirez F. Elastic and collagenous networks in vascular diseases. Cell Struct Funct. 2000;25:69–72. doi: 10.1247/csf.25.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barabas AP. Ehlers-Danlos syndrome type IV. N Engl J Med. 2000;343:366. doi: 10.1056/NEJM200008033430513. author reply 368. [DOI] [PubMed] [Google Scholar]
- 3.Baxter BT. Heritable diseases of the blood vessels. Cardiovasc Pathol. 2005;14:185–188. doi: 10.1016/j.carpath.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Blaker H, Funke B, Hausser I, Hackert T, Schirmacher P, Autschbach F. Pathology of the large intestine in patients with vascular type Ehlers-Danlos syndrome. Virchows Arch. 2007;450:713–717. doi: 10.1007/s00428-007-0415-6. [DOI] [PubMed] [Google Scholar]
- 6.Bornstein P. The NH(2)-terminal propeptides of fibrillar collagens: highly conserved domains with poorly understood functions. Matrix Biol. 2002;21:217–226. doi: 10.1016/s0945-053x(02)00008-2. [DOI] [PubMed] [Google Scholar]
- 7.Collins MH, Schwarze U, Carpentieri DF, Kaplan P, Nathanson K, Meyer JS, Byers PH. Multiple vascular and bowel ruptures in an adolescent male with sporadic Ehlers-Danlos syndrome type IV. Pediatr Dev Pathol. 1999;2:86–93. doi: 10.1007/s100249900095. [DOI] [PubMed] [Google Scholar]
- 8.Dahi S, Lee JG, Lovett DH, Sarkar R. Differential transcriptional activation of matrix metalloproteinase-2 and membrane type-1 matrix metalloproteinase by experimental deep venous thrombosis and thrombin. J Vasc Surg. 2005;42:539–545. doi: 10.1016/j.jvs.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 9.Deatrick KB, Eliason JL, Lynch EM, Moore AJ, Dewyer NA, Varma MR, Pearce CG, Upchurch GR, Wakefield TW, Henke PK. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–148. doi: 10.1016/j.jvs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Germain DP. Clinical and genetic features of vascular Ehlers-Danlos syndrome. Ann Vasc Surg. 2002;16:391–397. doi: 10.1007/s10016-001-0229-y. [DOI] [PubMed] [Google Scholar]
- 11.Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32. doi: 10.1186/1750-1172-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goergen CJ, Johnson BL, Greve JM, Taylor CA, Zarins CK. Increased anterior abdominal aortic wall motion: possible role in aneurysm pathogenesis and design of endovascular devices. J Endovasc Ther. 2007;14:574–584. doi: 10.1177/152660280701400421. [DOI] [PubMed] [Google Scholar]
- 13.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harkness ML, Harkness RD, McDonald DA. The collagen and elastin content of the arterial wall in the dog. Proc R Soc Lond B Biol Sci. 1957;146:541–551. doi: 10.1098/rspb.1957.0029. [DOI] [PubMed] [Google Scholar]
- 15.Jain D, Dietz HC, Oswald GL, Maleszewski JJ, Halushka MK. Causes and histopathology of ascending aortic disease in children and young adults. Cardiovasc Pathol. doi: 10.1016/j.carpath.2009.09.008. [published online ahead of print November 18, 2009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones JA, Spinale FG, Ikonomidis JS. Transforming growth factor-beta signaling in thoracic aortic aneurysm development: a paradox in pathogenesis. J Vasc Res. 2009;46:119–137. doi: 10.1159/000151766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, cranio-facial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 20.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 21.Maleszewski JJ, Miller DV, Lu J, Dietz HC, Halushka MK. Histopathologic findings in ascending aortas from individuals with Loeys-Dietz syndrome (LDS) Am J Surg Pathol. 2009;33:194–201. doi: 10.1097/PAS.0b013e31817f3661. [DOI] [PubMed] [Google Scholar]
- 22.Mathy JA, Lenton K, Nacamuli RP, Fong KD, Song HM, Fang TD, Yang GP, Longaker MT. FGF-2 stimulation affects calvarial osteoblast biology: quantitative analysis of nine genes important for cranial suture biology by real-time reverse transcription polymerase chain reaction. Plast Reconstr Surg. 2003;112:528–539. doi: 10.1097/01.PRS.0000070729.05978.BB. [DOI] [PubMed] [Google Scholar]
- 23.Parapia LA, Jackson C. Ehlers-Danlos syndrome—a historical review. Br J Haematol. 2008;141:32–35. doi: 10.1111/j.1365-2141.2008.06994.x. [DOI] [PubMed] [Google Scholar]
- 24.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680. doi: 10.1056/NEJM200003093421001. [DOI] [PubMed] [Google Scholar]
- 25.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M, Van Kien PK. Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur J Hum Genet. 2009;17:1411–1416. doi: 10.1038/ejhg.2009.76. [published online ahead of print May 20, 2009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4) −/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43:1045–1054. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 28.Rahkonen O, Su M, Hakovirta H, Koskivirta I, Hormuzdi SG, Vuorio E, Bornstein P, Penttinen R. Mice with a deletion in the first intron of the Col1a1 gene develop age-dependent aortic dissection and rupture. Circ Res. 2004;94:83–90. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 29.Ramot Y, Manno RA, Okazaki Y, Krakovsky M, Lamensdorf I, Meiron M, Toren A, Zehavi-Goldstein E, Vezzali E, Nyska A. Spontaneous aortitis in the Balb/c mouse. Toxicol Pathol. 2009;37:667–671. doi: 10.1177/0192623309338384. [DOI] [PubMed] [Google Scholar]
- 30.Schlatmann TJ, Becker AE. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol. 1977;39:13–20. doi: 10.1016/s0002-9149(77)80004-0. [DOI] [PubMed] [Google Scholar]
- 31.Schlatmann TJ, Becker AE. Pathogenesis of dissecting aneurysm of aorta. Comparative histopathologic study of significance of medial changes. Am J Cardiol. 1977;39:21–26. doi: 10.1016/s0002-9149(77)80005-2. [DOI] [PubMed] [Google Scholar]
- 32.Schwarze U, Schievink WI, Petty E, Jaff MR, Babovic-Vuksanovic D, Cherry KJ, Pepin M, Byers PH. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989–1001. doi: 10.1086/324123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeland U, Selejan S, Engelhardt S, Muller P, Lohse MJ, Bohm M. Interstitial remodeling in beta1-adrenergic receptor transgenic mice. Basic Res Cardiol. 2007;102:183–193. doi: 10.1007/s00395-006-0635-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh RJ, Mason JC, Lidington EA, Edwards DR, Nuttall RK, Khokha R, Knauper V, Murphy G, Gavrilovic J. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. 2005;67:39–49. doi: 10.1016/j.cardiores.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 35.Stella A, Gessaroli M, Cifiello BI, Mirelli M, Loperfido V, Riva R, Guizzardi S. Sack-Barabas syndrome (Ehlers-Danlos IV type) (clinic and histopathologic ultrastructural correlations) Vasc Surg. 1986;20:67–73. [Google Scholar]
- 36.Stevenson K, Kucich U, Whitbeck C, Levin RM, Howard PS. Functional changes in bladder tissue from type III collagen-deficient mice. Mol Cell Biochem. 2006;283:107–114. doi: 10.1007/s11010-006-2388-1. [DOI] [PubMed] [Google Scholar]
- 37.Towbin JA, Casey B, Belmont J. The molecular basis of vascular disorders. Am J Hum Genet. 1999;64:678–684. doi: 10.1086/302303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Eys GJ, Niessen PM, Rensen SS. Smoothelin in vascular smooth muscle cells. Trends Cardiovasc Med. 2007;17:26–30. doi: 10.1016/j.tcm.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe A, Shimada T. Vascular type of Ehlers-Danlos syndrome. J Nippon Med Sch. 2008;75:254–261. doi: 10.1272/jnms.75.254. [DOI] [PubMed] [Google Scholar]
- 40.Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]