

Abstract
2,4(1H)-Diarylimidazoles have been previously shown to inhibit hNaV1.2 sodium (Na) channel currents. Since many of the clinically used anticonvulsants are known to inhibit Na channels as an important mechanism of their action, these compounds were tested in two acute rodent seizure models for anticonvulsant activity (MES and scMet) and for sedative and ataxic side effects. Compounds exhibiting antiepileptic activity were further tested to establish a dose response curve (ED50). The experimental data identified four compounds with anticonvulsant activity in the MES acute seizure rodent model (compound 10, ED50 = 61.7 mg/kg; compound 13, ED50 = 46.8 mg/kg, compound 17, ED50 = 129.5 mg/kg and compound 20, ED50 = 136.7 mg/kg). Protective indexes (PI = TD50/ED50) ranged from 2.1 (compound 10) to greater than 3.6 (compounds 13, 17 and 20). All four compounds were shown to inhibit hNaV1.2 in a dose dependant manner. Even if a correlation between sodium channel inhibition and anticonvulsant activity was unclear, these studies identify four Na channel antagonists with anticonvulsant activity, providing evidence that these derivatives could be potential drug candidates for development as safe, new and effective antiepileptic drugs (AEDs).
Keywords: sodium channel blockers, epilepsy, seizure models, hNaV1.2
1. Introduction
Epilepsy is a chronic neurological disorder that affects 0.5–1.0% of the population. It is characterized by recurrent unprovoked seizures that can involve both brain hemispheres (generalized) or localized region of the brain (partial). There are many different types of seizures, that depend on which part and how much of the brain is affected by the electrical disturbance. At the present time there is no cure for epilepsy; current treatment options involve seizure suppression through invasive surgery, direct or indirect electrical stimulation, and/or the use of a myriad of antiepileptic drugs (AEDs). Unfortunately, even with optimal treatment paradigms at least 30% of patients continue to experience seizures and many other patients suffer from medication induced side effects.1,2 Thus, there is an urgent need for more effective and better tolerated AEDs. Seizure disorders come with very high associated costs to society in terms of lost wages, productivity and patient care. This provides an important rationale for the continued search for improved therapeutic targets and the design and development of novel ligands for those targets. Due to their fundamental role in establishing and regulating the excitability of neurons within the central nervous system (CNS), voltage-gated sodium channels (VGSCs) are of great importance for the suppression of seizures.3 In fact, many clinically used AEDs are known to target Na channels as an important mechanism of their action.4 In the mammalian brain, the fast, transient Na current responsible for generating the action potential is mediated by NaV1.1, NaV1.2, NaV1.3 and NaV1.6 Na channel isoforms. These channels also mediate the persistent, resurgent, or late Na current, which may play a significant role in epilepsy.5-8 In particular, the NaV1.2 isoform is important for drug targeting since it is abundantly expressed within the CNS with specific expression along the axon initial segment, an area important for the generation of action potentials.9 Furthermore, mutations in SCN2A, the gene encoding the Nav1.2 isoform, has been identified in patients with generalized epilepsy10 and many commercial AEDs, including lamotrigine, phenytoin, carbamazepine and oxcarbazepine, are sodium channel antagonists and have been shown to inhibit NaV1.2 Na channel currents (Figure 1).5,11–15
Figure 1.

Na channel blockers.
Recent studies from our group have identified analogues of 2,4(1H)-diarylimidazoles as being potent Na channel antagonists with greater inhibition of hNaV1.2 sodium channel currents than lamotrigine and phenytoin, two clinically used AEDs. A number of these analogues had IC50 values within the nanomolar-micromolar range.16,17 To determine if these novel Na channel antagonists exhibited anticonvulsant activity the molecules were tested in two rodent acute seizure models (MES: Maximal Electroshock Seizure test and scMet: subcutaneous Metrazol Seizure test). Compounds were also tested for sedative and ataxic side effects.
2. Results and Discussion
2.1 Chemical Synthesis
The compounds reported in Table 1 (3–29) were prepared through parallel synthesis, starting from the appropriate phenylglyoxal 1, benzaldehyde 2, ammonium acetate as ammonia source and a polar protic solvent, such as methanol at room temperature, to afford the best yields of 2,4(1H)-diarylimidazoles 3–29 (Scheme 1).16,17,18
Table 1.
Anticonvulsant Activity and Toxicity of Compounds 3–29 Administered Intraperitoneally to Mice
| Comp. | Structure | Dose | MES(ip)a | ScMetb | Toxc | |||
|---|---|---|---|---|---|---|---|---|
| mg/kg | 0.5hd | 4hd | 0.5hd | 4hd | 0.5hd | 4hd | ||
| 3 |  |
mg/kg | 100–300 | >300 | >300 | >300 | 100–300 | >300 |
| 4 |  |
mg/kg | 100–300 | >300 | >300 | >300 | 100–300 | >100 |
| 5 |  |
mg/kg | 100–300 | 100–300 | >300 | >300 | 100–300 | 100–300 |
| 6 |  |
mg/kg | >300 | >300 | >300 | >300 | >300 | >300 |
| 7 | 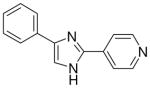 |
mg/kg | 100–300 | 100–300 | >300 | >300 | 100–300 | 100–300 |
| 8 | 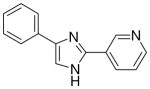 |
mg/kg | 100–300 | 100–300 | 100–300 | >300 | 100–300 | 100–300 |
| 9 | 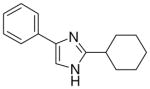 |
mg/kg | >300 | >300 | >300 | >300 | 100–300 | >300 |
| 10 | 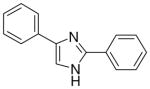 |
mg/kg | >30e | 100–300 | >300 | >300 | 100–300 | 100–300 |
| 11 | 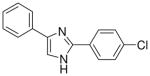 |
mg/kg | >300 | >300 | >300 | >300 | >300 | >300 |
| 12 | 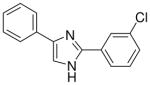 |
mg/kg | 100–300 | 100–300 | 100–300 | >300 | 100–300 | >300 |
| 13 | 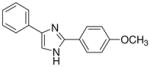 |
mg/kg | >30e | 100–300 | 100–300 | >300 | 100–300 | 100–300 |
| 14 | 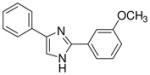 |
mg/kg | >30e | 100–300 | >300 | >300 | >30e | >300 |
| 15 | 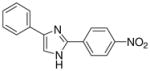 |
mg/kg | 100–300 | 100–300 | >300 | 100–300 | >300 | >300 |
| 16 | 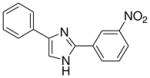 |
mg/kg | 100–300 | 100–300 | >300 | >300 | >300 | >300 |
| 17 | 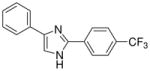 |
mg/kg | 100–300 | 100–300 | >300 | >300 | >300 | >300 |
| 18 | 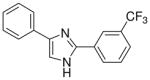 |
mg/kg | >300 | 100–300 | >300 | >300 | 100–300 | >300 |
| 19 | 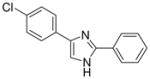 |
mg/kg | >30e | 100–300 | >300 | >300 | 100–300 | >300 |
| 20 | 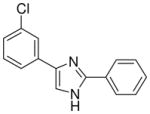 |
mg/kg | >30e | 100–300 | >300 | >300 | 100–300 | >300 |
| 21 | 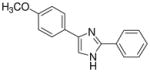 |
mg/kg | >30e | 100–300 | 100–300 | >300 | 100–300 | 100–300 |
| 22 | 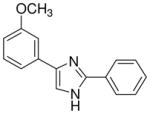 |
mg/kg | >30e | >300 | >300 | >300 | 100–300 | >300 |
| 23 | 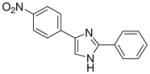 |
mg/kg | 100–300 | 100–300 | >300 | >300 | >300 | >300 |
| 24 | 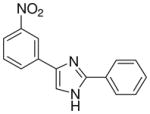 |
mg/kg | 100–300 | 100–300 | >300 | >300 | >300 | >300 |
| 25 | 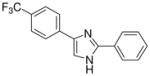 |
mg/kg | 100–300 | >300 | >300 | 100–300 | 100–300 | 100–300 |
| 26 | 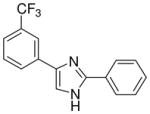 |
mg/kg | >30e | 100–300 | >300 | >300 | 100–300 | 100–300 |
| 27 |  |
mg/kg | >300 | >300 | >300 | >300 | >300 | >300 |
| 28 |  |
mg/kg | >300 | 100–300 | >300 | >300 | >300 | >300 |
| 29 |  |
mg/kg | >300 | 100–300 | >300 | >300 | >300 | >300 |
Maximal electroshock test (administered intraperitoneally to mice at doses ranging from 30 to 300 mg/Kg).
Subcutaneous metrazol test (administered intraperitoneally to mice at doses ranging from 30 to 300 mg/Kg).
Neurotoxicity (administered intraperitoneally to mice at doses ranging from 30 to 300 mg/Kg; no neurotoxicity detected for oral administration to rats).
Time of test after drug administration. Nd: Not determined.
MES activities > 30 means that these compounds show activities between 30–100 mg/kg.
Scheme 1.

Synthesis of the 2,4(1H)-diarylimidazoles 3–29
Reagents and Conditions: 1 equiv. 1 in 3.8 mL CH3OH; 1 equiv. 2 and 4 equiv. CH3COONH4 in 3.5 mL CH3OH. Overnight at r.t..
2.2 Anticonvulsant Screening
The biological evaluation of the 2,4(1H)-diarylimidazoles 3–29 was performed following the procedures proposed by the National Institute of Neurological Disorders and Stroke (NINDS), via the Anticonvulsant Screening Program (ASP). The primary qualitative evaluations performed in mice involved two convulsant tests (MES: Maximal Electroshock Seizure test and scMet: subcutaneous Metrazol Seizure test) along with an assessment of toxicity using the rotarod test. In the initial screening, candidate compounds were administered via intraperitoneal (i.p.) route. It is generally accepted that the MES model, which uses an electrical stimulus, induces generalized tonic-clonic seizures. Through electrical induction, it is used to help identify those compounds which prevent seizure spread. The scMet is a model that induces myoclonic seizures by chemical induction. It helps in identifying those compounds that might act by raising seizure threshold. Toxicity induced by the compounds under study is detected through the standardized rotorod test.19 In this model, a compound is administered i.p. at doses of 30, 100 and 300 mg/kg and groups of mice are tested at different time points (i.e. 0.5 and 4 hours) post administration of the test candidate. A subset of the active and non-toxic candidate compounds obtained were administered orally to rats. In these assessments quantitative evaluation of the anticonvulsant activity, expressed as an ED50 were performed. Quantitative measures included taking groups of 8 animals and administering various doses of test compound until at least two points are established between the limit of 100% and 0% of either protection or minimal toxicity. Details of the evaluation of anticonvulsant activity and toxicity are given in the Experimental Section.
The data regarding the primary screening of all the 2,4(1H)-diarylimidazoles 3–29 are reported in Table 1 (see also Supplementary Data – Table 1) and they represent a qualitative evaluation of the anticonvulsant activity of the compounds under study at different doses and times. Compounds were designed to determine the effects of:
having different rings connected to the imidazole central core, with the aim to understand the best substitution and to find an interesting lead compound to further modify (3–10);
having different substituents in meta or para position of both the phenyl rings of the diphenylderivative 10, with the aim to vary the physico-chemical properties (ionization, lipophilicity) of the molecules.
The first set of molecules tested (3–10), having different rings connected to the imidazole core, showed anticonvulsant activity at 100 mg/kg (except compound 6) after intraperitoneally administration to mice (Table 1). From these set of compounds we identified two lead compounds (8 and 10) that showed efficacy and low toxicity. Due to the good safety margins found, compounds 8 and 10 were selected for further studies, to determine their activity and toxicity after oral administration to rats. Results from testing in the MES test, at 30 mg/kg are seen in Table 2 (also see Supplementary Data – Table 2). Both the derivatives tested were able to protect animals against generalized tonic-clonic seizures, but only compound 10 showed a linear decrease of the activity, retaining potency across species while keeping low toxicity. The second set of derivatives (11–29), are characterized by the presence of two substituted-phenyl rings in C2 and C4 of the central heterocycle. Some protection was seen for compounds 12, 15, 23, 25, 26 and 29 in the MES test in mice at a concentration of 100 mg/kg dose (Table 1). In contrast, compounds 11, 17, 18, 27 and 28 were inactive. All the derivatives from this chemical series showed little or no capacity to raise the seizure threshold level induced by subcutaneous administration of the chemoconvulsant metrazole. The remaining compounds presented in Table 1 showed good protection at the 100 mg/kg dose and most were devoid of toxicity in the rotorod at the dose tested. The introduction of a methoxy group (compounds 13, 14, 21 and 22) afforded derivatives with comparable activity to compound 10, at 100 mg/kg, even if the p-substitution seems to increase the safety margins. The chloro substitution led to compounds with slightly different behaviour, but all characterized by a very low toxicity: derivatives 19 and 20, having a chlorine on the phenyl ring in the 4 position of the imidazole, demonstrated good activity in mice at 100 mg/kg (protection in 3/3 animals tested at 0.5 h), while the introduction of the halogen on the other phenyl ring (11 and 12) was detrimental for the protection against electrical induced seizures in mice. The nitro-substitution afforded good protection, especially for the m-derivatives 16 and 24 (protection in 2/3 animals tested at 0.5 h). Finally, the introduction of the trifluoromethyl substituent led to a decrease of potency (17, 18, 25, 26, 28 and 29).
Table 2.
Anticonvulsant Activity and Toxicity of Compounds 8, 10, 13, 16, 17, 19–22, 24 Administered Orally (30 mg/kg) to Rats
| Compd | Structure | MESa | TOXb | ||||
|---|---|---|---|---|---|---|---|
| 0.25hc | 0.5hc | 1hc | 2hc | 4hc | |||
| 8 |  |
1/4 | 0/4 | 1/4 | 0/4 | 0/4 | 0/4 (−)d |
| 10 |  |
2/4 | 2/4 | 1/4 | 0/4 | 0/4 | 0/4 (−)d |
| 13 |  |
2/4 | 0/4 | 3/4 | 2/4 | 0/4 | 0/4 (−)d |
| 16 |  |
0/4 | 0/4 | 1/4 | 0/4 | 1/4 | 0/4(−)d |
| 17 |  |
0/4 | 1/4 | 2/4 | 1/4 | 2/4 | 0/4(−)d |
| 19 | 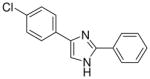 |
0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 (−)d |
| 20 | 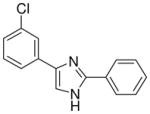 |
0/4 | 0/4 | 2/4 | 0/4 | 0/4 | 0/4 (−)d |
| 21 | 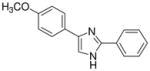 |
0/4 | 0/4 | 0/4 | 0/4 | 1/4 | 0/4 (−)d |
| 22 | 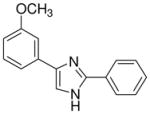 |
1/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 (−)d |
| 24 | 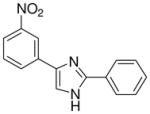 |
0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 (−)d |
Maximal electroshock test (number of animals protected/number of animals tested).
Neurotoxicity (number of animals protected/number of animals tested).
Time after drug administration.
(−) No neurotoxicity at doses tested.
From this initial screen we identified ten compounds that were further evaluated for oral availability using the MES acute seizure model in rats (Table 2). As previously reported, compounds 8 and 10, belonging to the first set, were also selected for evaluation of their anticonvulsant activity and toxicity after oral administration in rats (30 mg/kg), in a MES test (Table 2). This study was also conducted on the most promising molecules of the second set, such as the methoxy derivatives (13, 21 and 22), the chloro-substituted compounds on the phenyl in C4 of the imidazole (19 and 20) and the m-nitro derivatives (16 and 24). Compound 17 was added to the group because of its interesting behaviour at shorter time periods after i.p. administration in rats (see Supplementary Data Table 1: 2/3 at 0.25 h), whereas the active methoxy 14 was excluded due to its toxicity. Table 2 showed as the compounds characterized by the presence of a substituent on the phenyl ring in the 4 position of the central heterocycle (19, 21, 22 and 24) were not able to prevent seizure spread in the MES test after oral administration, and only the m-chloro derivative 20 demonstrated a marginal protection (2/4) 1 h after administration. A different behaviour was observed for the other compounds, where the anticonvulsant activity was retained across species, without any toxic effect and, once again, the p-methoxy derivative 13 demonstrated to be the most promising compound synthesized, having good protection at 0.25 h (2/4), 1 h (3/4) and 2 h (2/4), with no toxicity at all (Table 2).
From this second set of seizure tests we identified four compounds that were selected for a further quantitative in vivo anticonvulsant and toxicity evaluation to establish both effective (ED50) or lethal (TD50) doses (Table 3). For the unsubstituted diarylimidazole 10 separation between effective and toxic doses for the MES model were observed and a protective index (PI) of 2.1 was obtained. The separation was greater for the methoxy 13 which had a PI of 4.3. The largest difference between the effective and the toxic doses were observed for compounds 13, 17 and 20. The trifluoromethyl and the chloro compounds (17 and 20) had similar ED50 values around 130 mg/kg, but no toxicity was observed when tested up to 500 mg/kg, resulting in a PI value of at least 3.6.
Table 3.
Quantitative anticonvulsant activity (MES and scMet), toxicity in mice (i.p.; tested at 0.25h) and IC50 values for inhibition of hNaV1.2 Na currents of compounds 10, 13, 17 and 20
| Compd | MESa ED50b | PIc | scMetd ED50b | PIe | TD50f,b | hNav1.2 IC50 (μM) |
|---|---|---|---|---|---|---|
| 10 | 61.7g (51.9–71.4) | 2.1 | 160.9 (138.6–187.0) | 0.8 | 126.8 (107.6–140.2) | 40.6 |
| 13 | 46.8 (44.0–52.0) | 4.3 | 142.2 (103.6–203.4) | 1.4 | 200.8 (163.0–243.6) | 21.4 |
| 17i | 129.5 (101.5–163.3) | >3.8 | >300j | / | >500 | 10.1 |
| 20 | 136.7 (112.3–159.9) | >3.6 | >250 | / | >500 | 17.9 |
| Phenytoin21,22 | 9.5 (8.1–10.4) | 6.9 | / | / | 65.5 | >100 |
Maximal electroshock test.
The interval in parentheses stands for the 95% confidence interval.
Protective index (PI = TD50/ED50) in the MES test.
Subcutaneous metrazol test.
Protective index (PI = TD50/ED50) in the scMet test.
Neurotoxicity.
Dose response curves were generated for compounds 10, 13, 17 and 20 (Table 3, Figure 2 and also see Supplementary Data – Table 3) for inhibition of hNav1.2 Na channel currents. Example traces are shown in Figure 2. All four of the lead compounds inhibited hNav1.2 sodium channel currents, although a direct correlation between potency against hNav1.2 and anticonvulsant activity was difficult to make. For example, phenytoin did not exhibit great potency against hNav1.2, having an IC50 value greater than 100 μM and had a low TD50 value of 65.5. However phenytoin was very effective as an anticonvulsant with a PI of 6.9. In contrast, compound 17 had a 4 fold greater potency for hNav1.2 (IC50 = 10.1 μM) than phenytoin and also had no toxicity when tested up to 500 mg/kg. These findings suggest our lead compounds may be targeting other ion channel targets in addition to sodium channels or differentially targeting specific sodium channel gating modes such as the slow inactivation mode known to be important for AED activity and possibly better toxicity profiles.20
Figure 2.

Inhibition of hNav1.2 Na channel currents by active compounds. Na channel currents shown were elicited by a step depolarization to +10 mV from a holding potential of −60 mV for 12 ms.
In conclusion, this work demonstrates that our novel compounds have activity against the hNaV1.2 sodium channel isoform and also provide protection against seizures induced in the MES acute seizure model. Nevertheless, a direct correlation between sodium channel inhibition and anticonvulsant activity was unclear.
An initial qualitative evaluation for anticonvulsant activity of the compounds, after i.p. and oral administration, has provided evidence in the identification of a series possessing a novel anticonvulsant structure, characterized by the presence of two phenyl rings connected to an imidazole central core. Some of these molecules showed good evidence for inhibiting seizures (e.g compound 10 with an ED50 of 61.7 mg/kg and a PI of 2.1). Modifications of the basic structure resulted in the design and synthesis of the other potentially useful molecules (2,4(1H)-diarylimidazoles 13, 17 and 20). These compounds afforded protection in an acute model of generalized tonic-clonic seizures (MES; ED50 values of 46.8 mg/kg for 13, 129.5 mg/kg for 17 and 136.7 mg/kg for 20), with favourable side effects profiles (TD50 value of 200.8 mg/kg for 13 and >500 mg/kg for 17 and 20, administered i.p. in mice). These characteristics are encouraging, considering that phenytoin, a major clinically used AED, had similar actions on hNaV1.2 current at 10 μM (10.6% vs 30.1% of 13, 22.7% of 17 and 16.8% of 20)15 and had similar calculated PI value (6.9 vs 4.3 for 13 and greater than 3.6 for 17 and 20).
These results indicate the existence of promising anticonvulsants that with further optimization may provide advances in efficacy and side effect profiles. The introduction of substituents as Cl or CF3, even if on different position or on a different phenyl ring connected to the imidazole (17 and 20) led to compounds that were devoid of toxicity at the doses evaluated. But also the unsubstituted compound (10) and the molecule characterized by the presence of a methoxy group in the para position of the phenyl ring in C2 (13) showed interesting ED50, but the PI values were lower that those of the first two derivatives considered. It is likely these effects could be significantly improved with intensive structural optimization techniques. Even if a correlation between sodium channel inhibition and anticonvulsant activity was unclear, as it is also difficult to establish clear structure-activity relationships, the diarylimidazoles here presented can be considered as new prototype of anticonvulsant drugs by virtue of the favorable results obtained in the in vivo tests. It is justified to claim that the correct structural modifications made on these molecules could lead to new derivatives particularly useful for the treatment of epilepsy.
3. Experimental Section
3.1 General procedure for the synthesis of 2,4(1H)-diarylimidazoles 3–29
A solution of phenylglyoxal monohydrate (0.70 mmol) in methanol (3.8 mL) was added dropwise to a stirred suspension of arylaldehyde (0.70 mmol) and ammomium acetate (3.41 mmol) in methanol (3.5 mL). The reaction mixture was stirred overnight at room temperature, then the solvent was evaporated and the residue was partitioned between saturated aqueous NaHCO3 solution (20 mL) and methylene chloride (20 mL). The organic phase was dried over Na2SO4 and the solvent was removed in vacuo. The isolation of the target compounds from the crude reaction mixture was obtained using SCX-2 column (2g, 30–90 μm, loading 0.4 meq/g). The column was prewashed with DCM:methanol=1:1 (10 mL), the side products were eluted with methanol (10 mL) and then the desired 2,4(5)-arylimidazoles were eluted with a methanolic ammonia 5% w/w solution (10 mL). All the products were then crystallized as the hydrochloride salt from absolute ethanol/diethyl ether.
The characterization for purity for compounds 10–29 was previously described.17
3.1.1 3-(4(5)-phenylimidazol-2-yl)thiophene (3)
78% yield, mp. 276–277°C. 1H NMR (300 MHz, DMSO-d6): δ 7.44 (t, J = 7.5, 1H), 7.53 (t, J = 6.6, 2H), 7.85–7.88 (dd, J = 7.2, J = 3.0, 1H), 7.98 (bs, 3H), 8.20 (s, 1H), 8.73 (bs, 1H). MS (EI) 227 [M+]. Anal. Calcd for C13H10N2S·HCl·0.5H2O: C, 57.45; H, 4.45; N, 10.31. Found: C, 57.12; H, 4.52; N, 9.94.
3.1.2 3-(4(5)-phenylimidazol-2-yl)furan (4)
75% yield, mp. 275–276°C. 1H NMR (300 MHz, DMSO-d6): δ 7.37 (bs, 1H), 7.43 (t, J = 8.1, 1H), 7.53 (t, J = 8.1, 2H), 7.97 (bs, 3H), 8.20 (s, 1H), 8.81 (s, 1H). MS (EI) 211 [M+]. Anal. Calcd for C13H10N2O·HCl: C, 63.40; H, 4.50; N, 11.38. Found: C, 63.18; H, 4.48; N, 11.26.
3.1.3 2-(4(5)-phenylimidazol-2-yl)furan (5)
82% yield, mp. 254–255°C. 1H NMR (300 MHz, DMSO-d6): δ 6.87 (bs, 1H), 7.42 (t, J = 7.2, 1H), 7.51 (t, J = 6.6, 2H), 7.81 (d, J = 3.6, 1H), 8.00 (d, J = 7.2, 2H), 8.12 (s, 1H), 8.20 (s, 1H). MS (EI) 211 [M+]. Anal. Calcd for C13H10N2O·HCl·0.5H2O: C, 61.06; H, 4.73; N, 10.95. Found: C, 61.46; H, 4.73; N, 10.95.
3.1.4 2-(4(5)-phenylimidazol-2-yl)benzofuran (6)
81% yield, mp. 268–272°C. 1H NMR (300 MHz, DMSO-d6): δ 7.41 (bs, 2H), 7.48–7.53 (bs, 3H), 7.74 (d, J = 8.1, 1H), 7.85 (d, J = 7.5, 1H), 7.98 (bs, 3H), 8.20 (s, 1H). MS (EI) 261 [M+]. Anal. Calcd for C17H12N2O·HCl·H2O: C, 64.87; H, 4.80; N, 8.90. Found: C, 65.26; H, 4.60; N, 8.83.
3.1.5 4-(4(5)-phenylimidazol-2-yl)pyridine (7)
77% yield, mp. 237-241°C (dec.). 1H NMR (300 MHz, DMSO-d6): δ 7.35 (t, J = 7.4, 1H), 7.47 (t, J = 7.7, 2H), 8.01 (d, J = 7.4, 2H), 8.22 (s, 1H), 8.68 (d, J = 6.8, 2H), 8.96 (d, J = 6.8, 2H). MS (EI) 222 [M+]. Anal. Calcd for C14H11N3·2HCl·0.5H2O: C, 55.62; H, 4.67; N, 13.90. Found: C, 55.53; H, 5.03; N, 13.95.
3.1.6 3-(4(5)-phenylimidazol-2-yl)pyridine (8)
71% yield, mp. 260–264°C (dec.). 1H NMR (300 MHz, DMSO-d6): δ 7.45 (t, J = 7.4, 1H), 7.55 (t, J = 7.8, 2H), 7.80 (dd, J = 7.8, J = 5.77, 1H), 8.01 (d, J = 7.4, 2H), 8.31 (s, 1H), 8.74 (d, J = 8.5, 1H), 8.84 (dd, J = 7.0, J = 1.4, 1H), 9.42 (d, J = 1.8, 1H). MS (EI) 222 [M+]. Anal. Calcd for C14H11N3·2HCl: C, 57.16; H, 4.45; N, 14.28. Found: C, 57.14; H, 4.45; N, 14.24.
3.1.7 2-cyclohexyl-4(5)-phenylimidazole (9)
88% yield, mp. 154–155°C. 1H NMR (300 MHz, DMSO-d6): δ 1.21–1.40 (m, 3H), 1.63–1.72 (m, 3H), 1.76–1.83 (m, 2H), 1.99 (d, J = 10.2, 2H), 3.10 (tt, J = 12.0, J = 3.6, 1H), 7.40 (t, J = 7.5, 1H), 7.49 (t, J = 7.5, 2H), 7.89 (d, J = 7.5, 2H), 8.02 (s, 1H). MS (EI) 227 [M+]. Anal. Calcd for C15H18N2·HCl·H2O: C, 64.16; H, 7.54; N, 9.97. Found: C, 64.55; H, 7.16; N, 9.99.
3.2 Sodium channel electrophysiology
Human embryonic kidney cells (HEK) cells stably expressing human Nav1.2 were a kind gift from Dr. H.A. Hartmann (University of Baltimore, Maryland, USA) and were grown in DMEM/F12 media (Invitrogen, Corp, CA, USA) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml) and G418 (500 μg/ml; Sigma, MO, USA). Cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37 °C.
Sodium currents were recorded using the whole-cell configuration of the patch clamp recording technique with an Axopatch 200 amplifier (Molecular Devices, USA). All voltage protocols were applied using pCLAMP 9 software and a Digidata 1322A (Molecular Devices, USA). Currents were amplified and low pass filtered (2 kHz) and sampled at 33 kHz. Borosilicate glass pipettes were pulled using a Brown-Flaming puller (model P87, Sutter Instruments Co, Novato, CA) and heat polished to produce electrode resistances of 0.5–1.5 MΩ when filled with the following electrode solution (in mM); CsCl 130, MgCl2 1, MgATP 5, BAPTA 10, HEPES 5 (pH adjusted to 7.4 with CsOH). Cells were plated on glass coverslips and superfused with solution containing the following composition; (in mM) NaCl 130, KCl 4, CaCl2 1, MgCl2 5, HEPES 5, and glucose 5 (pH adjusted to 7.4 with NaOH).
Compounds were prepared as 10 mM stock solutions in dimethly sulfoxide (DMSO) and diluted to desired concentration in perfusion solution. The maximum DMSO concentration used had no effect on current amplitude. All experiments were performed at room temperature (20–22 °C). After establishing whole-cell, a minimum series resistance compensation of 75% was applied. Sodium currents were elicited by a depolarizing step from a holding potential of −100 mV to +10 mV for a duration of 12 ms at 15 s intervals. Test compounds were applied after a 3 min control period and continued until a steady state current amplitude was observed. Tonic block was assessed by comparing peak sodium current in drug free conditions to peak current when drug was present. Dose response data were fitted using the Hill equation:
Where C is the drug concentration, IC50 is the concentration that blocks 50% of the current and H is the hill coefficient. All data represent percentage mean block ± standard error of the mean (S.E.M.).
3.3 Description of animal testing methods for MES, ScMet and toxicity evaluations
Animals utilized in these experiments were either male albino CF1 mice (18–25 g, Charles River, Portage, MI) or male albino Sprague-Dawley rats (100–150 g; Charles River, Raleigh, NC) experimental animals. All animals were allowed free access to both food (Prolab RMH 3000) and water except when they were removed from their cages for testing. Animals were used only once. All rats and mice were housed, fed, and handled in a manner consistent with the recommendations in the National Research Council Publication, “Guide for the Care and Use of Laboratory Animals”. No insecticides capable of altering hepatic drug metabolism enzymes were used in the animal facilities. All animals were euthanized in accordance with the Institute of Laboratory Resources policies on the humane care of laboratory animals. Compounds were administered in 0.5% methylcellulose either orally (rat) or intraperitoneally (mice) (p.o. or i.p.) in a volume of 0.01 ml/g body weight in mice and 0.04 ml/10 g body weight in rats. In vivo anticonvulsant activity was established using both an electrical and chemoconvulsant seizure test which have been described previously. The electrical tests used were the maximal electroshock (MES) seizure test and the s.c.Met as the chemical test.
3.3.1 MES Test
The MES is a model for generalized tonic-clonic seizures and provides an indication of a compound’s ability to prevent seizure spread when all neuronal circuits in the brain are maximally active. These seizures are highly reproducible and are electrophysiologically consistent with human seizures. For the MES, a drop of anesthetic/electrolyte solution (0.5% tetracaine hydrochloride in 0.9% saline) was applied to the eyes of each animal prior to placement of the corneal electrodes. The electrical stimulus in the MES test was 50 mA, 60 Hz, for mice and 150 mA, 60 Hz, for rats delivered for 0.2 sec by an apparatus similar to that originally described by Woodbury and Davenport.24–27 Abolition of the hindleg tonic extensor component of the seizure was used as the endpoint. Mice are initially tested at various intervals following doses of 30, 100 and 300 mg/kg of test compound given by i.p. injection while rats are initially screened at a fixed dose of 30 mg/kg.
3.3.2 Subcutaneous Metrazol Seizure Threshold Test (scMET)
Subcutaneous injection of the convulsant Metrazol produces clonic seizures in laboratory animals. The scMET test detects the ability of a test compound to raise the seizure threshold of an animal and thus protect it from exhibiting a clonic seizure. Animals are pretreated with various doses of the test compound given by i.p. injection. At the previously determined TPE of the test compound, the dose of Metrazol which will induce convulsions in 97% of animals (CD97: 85 mg/kg mice) is injected into a loose fold of skin in the midline of the neck. The animals are placed in isolation cages to minimize stress28 and observed for the next 30 minutes for the presence or absence of a seizure. An episode of clonic spasms, approximately 3–5 seconds, of the fore and/or hindlimbs, jaws, or vibrissae is taken as the endpoint. Animals which do not meet this criterion are considered protected.
3.3.3 Acute Toxicity—Minimal Motor Impairment
To assess a compound’s undesirable side effects (toxicity), animals are monitored for overt signs of impaired neurological or muscular function. In mice, the rotorod19 procedure is used to disclose minimal muscular (MMI) or neurological impairment (MNI). When a mouse is placed on a rod that rotates at a speed of 6 rpm, the animal can maintain its equilibrium for long periods of time. The animal is considered toxic if it falls off this rotating rod three times during a 1 min period. In addition to MMI, animals may exhibit a circular or zigzag gait, abnormal body posture and spread of the legs, tremors, hyperactivity, lack of exploratory behavior, somnolence, stupor, catalepsy, loss of placing response and changes in muscle tone.
3.3.4 Determination of Median Effective (ED50) or Behavioral Toxic Dose (TD50)
All quantitative in vivo anticonvulsant efficacy/toxicity studies were conducted at the previously determined TPE. Groups of at least eight mice or rats were tested with various doses of the candidate drug until at least two points were established between the limits of 100% protection or minimal toxicity and 0% protection or minimal toxicity. The dose of drug required to produce the desired endpoint in 50% of animals (ED50 or TD50) in each test, the 95% confidence interval, the slope of the regression line, and the S.E.M. of the slope were then calculated by a computer program based on the method described by Finney.29
Supplementary Material
Acknowledgments
Financial support from Italian MIUR, Siena Biotech S.p.A. and the National Institutes of Health NINDS R21NS061069-02 (MKP) is gratefully acknowledged. We are grateful to the Centro Interdipartimentale Misure of the University of Parma for providing the NMR instrumentation.
Footnotes
Supplementary data are associated with this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- 1.Remy S, Beck H. Brain. 2006;129:18–35. doi: 10.1093/brain/awh682. [DOI] [PubMed] [Google Scholar]
- 2.Brodie MJ. Epilepsy Res. 2001;45:3–6. doi: 10.1016/s0920-1211(01)00203-0. [DOI] [PubMed] [Google Scholar]
- 3.Catterall WA. Neuron. 2000;26:13–26. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 4.Large CH, Kalinichev M, Lucas A, Carignani C, Bradford A, Garbati N, Sartori I, Austin NE, Ruffo A, Jones DNC, Alvaro G, Read KD. Epilepsy Research. 2009;85:96–106. doi: 10.1016/j.eplepsyres.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 5.Meldrum BS, Rogawski MA. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogawski MA, Löscher W. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 7.Ketelaars SOM, Gorter JA, van Vliet EA, Lopes da Silva FH, Wadman WJ. Neuroscience. 2001;105:109–120. doi: 10.1016/s0306-4522(01)00176-2. [DOI] [PubMed] [Google Scholar]
- 8.Ellerkmann RK, Remy S, Chen J, Sochivko D, Elger CE, Urban BW, Becker A, Beck H. Neuroscience. 2003;119:323–333. doi: 10.1016/s0306-4522(03)00168-4. [DOI] [PubMed] [Google Scholar]
- 9.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- 10.Meisler HM, Kearney JA. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 12.Yarov-Yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall WA. J Biol Chem. 2001;274:20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- 13.Liu G, Yarov-Yarovoy V, Nobbs M, Clare JJ, Scheuer T, Catterall WA. Neuropharmacology. 2003;44:413–422. doi: 10.1016/s0028-3908(02)00400-8. [DOI] [PubMed] [Google Scholar]
- 14.Taylor CP, Narasimhan SL. Adv Pharmacol. 1997;39:47–98. [PubMed] [Google Scholar]
- 15.Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White S. Epilepsy Research. 2009;83:1–43. doi: 10.1016/j.eplepsyres.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Rivara M, Baheti AR, Fantini M, Cocconcelli G, Ghiron C, Kalmar CL, Singh N, Merrick EC, Patel MK, Zuliani V. Bioorg & Med Chem Lett. 2008;18:5460–5462. doi: 10.1016/j.bmcl.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 17.Fantini M, Rivara M, Zuliani V, Kalmar CL, Vacondio F, Silva C, Baheti AR, Singh N, Merrick EC, Katari RS, Cocconcelli G, Ghiron C, Patel MK. Bioorg & Med Chem. 2009;17:3642–3648. doi: 10.1016/j.bmc.2009.03.067. [DOI] [PubMed] [Google Scholar]
- 18.Zuliani V, Cocconcelli G, Fantini M, Ghiron C, Rivara M. J Org Chem. 2007;72:4551–4553. doi: 10.1021/jo070187d. [DOI] [PubMed] [Google Scholar]
- 19.Dunham MS, Miya TA. J Amer Pharmac Assoc Sci Edit. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- 20.Jones PJ, Merrick EC, Batts TW, Hargus NJ, Wang Y, Stables JP, Bertram EH, Brown ML, Patel MK. J Pharmacol Exp Ther. 2009;328:201–212. doi: 10.1124/jpet.108.144709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Sun XY, Chai KY, Lee JS, Song MS, Quan ZS. Bioorg Med Chem. 2007;15:6775–6781. doi: 10.1016/j.bmc.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Cui XS, Chen J, Chai KY, Lee JS, Quan ZS. Med Chem Res. 2009;18:49–58. [Google Scholar]
- 23.Baker GA, Jacoby A, Buck D, Stalgis C, Monnet D. Epilepsia. 1997;38:353–362. doi: 10.1111/j.1528-1157.1997.tb01128.x. [DOI] [PubMed] [Google Scholar]
- 24.Woodbury LA, Davenport VD. Arch Int Pharmacodyn Ther. 1952;92:97–104. [PubMed] [Google Scholar]
- 25.Swinyard EA, Woodhead JH, White HS, Franklin MR. In: Antiepileptic Drugs. Levy RHM, Melrum B, Penry JK, Dreifuss FE, editors. Raven Press; New York: 1989. pp. 85–102. [Google Scholar]
- 26.White HS, Johnson M, Wolf HH, Kupferberg HJ. Ital J Neurol Sci. 1995;16:73–77. doi: 10.1007/BF02229077. [DOI] [PubMed] [Google Scholar]
- 27.White HS, Woodhead JH, Franklin MR. In: Antiepileptic Drugs. Levy RHM, Melrum B, Penry JK, Dreifuss FE, editors. Raven Press; New York: 1995. pp. 99–110. [Google Scholar]
- 28.Swinyard EA, Clark LD, Miyahara JT, Wolf HH. J Physiol. 1961;132:97–102. [PubMed] [Google Scholar]
- 29.Finney DJ. Probit Analysis. 3. London: Cambridge University Press; 1971. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.