

Abstract
In the earliest stages of metastasis, breast cancer cells must reorganize the cytoskeleton to affect cell shape change and promote cell invasion and motility. These events require the cytoskeletal regulators Cdc42 and Rho, their effectors, such as N-WASp/WAVE, and direct inducers of actin polymerization such as Arp2/3. Little consideration has been given to molecules that shape the cell membrane. The F-BAR proteins CIP4, TOCA-1, and FBP17 generate membrane curvature and act as scaffolding proteins for activated Cdc42 and N-WASp. We found that expression of CIP4, but not TOCA-1 or FBP17, was increased in invasive breast cancer cell lines in comparison to weakly or non-invasive breast cancer cell lines. Endogenous CIP4 localized to the leading edge of migrating cells and to invadopodia in cells invading gelatin. Because CIP4 serves as a scaffolding protein for Cdc42, Src, and N-WASp, we tested whether loss of CIP4 could result in decreased N-WASp function. Interaction between CIP4 and N-WASp was EGF-responsive, and CIP4 silencing by siRNA caused decreased tyrosine phosphorylation of N-WASp at a Src-dependent activation site (Y256). CIP4 silencing also impaired the migration and invasion of MDA-MB-231 cells and was associated with decreased formation of invadopodia and gelatin degradation. This study presents a new role for CIP4 in the promotion of migration and invasion of MDA-MB-231 breast cancer cells and establishes the contribution of F-BAR proteins to cancer cell motility and invasion.
Keywords: breast cancer, invasion, Src kinases, Rho GTPases, invadopodia
INTRODUCTION
Invasive breast cancer cells acquire a migratory phenotype atypical of epithelial-type cells. This phenotype correlates with the formation of specialized structures such as lamellipodia and invadopodia, which are formed by a highly dynamic cytoskeleton. These structures are largely under the control of Src kinases, Rho GTPases, and their effector proteins. Rho GTPases (Rho, Rac, and Cdc42) comprise a group of small G proteins that cycle between inactive, GDP-bound and active, GTP-bound states. Once activated, they regulate a variety of effector proteins involved in cytoskeletal reorganization. Activation of RhoA causes bundling of existing actin filaments into stress fibers and the activation of myosin to generate contractile forces. In contrast, Rac and Cdc42 stimulate the production of highly branched actin filaments that are characteristic of lamellipodia (1).
N-WASp is a ubiquitously expressed form of the Wiskott-Aldrich Syndrome protein (WASp) and links Cdc42 to the Arp2/3 complex to stimulate actin polymerization (2). N-WASp activation is complex and includes the binding of PI(4,5)P2 to a conserved basic region. Association of GTP-bound Cdc42 with N-WASp causes it to change its configuration (3). Once in its open conformation, N-WASp can then bind to Arp2/3, promoting actin nucleation (3–6). N-WASp is also subject to phosphorylation by Src family kinases at tyrosine 256, which occurs only when N-WASp is in an open conformation and provides a mechanism for “memory” of activation (7). N-WASp also contains a proline-rich motif that serves as a docking site for SH3 containing proteins such as the Cdc42-interacting protein 4 (CIP4) (8, 9).
CIP4 was originally identified in yeast-2-hybrid screens for proteins that interacted with the activated forms of Cdc42 or Src kinase (8, 9). It was initially classified as a member of the PCH (pombe Cdc15 homology) family of proteins because of its N-terminal FER-CIP4 homology (FCH) domain, coiled-coil region, and C-terminal SH3 domain (8–11). The coiled-coil region is classified as a BAR (Bin-Amphiphysin-Rvs) domain. In tandem, the FCH and BAR regions comprise the F-BAR domain, which is shared in the family of CIP4, FBP17, and TOCA-1. The F-BAR region, the site of dimerization between CIP4 molecules, forms a curved structure that is associated with the tubulation of membranes and membrane concavity (12) (13). The cellular function of the F-BAR proteins has been most often described in conjunction with the formation of endocytic vesicles (14–18), where proteins such as CIP4 interact with N-WASp and dynamin to promote the curvature and scission of the budding vesicle (15, 17).
Cytoskeletal rearrangement is required for the migration and invasion of malignant cells (19). Invasive cancer cells escape from the primary tumor by forming invadopodia, protrusions into the surrounding matrix associated with concentrated matrix degradation and complex rearrangement of the actin cytoskeleton (20, 21). In contrast to the flat, branched structure of lamellipodia, invadopodia are finger-like projections supported by bundles of actin, similar to the podosomes of hematopoietic cells (20, 21). Several cytoskeletal-associated proteins are enriched at invadopodia during the early stages of their development, including Cdc42 and N-WASp (22, 23). Signaling and adhesion molecules such as Src, Epidermal Growth Factor (EGF) Receptor, Focal Adhesion Kinase, and integrins are also found at invadopodia (21). These structures function as sites of concentrated protease secretion, allowing them to degrade the extracellular matrix and facilitate cellular migration through a basement membrane (24–26). The in vitro formation of invadopodia correlates with metastasis in vivo and can be visualized by growing cells on a gelatin matrix (24, 27). Expression and activation of both Cdc42 and N-WASp are critical to the formation of invadopodia (22, 28). EGF stimulation promotes the formation of invadopodia, presumably through this Cdc42-N-WASp pathway (22, 28). Although GTP-Cdc42 alone is an activator of N-WASp (3), scaffolding proteins such as TOCA-1 are able to promote their interaction and stimulate more potent N-WASp activation (16, 29).
Invadopodia formation in the basal-type breast cancer cell line MDA-MB-231 has been well documented (20, 21). In this study we show that the strongly invasive MDA-MB-231 breast cancer cells express unusually high levels of CIP4 and that in vitro migration and invasion are strictly dependent on CIP4 expression. Furthermore, because CIP4 overexpression was not observed in other weakly invasive breast cancer cell lines, these data suggest that CIP4 overexpression may contribute to the metastatic phenotype. This work advances a novel role for CIP4 in promoting the invasive capacity of breast cancer cells, suggesting a more complex role for F-BAR proteins in cytoskeletal reorganization.
MATERIALS AND METHODS
Cell Culture
Cell lines were provided by Drs. Gordon Mills and Janet Price from MD Anderson Cancer Center. All cell lines were grown at 37° C and 5% CO2. MDA-MB-231 cells were maintained in DMEM/F12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal-calf serum (Hyclone, Logan, UT), 100 U/mL penicillin, and 100 µg/mL streptomycin. MCF7, T47D, SKBR3, Hs578T, and 293T cell lines were maintained in high-glucose DMEM media (Invitrogen) supplemented with 10% fetal-calf serum, 100 U/mL penicillin, and 100 µg/mL streptomycin. MCF-10a cells were cultured as previously described (30). MDA-MB-231 cell line was validated by STR DNA fingerprinting using the AmpFℓSTR Identifiler kit according to manufacturer instructions (Applied Biosystems cat 4322288). The STR profiles were compared to known ATCC fingerprints (ATCC.org), to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/) (Nucleic Acids Research 37:D925-D932 PMCID: PMC2686526) and to the MD Anderson fingerprint database. The STR profiles matched known DNA fingerprints or were unique.
Antibodies and Reagents
Commercially available antibodies used were: CIP4 (Becton Dickenson), FBP17 (Abcam), GAPDH (Cell Signaling, Danvers, MA), Cortactin (Upstate, Lake Placid, NY), N-WASp (ECM Biosciences and Santa Cruz), TRITC-phalloidin (Molecular Probes, Eugene, OR), and Cy3 and Cy5 secondary antibodies (Jackson ImmunoResearch, West Grove, PA). The TOCA-1 monoclonal antibody was provided by Dr. Giorgio Scita (Milan, Italy) (31, 32). Recombinant human EGF was purchased from Invitrogen (Carlsbad, CA).
Real time PCR
mRNA was extracted from sub-confluent cells with Trizol reagent (Invitrogen), treated with DNase (Invitrogen), and transcribed to cDNA with an iScript cDNA Synthesis Kit (BioRad). Real time PCR amplification reactions were carried out with iQ SYBR Green reagent (BioRad) and a MyiQ Color Real-Time PCR Detection System (BioRad). Values for each gene were normalized to actin. Probes specific to human CIP4, TOCA-1, FBP17, and actin were designed as follows: CIP4 (F: 3’-CAGCGAAAACGGCTTCAA-5’; R: 5’-GTCCCCCATCTGAGGTGTCT -3’), TOCA-1 (F: 3’-TGGATGCCAAAACCACAGTA -5’; R: 5’-CTGGTGGCAGATGACTGAAA -3’), FBP17 (F: 3’-GGAAGTGCCTGGATGGAATA -5’; R: 5’-CGCTTCATTGGCTGAGTGTA -3’), actin (F: 3’-ATAGCACAGCCTGGATAGCAA-5’; R: 5’-CACCTTCTACAATGAGCTGCG-3’).
RNAi mediated knockdown
MDA-MB-231 or 293T cells were transiently transfected with 10 nM of CIP4- or N-WASp-directed or non-targeting siRNA (Qiagen) using the HiPerFect transfection reagent (Qiagen). Both CIP4-directed siRNAs used target all known isoforms of CIP4. 72 hours, cells were replated for the appropriate assay.
Immunoprecipitation and Immunoblotting
Cells were grown in complete media overnight, treated with 100 ng/mL of human EGF for 1–30 minutes at 37° C, washed with cold PBS, collected by scraping, and lysed (1% NP-40 buffer with 10% glycerol, 50mM TrisCl, 150mM NaCl, 50mM NaF) supplemented with 1mM EDTA, 1mM PMSF, 1mM Na3VO4, and protease inhibitor cocktail (Sigma). After standardizing protein concentration, as determined by a Bradford protein assay, samples were either prepared for loading on SDS-PAGE or incubated with the appropriate antibody and immunoprecipitated. Protein G-Sepharose beads (Sigma) overnight. Immune complexes were denatured by boiling in Laemmli buffer, resolved by SDS-PAGE, and transferred to Immobilon-P Transfer Membranes (Millipore Corp., Bedford, MA). Membranes were blocked overnight with 5% nonfat milk or BSA in PBS with 0.1% Tween-20. Blots were incubated with primary and then secondary antibodies for 1 hour each at room temperature or 37° C. Immunoreactive bands were visualized using enhanced chemiluminescence (Amersham, Piscataway, NJ). All membranes were then incubated with stripping buffer (Pierce, Rockford, IL) for 30 minutes at 37°C, reblocked, and reprobed for GAPDH as a loading control. Densitometric analysis was performed using the NIH software, ImageJ (Mac platform, Bethesda, MD), for normalization.
Immunofluorescence
Cells were grown on glass coverslips, fixed in 3.7% formaldehyde (30 minutes), permeabilized with 0.1% TritonX-100 (5 minutes), and blocked in 1% BSA (30 minutes), each at room temperature. Samples were incubated with mouse α-CIP4 antibody overnight at 4° C, followed by Cy3- or Cy5-conjugated anti-mouse and TRITC-phalloidin for 1 hour at room temperature. Cells were re-fixed in 3.7% formaldehyde. Slides were prepared using ProLong Antifade with DAPI mounting media (Molecular Probes), and imaged with either a Nikon Eclipse TE2000U microscope with MetaMorph imaging software (Molecular Devices, Sunnyvale, CA) or Nikon A1 Confocal Microscope with Nikon NIS-Elements software (Melville, NY).
Invadopodia Formation Assay
Gelatin was labeled with FITC (Sigma) in 0.1 M bicarbonate buffer (pH 9.0), dialyzed extensively against PBS, and stored at 4° C. FITC-gelatin was crosslinked with 0.5% glutaraldehyde (10 minutes on ice and 30 minutes at room temperature) and reduced with 1 mg/mL sodium borohydride (5 minutes at room temperature). Cells were plated on FITC-gelatin-coated dishes in complete media and allowed to invade for 16–20 hours, then processed as described above. Samples were stained with TRITC-phalloidin. Invadopodia were counted from 6 to 10 random fields in each sample and averaged over multiple experiments. Gelatin degradation was quantified using the Macnification software (Orbicule BVBA, Heverlee, Belgium).
Cell Migration and Invasion
Migration was measured by wound healing assay, in which cells were grown to 80% confluence, streaked with a sterile pipet tip, and the wound width was measured (pixels). Cells were allowed to recover for 24 hours in complete media, after which the remaining wound width was measured. Invasion assays were performed using Matrigel invasion chambers (BD Biosciences, Bedford, MA) seeded with 2.5 × 104 cells per well. Complete media was used in both the upper and lower chambers, with or without 100 ng/mL EGF added only to the lower chamber. Cells were allowed to invade for 24 hours through the Matrigel and 8 µm-pore membrane, at which point the inserts were removed, the upper side of the membranes were scrubbed. The cells were then fixed in methanol, rinsed in distilled water (1 minute each), air-dried, and mounted on slides with ProLong Antifade with DAPI (Molecular Probes). Membranes were visualized with Zeiss epifluorescence microscope to count the number of invading cells in 9 random fields per sample.
FRET assay
Acceptor photobleaching FRET (apFRET) was used to quantify the EGF-dependent interactions between CIP4/N-WASp. All images were recorded using a Zeiss LSM 510 confocal microscope using a 63X, 1.3 NA objective. apFRET was applied largely as described previously where bleach settings are defined empirically (33, 34). Briefly, a cell (MDA-MB-231) expressing YFP-CIP4 and CFP-N-WASp was selected and 3 regions of interest (ROIs) were drawn for YFP bleaching and controls. The large ROI (ROI 3) was scanned 10 times using routine scanning settings. The bleach ROIs (ROI1) region was then bleached at 100% intensity for 1000 iterations or approximately 3 min. One ROI (ROI 2) was drawn to measure any potential for off-target bleaching, which accounted for less than 1% of changes in YFP and CFP. Following the bleach, 10 more scans were taken using the prebleach settings. The FRET efficiency (increase in CFP upon YFP bleaching) was calculated using the following equation:
Control experiments were performed to validate the apFRET protocol. These included vector controls (CFP, YFP) co-transfected with either YFP-CIP4 or CFP-N-WASp, respectively. In these control experiments, the false positive increase in CFP or CFP-N-WASp upon bleaching YFP-CIP4 or YFP, respectively, ranged from 1–6%.
RESULTS
CIP4 is highly expressed in invasive breast cancer cell lines
To determine the relative expression levels of CIP4 in invasive versus non-invasive breast cancer cell lines, we examined whole cell lysates of five well-characterized cell lines. By previous studies, MDA-MB-231 and Hs578T cells are highly invasive through in vitro Boyden chamber assays, whereas MCF7, T47D, and SKBR3 cells are only weakly invasive (35). Immunoblotting showed that CIP4 protein expression was higher in the invasive cell lines (Figure 1A). CIP4 expression in the non-cancerous mammary epithelial cells line MCF10a was comparable to the low levels in MCF7 and T47D cells (data not shown). There was no increase in FBP17 or TOCA-1 expression with increasing breast cancer invasiveness. Densitometric quantification comparing MDA-MB-231 with MCF7 and T47D showed significantly more CIP4 in the invasive line (Figure 1B, p=0.004 and p=0.046, respectively). Multiple isoforms of CIP4 that lack a functional SH3 domain have been identified in other cell lines (9, 11). Western blotting indicates that CIP4a, the ubiquitously expressed isoform, is the predominant protein found in the breast cancer cell lines in this study, although there is evidence of alternative splice forms by RT-PCR (data no shown).
Figure 1.

High CIP4 expression occurs in invasive breast cancer cell lines. A) Whole cell lysates were tested for CIP4, FBP17, TOCA-1 and GAPDH expression by immunoblotting. B) CIP4 expression plotted as average densitometric quantification of at least 3 immunoblots. Error bars = SD. ** indicates p<0.05. C) mRNA levels of CIP4, FBP17 and TOCA-1 in each cell line were quantified by real time PCR. Results were standardized to actin expression. Average of 4 experiments +/− SEM. ** indicates p<0.05, *** indicates p<0.001. D) Unstimulated MDA-MB-231, MCF7, and MCF10a cells were stained for endogenous CIP4 localization by immunofluorescence. Arrows indicate CIP4 accumulation at the cell membrane. 30 µm scale bars.
We corroborated the elevated CIP4 protein level by quantifying the transcript levels of each CIP4 family member using quantitative PCR. CIP4 mRNA was also significantly increased in the MDA-MB-231 cells (Figure 1C). Average CIP4 transcript expression was 6.3-fold higher in MDA-MB-231 cells compared with MCF-7 (p=0.009) and 2.8-fold higher versus T47D (p=0.026). Transcript levels of TOCA-1 and FBP17 did not correlate with the protein expression profiles. Therefore, we chose to investigate the unique profile of CIP4 expression with respect to cellular invasion. The MDA-MB-231 cell line is classified as triple-negative, lacking the estrogen receptor (ER), progesterone receptor (PR), and HER2, which is associated with more aggressive, invasive disease (36). Bioinformatic analysis of 46 well-characterized breast cancer cell lines (35) showed that CIP4 was increased in triple negative cell lines versus breast cancer cells of other biomarker status (Supplemental Figure 1, p=1.03×10−7). No significant difference was found in the expression of FBP17 (p=0.098) or TOCA-1 (p=0.983).
CIP4 associates with the plasma membrane via its F-BAR domain, which contributes to its function. We compared the localization of endogenous CIP4 in the highly invasive MDA-MB-231 cell line with its localization in the weakly invasive MCF7 and non-cancerous MCF-10a cell lines (Figure 1D). Although all three lines showed diffuse cytoplasmic staining, only the MDA-MB-231 cells accumulated CIP4 at the cell membrane. This was especially evident at lamellipodia (arrows). MCF-7 and MCF10a cells had very little or no CIP4 at the cell periphery.
CIP4 promotes cellular migration
Because CIP4 controls cytoskeletal organization, we next examined whether its expression was critical to the motile phenotype of MDA-MB-231 cells. In actively migrating cells, the endogenous CIP4 was enriched at the leading edge of the cell, suggesting a role in cell migration (Figure 2A). Using siRNA directed at CIP4, we successfully inhibited the expression of CIP4 after 72 hours (Figure 2B). The siRNA sequences used in this manuscript target all known isoforms of CIP4 (data not shown). We then performed wound-healing assays as a measure of 2D migration. By measuring the width of the wound at to and t24h, we found that MDA-MB-231 cells treated with CIP4-directed siRNA had a significant loss of migration compared to cells treated with a non-targeting, control siRNA (Figure 2C, p<0.0001). Effects of CIP4 knockdown on MDA-MB-231 migration were not due to incidental changes in cell proliferation (Supplemental Figure 2). Thus, these data indicate that CIP4 expression is required for MDA-MB-231 motility.
Figure 2.

CIP4 promotes cellular migration. A) Staining of endogenous CIP4 in migrating MDA-MB-231 cell. Dashed arrow indicates direction of migration. Solid arrowheads indicate CIP4 at leading edge. B) MDA-MB-231 cells were transfected with non-targeting (control) or CIP4-specific siRNA and whole cell lysates were immunoblotted for CIP4 at 48 and 72 hours after transfection. Blots were reprobed for GAPDH. C) MDA-MB-231 cells were transfected with siRNA (Control vs. CIP4), grown to confluence (72 hours), wounded, and allowed to recover for 24 additional hours. Borders of wound are delineated in black. Images are representative of multiple independent experiments. Average width of wound was quantified at t0 and t24h after wounding. Average of 3 replicates; error bars represent SD. *** = p<0.0001.
CIP4 controls N-WASp activation in response to EGF
Cdc42-mediated activation of N-WASp can occur downstream of EGF stimulation (37, 38). Because CIP4 interacts with Cdc42 (8) and localizes with EGFR-containing vesicles (32), we investigated the role of CIP4 in the EGF-dependent activation of N-WASp. After EGF stimulation of MDA-MB-231 cells, we observed a sharp increase in the interaction between endogenous CIP4 and N-WASp by co-immunoprecipitation occurring at a very short time point (around 1 minute) (Supplemental Figure 3A). To confirm this effect and investigate the dynamics at faster time points, we used acceptor photobleaching FRET (apFRET) to analyze MDA-MB-231 cells transfected with YFP-CIP4 and CFP-N-WASp. apFRET uses photobleaching to eliminate energy transfer from donor (CFP-N-WASp) to acceptor (YFP-CIP4). Subsequently, the intensity of CFP-N-WASp increases indicating the magnitude of donor radiation at the acceptor frequency and the extent of interaction between the two fluorophores. By this method, we also observed a rapid, transient increase in FRET efficiency between CIP4 and N-WASp after EGF stimulus (Figure 3A and Supplemental Figure 3B). Because the Y256 site of N-WASp, which is analogous to the Y291 in WASp, can only be phosphorylated in the open conformation (7, 39, 40), we used the phosphorylation status of this site as a surrogate marker for N-WASp activation. In control cells, EGF stimulus increased the phosphorylation of N-WASp at this site at 1 minute (Figure 3B). Knockdown of CIP4 decreased the basal level of N-WASp phosphorylation in both MDA-MB-231 cells (Figure 3C) and EGF-stimulated 293T cells (Supplemental Figure 4). This indicates that CIP4 contributes to EGF-dependent activation of N-WASp.
Figure 3.
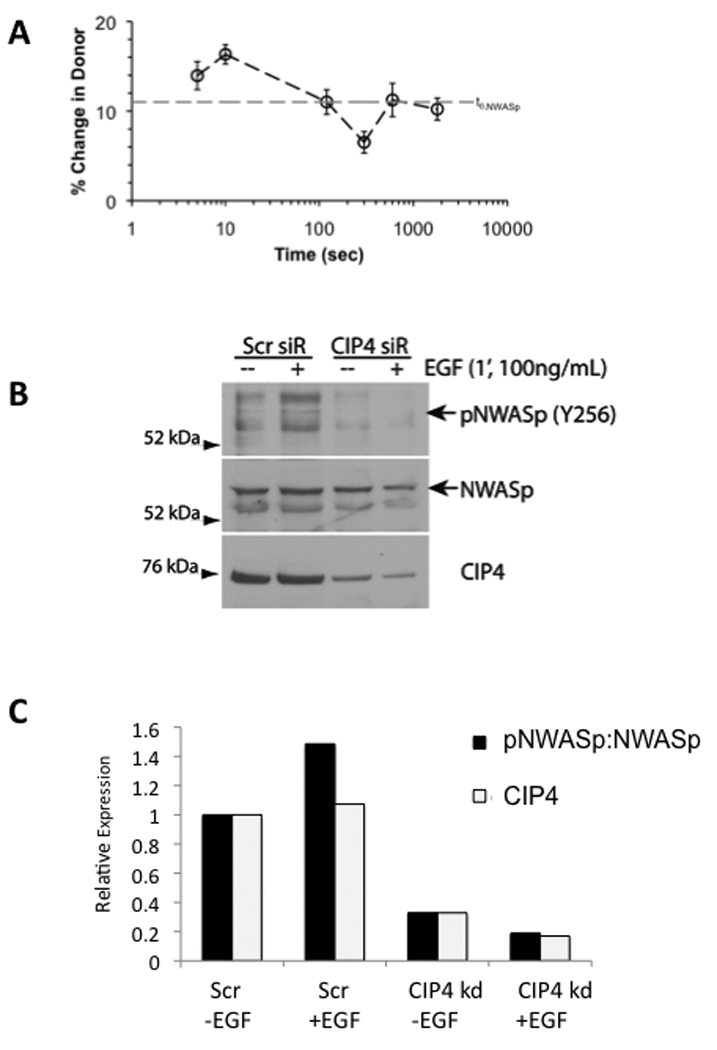
CIP4 promotes the EGF-dependent activation of N-WASp in MDA-MB-231 cells. A) MDA-MB-231 cells were transfected with YFP-CIP4 and CFP-NWASp and treated with EGF from 0 to 1800 seconds (n=11, mean ± sem.). Dynamic protein-protein interactions were quantified using apFRET. Upon photobleaching YFP-CIP4 in the plasma membrane compartment, interactions with CFP-N-WASp were denoted by increases in CFP fluorescence intensities. The dashed line (t0, N-WASp) indicates the basal FRET detected before EGF stimulation. B) Cells transfected with control or CIP4-siRNA were treated with EGF for 1 minute, lysed, and probed for phospho-N-WASp (Y256), as indicated by the arrow, then reprobed for total N-WASp and CIP4. Blot is representative of 2 independent experiments.
CIP4 is required for maximal invadopodia formation
N-WASp has previously been shown to control invadopodia formation, which is thought to be a critical to the ability of cells to traverse the extracellular matrix (22, 41, 42). Because CIP4 controlled N-WASp activation, we assessed whether it was also important for invadopodia formation. MDA-MB-231 cells were plated on thin layers of FITC-labeled gelatin and incubated overnight to allow invadopodia formation. After incubation, the cells were stained for endogenous CIP4 and actin to visualize the invadopodia (Figure 4A). Colocalization of punctate actin structures and areas of gelatin-degradation indicated an active invadopodia. We found that CIP4 was also enriched at these invadopodia. Z-stack projections of the structures demonstrate the localization of CIP4 to the base of the invadopodia (Figure 4B), compared with the long projection of actin through the degraded gelatin. Similarly, previous reports have localized N-WASp to the base of the invadopodia (22).
Figure 4.

CIP4 localizes to invadopodia. A–B) MDA-MB-231 cells were plated on a thin-layer of FITC-gelatin and allowed to invade for 16 hours before being stained for endogenous CIP4 and actin. A) Top: CIP4 and actin staining of untreated MDA-MB-231 cells. Scale bar represents 10 µm. Bottom: CIP4 (red) and actin (blue) colocalized with holes in FITC-gelatin monolayer (green) in XY-projection, indicated by arrows. B) Z-stack projections of FITC-gelatin alone, and with CIP4 or actin. Overlay of gelatin (green), CIP4 (red), and actin (blue) in Z-stack projection shows active invadopodia.
To determine whether CIP4 played a critical role in invadopodia formation, we first quantified the percentage of cells with invadopodia after treatment with control or CIP4 siRNA. By this metric there was no significant effect of CIP4 knockdown (data not shown). However, when we measured the distribution of the number of invadopodia per cell in the control versus CIP4- or N-WASp-siRNA treated cells (Figure 5A), we observed that the cells with CIP4 knockdown had significantly fewer invadopodia per cell (Figure 5B, p<0.05), a decrease comparable to that seen with knockdown of N-WASp. To measure the size and function of invadopodia, we quantified the area of gelatin degradation per cell after CIP4 and N-WASp knockdown and found significantly less gelatin lost after treatment (Figure 5C, p<0.001).
Figure 5.
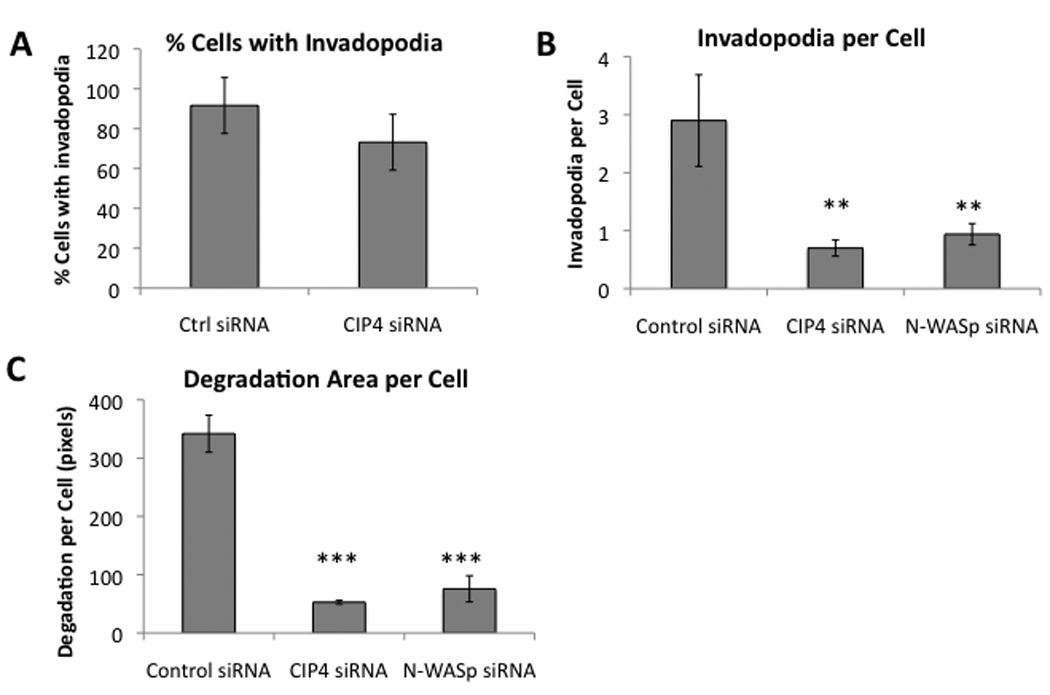
CIP4 is required for maximal invadopodia formation. A) siRNA-mediated knockdown of N-WASp in MDA-MB-231, lysed and immunoblotted 72 hours after transfection. B) Cells transfected with control or CIP4 siRNA were plated on FITC-gelatin, incubated overnight, and fixed and stained for cortactin. Colocalized points of cortactin staining and gelatin-degradation were quantified for 10 random fields per sample. Error bars represent SD. C) Cells transfected with control, CIP4, or N-WASp siRNA were plated on FITC-gelatin overnight, fixed, and stained with TRICT-phalloidin. Gelatin-degradation areas were counted and measured, normalized for cell number, and averaged over replicates from 3 independent experiments. Error bars represent SEM. ** = p<0.05, *** = p<0.001.
CIP4 promotes invasion of MDA-MB-231 cells in vitro
Having demonstrated that CIP4 was critical to migration and affected invadopodia stability, we next examined whether loss of CIP4 would affect the invasiveness of MDA-MB-231 cells in a Boyden chamber assay. Seventy-two hours after siRNA transfection, the cells were replated onto Matrigel-coated membranes and incubated in complete media for an additional 24 hours. With two independent CIP4-directed siRNA sequences, we found a significant loss of invasion (p<0.05 and p<0.001) as compared with the control (non-targeting siRNA) samples (Figure 6A). This loss of invasion was comparable to the loss seen with knockdown of N-WASp (p<0.05) and dasatinib-mediated inhibition of Src kinases (43), which are both critical components of cellular invasion. While MDA-MB-231 cells invade efficiently in the absence of any chemoattractant, their rate of invasion is significantly increased by the presence of EGF in the lower chamber. Thus, we also measured the effect of CIP4 knockdown on EGF-stimulated Matrigel invasion. In these assays, we also observed a nearly complete inhibition of EGF response (Figure 6B).
Figure 6.
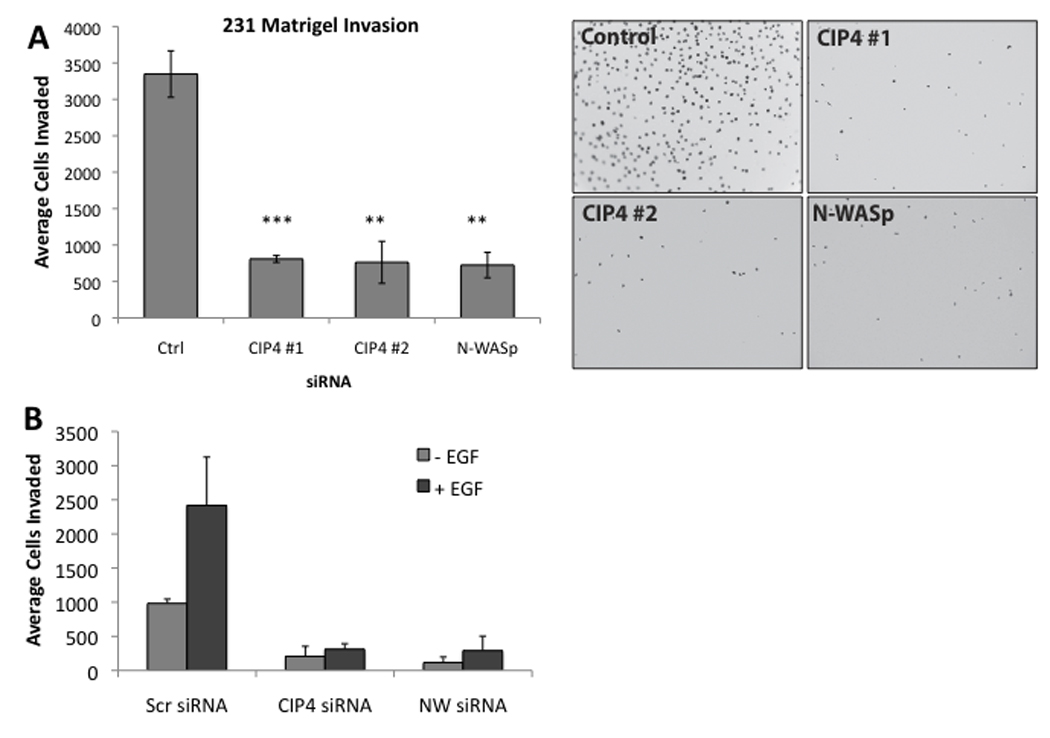
CIP4 expression is required for invasion of MDA-MB-231 cells in vitro. A) Cells were transfected with control (non-targeting) siRNA or siRNA specific for CIP4 or N-WASp, grown for 72 hours, replated on Matrigel-coated Boyden chambers, and allowed to invade for an additional 24 hours before membranes were collected and invading cells counted in 9 fields per membrane. Representative images are shown. B) Matrigel invasion assay was performed with or without EGF (100 ng/mL)-supplemented media in the lower chamber. Average of 2 independent experiments; error bars represent SD. ** = p<0.05, *** = p<0.001.
DISCUSSION
The acquisition of an invasive phenotype is a hallmark of metastatic cancer and portends a poorer prognosis (44). Diagnosis of advanced breast cancer represents a severe decrease in 5-year survival (23.3%) compared with localized forms of the disease (98.3%) (45). “Triple-negative” type breast cancers, which are associated with a more aggressive disease, are refractory to current targeted therapies against HER2 (36, 46). Additionally, these tumors often over-express the EGF Receptor (EGFR), which has been associated with invasiveness and poor patient prognosis (47). Therefore, therapeutic targeting of cellular invasion could significantly impact the treatment of advanced breast cancer. Several proteins have been associated with a pro-invasive phenotype, many of which are linked to the cytoskeletal system, including Src, Cdc42, and N-WASp (43, 48, 49). The data presented here demonstrate that the F-BAR protein CIP4 contributes to the invasive phenotype of MDA-MB-231 breast cancer cells in vitro.
To investigate a potential pro-invasive role for CIP4, we evaluated a panel of breast cancer cell lines and demonstrated an increased expression of CIP4, both at the mRNA and protein levels, in the most invasive cell lines (MDA-MB-231 and Hs578T) (Figure 1), representative of the “triple-negative” breast cancer sub-type (35, 46). Gene expression of a larger dataset confirmed a statistically significant increase in CIP4 expression (but not FBP17 or TOCA-1) in triple negative breast cancer cell lines (Supplemental Figure 1). In contrast to the non-invasive (MCF-7) and non-cancerous (MCF-10a) cell lines, the invasive MDA-MB-231 cells demonstrated a high concentration of CIP4 at the plasma membrane, especially at the dynamic ruffled edges of both resting and actively migrating cells (Figures 1 and 2). Furthermore, depletion of CIP4 in MDA-MB-231 cells significantly impaired cellular migration across a cell-culture surface (Figure 2), supporting our hypothesis that CIP4 promotes cell motility.
The F-BAR family of proteins, which are characterized by their curved, amphipathic helix, or BAR domains (13) (12, 13) has not been previously implicated in the migration or invasion of cancer cells. While the F-BAR domain of CIP4 mediates its interaction with the plasma membrane, CIP4 also interacts with the cytoskeletal regulatory proteins Cdc42 (8), N-WASp (15), and Src family kinases (9). As a scaffolding protein, CIP4 may provide a link between areas of membrane curvature and localized actin remodeling (8). By demonstrating CIP4’s role in N-WASp phosphorylation, we propose a mechanism by which CIP4 targets N-WASp activation to segments of curved membrane to induce or reinforce actin remodeling at endocytic pits, lamellipodia, and invadopodia.
N-WASp serves as a major effector for the Rho GTPase Cdc42, mediating Cdc42-dependent Arp2/3 activation and actin polymerization (2–6). Previously, Ho et al demonstrated that TOCA-1, a related F-BAR protein, strengthens the Cdc42-mediated activation of N-WASp, resulting in increased actin polymerization in fluorescence-based, cell-free assays (50). We hypothesized that CIP4 may function through a similar mechanism, stabilizing the activation of N-WASp to promote cellular motility. Because N-WASp activation at the plasma membrane is increased downstream of EGF (22), we investigated whether the interaction between CIP4 and N-WASp might also be responsive to EGF stimulation. Through co-immunoprecipitation and apFRET microscopy, we determined the EGF-dependent interaction kinetics between CIP4 and N-WASp. Consistent with immunoprecipitation results (Figure 3), apFRET specifically detected a constitutive interaction between N-WASp and CIP4 that was rapidly increased (< 1 minute) by EGF (IFRET =+5%) with a monotonic decrease in interaction (IFRET= −9%), eventually returning to basal levels (Supplemental Figure 3B). Recent studies have indicated periodic activity of N-WASp during clathrin-mediated endocytosis (51) that coincides with rapid activation of N-WASp, as observed by an N-WASp FRET biosensor (22). We further demonstrated that CIP4 stabilizes the phosphorylation of N-WASp (Y256), indicative of N-WASp activation, both basally and in response to EGFR activation (Figure 3). Although phosphorylation on Y256 is not a direct measure of N-WASp activation, this site is only accessible to Src kinases when N-WASp is in the open, or active, conformation. Previous studies have demonstrated a close correlation between Y256 phosphorylation and N-WASp activation (7, 29, 40). One possible mechanism is that CIP4 relieves the inhibition of N-WASp, as has been shown for Toca-1.(50)
Both N-WASp and Cdc42 are critical to the formation of invadopodia, which are specialized cytoskeletal structures that combine localized actin protrusion with matrix-metalloprotease secretion to degrade extracellular matrices and allow invasion (21, 28). The formation of these invasive structures has been linked to localized EGFR activation and presumably requires the Cdc42-N-WASp pathway (37, 42). As a potential mechanism for CIP4 in promoting the pro-invasive phenotype, we examined whether CIP4 was also involved in the structure or function of these invasive structures. We demonstrated that CIP4 is localized to active invadopodia through immunofluorescence microscopy of MDA-MB-231 cells grown over a thin FITC-gelatin layer (Figure 4A and 4B). No members of the F-BAR family have previously been implicated in the formation of invadopodia.
Although CIP4 depletion did not significantly affect the number of invadopodia-positive cells, the average number of invadopodia within individual cells was decreased, suggesting a role for CIP4 in the formation or stability of invadopodia (Figure 5A). Additionally, we observed a decrease in gelatin degradation per cell, a measure of invadopodial size, with loss of CIP4 (Figure 5B). Stable, mature invadopodia degrade large holes in the gelatin layer, whereas small, unoccupied holes indicate the formation and dissolution of an unstable invadopodia. The decrease in degradation suggests impairment in the function of invadopodia in the absence of CIP4. Using an ELISA assay, we did detect a 28% decrease in the secretion of MMP-9, a gelatin-specific metalloprotease associated with invadopodia, upon depletion of CIP4 (data not shown). This effect, however, was small in comparison to the loss of invadopodia and unlikely to account for the overall defect in invadopodia formation.
Although CIP4 promotes the formation of invadopodia, it was critical that we establish a functional role for CIP4 in cellular invasion. Therefore, we investigated whether CIP4 depletion affected the invasiveness of MDA-MB-231 cells through Matrigel-coated Boyden chamber assays. We found that CIP4 loss severely abrogated the spontaneous invasion (80% decrease) of MDA-MB-231 cells in vitro (Figure 6A), comparable to the degree of inhibition seen with N-WASp depletion. Additionally, loss of CIP4 prevented any significant increase in invasion towards EGF (Figure 6B) in the same Boyden assay, supporting the hypothesis that CIP4 promotes invasion through interaction with the Cdc42-N-WASp pathway.
Many previous studies have demonstrated the association between membrane curvature and F-BAR-containing proteins (13, 17, 52–55). Takano et al (54) proposed a mechanism for Toca-1 and FBP17 by which F-BAR proteins facilitate actin polymerization in a membrane curvature-dependent manner. Their model demonstrates electrostatic repulsion between the membrane-bound F-BAR domain and acidic residues adjacent to the SH3 domain confer, which creates a spring-like scaffolding structure and facilitates the activation of N-WASp. Based the results we present in this manuscript, we propose a similar model for CIP4 in which CIP4 stabilizes the open conformation of N-WASp specifically at areas of membrane curvature. Through this mechanism, CIP4 could promote the formation of invadopodia and the resulting cellular invasion.
In summary, this study demonstrates a novel role for F-BAR proteins in the migration and invasion of cancer cells. Our work demonstrates that CIP4 is required for the phosphorylation of N-WASp in response to EGF, which would regulate the activation of Arp2/3 and subsequent actin polymerization. We have shown that CIP4 not only localizes to invadopodia, but also affects their formation and function in invasive breast cancer cells, leading to cellular migration and invasion in vitro. CIP4’s structure suggests that it functions as a scaffolding protein to target actin polymerization to areas membrane curvature by integrating N-WASp regulation with F-BAR mediated membrane curvature. Through this mechanism, CIP4 overexpression may contribute to the invasive phenotype of triple-negative, EGFR-positive breast cancers. These results support the further investigation of CIP4 as a potential biomarker for invasive breast cancers.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Giorgio Scita for providing anti-TOCA-1 antibody. This work was supported by a Grant-in-Aid from the American Heart Association, Charlotte Geyer Foundation, NIH RO1HL080052 to S.J.C; NIH 5R01CA116356 to J.A.F.; St. Baldricks Fellowship to C.A.; and a Department of Defense Breast Cancer Pre-Doctoral Training Fellowship and award by the University of Texas Health Sciences Center Graduate School of Biomedical Sciences to C.S.P. STR DNA fingerprinting was done by the MD Anderson Cancer Center Support grant funded Characterized Cell Line core, NCI-CA16672.
REFERENCES
- 1.Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002;84:57–80. doi: 10.1016/s0065-230x(02)84003-9. [DOI] [PubMed] [Google Scholar]
- 2.Rohatgi R, Ho HY, Kirschner MW. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J Cell Biol. 2000;150:1299–1310. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prehoda KE, Scott JA, Mullins RD, Lim WA. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- 4.Fukuoka M, Suetsugu S, Miki H, Fukami K, Endo T, Takenawa T. A novel neural Wiskott-Aldrich syndrome protein (N-WASP) binding protein, WISH, induces Arp2/3 complex activation independent of Cdc42. J Cell Biol. 2001;152:471–482. doi: 10.1083/jcb.152.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higgs HN, Pollard TD. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu Rev Biochem. 2001;70:649–676. doi: 10.1146/annurev.biochem.70.1.649. [DOI] [PubMed] [Google Scholar]
- 6.Millard TH, Sharp SJ, Machesky LM. Signalling to actin assembly via the WASP (Wiskott-Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem J. 2004;380:1–17. doi: 10.1042/BJ20040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres E, Rosen MK. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol Cell. 2003;11:1215–1227. doi: 10.1016/s1097-2765(03)00139-4. [DOI] [PubMed] [Google Scholar]
- 8.Aspenstrom P. A Cdc42 target protein with homology to the non-kinase domain of FER has a potential role in regulating the actin cytoskeleton. Curr Biol. 1997;7:479–487. doi: 10.1016/s0960-9822(06)00219-3. [DOI] [PubMed] [Google Scholar]
- 9.Dombrosky-Ferlan P, Grishin A, Botelho RJ, et al. Felic (CIP4b), a novel binding partner with the Src kinase Lyn and Cdc42, localizes to the phagocytic cup. Blood. 2003;101:2804–2809. doi: 10.1182/blood-2002-03-0851. [DOI] [PubMed] [Google Scholar]
- 10.Tsuji E, Tsuji Y, Fujiwara T, Ogata S, Tsukamoto K, Saku K. Splicing variant of Cdc42 interacting protein-4 disrupts beta-catenin-mediated cell-cell adhesion: expression and function in renal cell carcinoma. Biochem Biophys Res Commun. 2006;339:1083–1088. doi: 10.1016/j.bbrc.2005.11.117. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Rudert WA, Grishin A, et al. Identification and genetic analysis of human and mouse activated Cdc42 interacting protein-4 isoforms. Biochem Biophys Res Commun. 2002;293:1426–1430. doi: 10.1016/S0006-291X(02)00398-4. [DOI] [PubMed] [Google Scholar]
- 12.Ayton GS, Lyman E, Krishna V, et al. New insights into BAR domain-induced membrane remodeling. Biophys J. 2009;97:1616–1625. doi: 10.1016/j.bpj.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frost A, Perera R, Roux A, et al. Structural basis of membrane invagination by F-BAR domains. Cell. 2008;132:807–817. doi: 10.1016/j.cell.2007.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bu W, Chou AM, Lim KB, Sudhaharan T, Ahmed S. The Toca-1-N-WASP complex links filopodial formation to endocytosis. J Biol Chem. 2009;284:11622–11636. doi: 10.1074/jbc.M805940200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartig SM, Ishikura S, Hicklen RS, et al. The F-BAR protein CIP4 promotes GLUT4 endocytosis through bidirectional interactions with N-WASp and Dynamin-2. J Cell Sci. 2009;122:2283–2291. doi: 10.1242/jcs.041343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamioka Y, Fukuhara S, Sawa H, et al. A novel dynamin-associating molecule, formin-binding protein 17, induces tubular membrane invaginations and participates in endocytosis. J Biol Chem. 2004;279:40091–40099. doi: 10.1074/jbc.M404899200. [DOI] [PubMed] [Google Scholar]
- 17.Shimada A, Niwa H, Tsujita K, et al. Curved EFC/F-BAR-domain dimers are joined end to end into a filament for membrane invagination in endocytosis. Cell. 2007;129:761–772. doi: 10.1016/j.cell.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 18.Tsujita K, Suetsugu S, Sasaki N, Furutani M, Oikawa T, Takenawa T. Coordination between the actin cytoskeleton and membrane deformation by a novel membrane tubulation domain of PCH proteins is involved in endocytosis. J Cell Biol. 2006;172:269–279. doi: 10.1083/jcb.200508091. %R 101083/jcb200508091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vignjevic D, Montagnac G. Reorganisation of the dendritic actin network during cancer cell migration and invasion. Semin Cancer Biol. 2008;18:12–22. doi: 10.1016/j.semcancer.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–117. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 22.Lorenz M, Yamaguchi H, Wang Y, Singer RH, Condeelis J. Imaging Sites of N-WASP Activity in Lamellipodia and Invadopodia of Carcinoma Cells. Current Biology. 2004;14:697. doi: 10.1016/j.cub.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 23.Sarmiento C, Wang W, Dovas A, et al. WASP family members and formin proteins coordinate regulation of cell protrusions in carcinoma cells. J Cell Biol. 2008;180:1245–1260. doi: 10.1083/jcb.200708123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440–4449. doi: 10.1038/sj.onc.1202827. [DOI] [PubMed] [Google Scholar]
- 25.Clark ES, Weaver AM. A new role for cortactin in invadopodia: Regulation of protease secretion. Eur J Cell Biol. 2008 doi: 10.1016/j.ejcb.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desai B, Ma T, Chellaiah MA. Invadopodia and matrix degradation, a new property of prostate cancer cells during migration and invasion. J Biol Chem. 2008;283:13856–13866. doi: 10.1074/jbc.M709401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coopman PJ, Do MT, Thompson EW, Mueller SC. Phagocytosis of cross-linked gelatin matrix by human breast carcinoma cells correlates with their invasive capacity. Clin Cancer Res. 1998;4:507–515. [PubMed] [Google Scholar]
- 28.Yamaguchi H, Lorenz M, Kempiak S, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. %R 101083/jcb200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torres E, Rosen MK. Protein-tyrosine kinase and GTPase signals cooperate to phosphorylate and activate Wiskott-Aldrich syndrome protein (WASP)/neuronal WASP. J Biol Chem. 2006;281:3513–3520. doi: 10.1074/jbc.M509416200. [DOI] [PubMed] [Google Scholar]
- 30.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 31.Giuliani C, Troglio F, Bai Z, et al. Requirements for F-BAR proteins TOCA-1 and TOCA-2 in actin dynamics and membrane trafficking during Caenorhabditis elegans oocyte growth and embryonic epidermal morphogenesis. PLoS Genet. 2009;5:e1000675. doi: 10.1371/journal.pgen.1000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J, Troglio F, Mukhopadhyay A, et al. F-BAR-containing adaptor CIP4 localizes to early endosomes and regulates Epidermal Growth Factor Receptor trafficking and downregulation. Cell Signal. 2009;21:1686–1697. doi: 10.1016/j.cellsig.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Greeson JN, Organ LE, Pereira FA, Raphael RM. Assessment of prestin self-association using fluorescence resonance energy transfer. Brain Res. 2006;1091:140–150. doi: 10.1016/j.brainres.2006.02.084. [DOI] [PubMed] [Google Scholar]
- 34.Karpova TS, Baumann CT, He L, et al. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J Microsc. 2003;209:56–70. doi: 10.1046/j.1365-2818.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- 35.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irvin WJ, Jr, Carey LA. What is triple-negative breast cancer? Eur J Cancer. 2008;44:2799–2805. doi: 10.1016/j.ejca.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 37.El-Sibai M, Nalbant P, Pang H, et al. Cdc42 is required for EGF-stimulated protrusion and motility in MTLn3 carcinoma cells. J Cell Sci. 2007;120:3465–3474. doi: 10.1242/jcs.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward ME, Wu JY, Rao Y. Visualization of spatially and temporally regulated N-WASP activity during cytoskeletal reorganization in living cells. Proc Natl Acad Sci U S A. 2004;101:970–974. doi: 10.1073/pnas.0306258101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cory GO, Garg R, Cramer R, Ridley AJ. Phosphorylation of tyrosine 291 enhances the ability of WASp to stimulate actin polymerization and filopodium formation. Wiskott-Aldrich Syndrome protein. J Biol Chem. 2002;277:45115–45121. doi: 10.1074/jbc.M203346200. [DOI] [PubMed] [Google Scholar]
- 40.Park H, Cox D. Cdc42 regulates Fc gamma receptor-mediated phagocytosis through the activation and phosphorylation of Wiskott-Aldrich syndrome protein (WASP) and neural-WASP. Mol Biol Cell. 2009;20:4500–4508. doi: 10.1091/mbc.E09-03-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 2009;28:137–149. doi: 10.1007/s10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- 42.Desmarais V, Yamaguchi H, Oser M, et al. N-WASP and cortactin are involved in invadopodium-dependent chemotaxis to EGF in breast tumor cells. Cell Motil Cytoskeleton. 2009;66:303–316. doi: 10.1002/cm.20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pichot CS, Hartig SM, Xia L, et al. Dasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cells. Br J Cancer. 2009;101:38–47. doi: 10.1038/sj.bjc.6605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 45.Horner M, Ries L, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2006. Bethesda, MD: National Cancer Institute; 2009. [Google Scholar]
- 46.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magkou C, Nakopoulou L, Zoubouli C, et al. Expression of the epidermal growth factor receptor (EGFR) and the phosphorylated EGFR in invasive breast carcinomas. Breast Cancer Res. 2008;10:R49. doi: 10.1186/bcr2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 49.Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379–386. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho HY, Rohatgi R, Lebensohn AM, et al. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 51.Merrifield CJ, Perrais D, Zenisek D. Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells. Cell. 2005;121:593–606. doi: 10.1016/j.cell.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 52.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Kakimoto T, Katoh H, Negishi M. Regulation of neuronal morphology by Toca-1, an F-BAR/EFC protein that induces plasma membrane invagination. J Biol Chem. 2006;281:29042–29053. doi: 10.1074/jbc.M604025200. [DOI] [PubMed] [Google Scholar]
- 54.Takano K, Toyooka K, Suetsugu S. EFC/F-BAR proteins and the N-WASP-WIP complex induce membrane curvature-dependent actin polymerization. Embo J. 2008;27:2817–2828. doi: 10.1038/emboj.2008.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toguchi M, Richnau N, Ruusala A, Aspenstrom P. Members of the CIP4 family of proteins participate in the regulation of platelet-derived growth factor receptor-beta-dependent actin reorganization and migration. Biol Cell. 2010;102:215–230. doi: 10.1042/BC20090033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.