

Abstract
There is increasing evidence that the incidence of Alzheimer's disease (AD) is significantly influenced by cardiovascular risk factors in association with a cluster of metabolic diseases including diabetes and atherosclerosis. The shared risk is also reflected in the dietary and lifestyle links to both metabolic disorders and AD-type cognitive dysfunction. Recent studies with genetic and diet-induced animal models have begun to illuminate convergent mechanisms and mediators between these two categories of disease conditions with distinct tissue-specific pathologies. While it is clear that peripheral inflammation and insulin resistance are central to the pathogenesis of the disorders of metabolic syndrome, it seems that the same mechanisms are also in play across the blood brain barrier (BBB) that lead to AD-like molecular and cognitive changes. This review will highlight these convergent mechanisms and discuss the role of cerebrovascular dysfunction as a conduit to brain emergence of these pathogenic processes that might also represent future therapeutic targets in AD in common with metabolic disorders.
Keywords: Alzheimer's disease, metabolic syndrome, diabetes, atherosclerosis, diet, insulin resistance, inflammation, cerebrovascular dysfunction
“To lipids my brain might succumb
They make synapses slower and numb
I think fatty foods
Are good for my moods
But my brain suffers, they make me... dumb”
-a limerick recently heard on the National Public Radio (NPR) program, ‘Wait Wait... Don't Tell Me’.
The frequent warnings against unhealthy lifestyle rife in the media and elsewhere certainly portray increasing awareness along with real concern (and, rightly so) regarding the widespread epidemic of modern diseases with endocrine, metabolic and cardiovascular underpinnings that are largely attributable to the dual threat of sedentary lifestyle choices and unhealthy food habits. In particular, ‘Westernized’ high caloric diets rich in saturated, trans and omega-6 fatty acids, refined carbohydrates and cholesterol are now known to be the main cause of several metabolic disorders including obesity, type-2 diabetes and atherosclerosis as part of the disease cluster called ‘metabolic syndrome’. Recent findings indicate that our brain might also ‘succumb’ to these lipids and carbohydrates when ingested in excess pointing to convergent mechanisms through which comorbid metabolic disorders may promote brain dysfunction and dementia. One of the convergent mechanisms is inflammation. While systemic inflammation mediated by activated peripheral immune system (along with its byproduct i.e., insulin resistance) defines the primary pathogenic process underlying the metabolic disorders, neuroinflammation may play a parallel role in the progression, if not also the pathogenesis, of a number of neurodegenerative diseases including Alzheimer’ disease (AD). It is also being realized that despite the presence of a physical and functional barrier between brain and the peripheral systems, an active neuro-immune cross talk maintains an important homeostatic role so that any changes in the systemic immune status will have a significant impact on brain pathology and repair (Banks and Erickson 2010; McAllister and van de Water 2009). Although the blood brain barrier (BBB) is minimally permeable (- except in conditions such as stroke and injury and in primary inflammatory diseases such as multiple sclerosis where in the disrupted barrier facilitates macrophage infiltration), there is new evidence for an increased influx of exogenous monocytes into the CNS under conditions of acute (i.e., infection) or chronic peripheral inflammation (Audoy-Remus et al. 2008; Getts et al. 2008; D'Mello et al. 2009). Therefore, it is likely that even the low-grade inflammation due to metabolic disorders or disturbances will have significant influence on the brain. Recent studies with genetic and diet-induced animal models of diabetes and atherosclerosis/hypercholesterolemia have begun to shed light on the convergent mechanisms shared between such metabolic disorders and sporadic AD. A key link may lie in the cerebrovascular dysfunction with altered barrier and transport properties due to ‘metabolic inflammation’ and insulin resistance, the two related processes associated with the components of metabolic syndrome. This review will propose and develop the idea that the same two processes re-emerge across the broken barrier to initiate AD-like molecular and cognitive changes. The intent is not to provide a comprehensive account of all the clinicopathological and epidemiological features of the metabolic syndrome and AD [-which are extensively covered in several cited reviews], but highlight the pathways and mediators that mechanistically link the two conditions as guided by recent progress in the field.
Atherosclerosis, diabetes and the risk of AD
AD is characterized by a progressive cognitive and behavioral decline due to selective loss or dysfunction of neurons in specific brain regions/neural circuits including the neocortex, hippocampus and basal forebrain (Braak et al. 1993; Morrison and Hof 1997). The pathological hallmarks of AD are intracellular neurofibrillary tangles (NFT) consisting of hyperphosphorylated tau and extracellular plaques that contain the amyloidogenic peptide, Aβ (Selkoe 1999; Hardy and Selkoe 2002), a cleaved product of its precursor protein (APP). Genetic mutations in APP or APP cleaving proteolytic enzymes i.e., presenilin (PS)1/2 account for most of the familial cases of AD. However, the vast majority (over 95%) of AD cases fall into the late-onset disease (LOAD) category, within which heterogeneity exists with regard to risk factors, pathogenic and pathological characteristics. Besides age, presence of an apolipoprotein allele, e4 (ApoE4) is a clear risk for LOAD (Roses 1996). Recent genome-wide association studies have identified three new susceptibility loci (CLU, PICALM and CR1) for LOAD that may be involved in Aβ clearance, a role shared by ApoE (Kim et al. 2009b; van Es and van den Berg 2009). As per the amyloid cascade hypothesis, an imbalance between Aβ production and clearance plays a critical role in the progression of all AD (Hardy and Selkoe 2002). It is thought that increased levels of Aβ oligomers cause neuronal injury and synaptic dysfunction/disruption both directly and indirectly via inflammatory and oxidative stress involving an activation of microglia, the brain innate immune cells.
The Apo E4 connection to LOAD is particularly significant in terms of its suspected role in atherosclerosis and other cardiovascular diseases. It should be noted however, that the higher risk of developing AD in carriers of one or two copies of E4 isoforms compared with carriers of other combinations (E2/E3) may or may not depend on the cholesterol transporter function of the protein. Instead, the greater impact of apo E4 on brain dysfunction (-both in AD and vascular dementia) relative to peripheral effects is thought to derive from its pleiotropic functions including amyloid generation and clearance, maintenance of synaptic and cerebrovascular integrity, etc. Nonetheless, a number of findings (genetic, experimental, and epidemiological) suggest a link between hypercholesterolemia and other vascular risk factors and the development of AD (Puglielli et al. 2003; Casserly and Topol 2004; Sambamurti et al. 2004; Cechetto et al. 2008; de la Torre 2009). First, several AD-associated genes, besides ApoE (i.e., cyp46, ABCA1) that show disease-specific polymorphisms, are otherwise normal participants in cholesterol metabolism (Wollmer 2010). Second, clinical studies indicate that middle-aged individuals with increased cholesterol are more susceptible to AD and that elevated levels of low-density lipoprotein (LDL) cholesterol and reduced HDL/apoA-I correlate well with disease incidence compared to asymptomatic cases (Merched et al. 2000; Puglielli et al. 2003). Third, animal studies using New Zealand white rabbits (Sparks et al. 2000) and transgenic mouse models of AD (Refolo et al. 2000; Levin-Allerhand et al. 2002) demonstrate that diet-induced hypercholesterolemia enhances brain Aβ accumulation. Cholesterol has also been shown to directly modulate APP processing in neuronal cell cultures (Sambamurti et al. 2004). Many such, but not all, observations have suggested that cholesterol may play a prominent role in AD pathogenesis and that lowering of it may benefit disease prognosis. In fact, retrospective studies have demonstrated that cholesterol-lowering drugs i.e., statins, could reduce the incidence of AD (Wolozin et al. 2000). However, the statin activity most likely involves mechanisms other than inhibiting cholesterol synthesis, a prominent one being the drug's inhibitory effects on inflammation (Liao and Laufs 2005; Wolozin et al. 2006), now considered a central player in atherosclerosis (Rocha and Libby 2009).
Substantial amount of clinical studies implicates diabetes mellitus, both type 1 (T1DM) and type 2 (T2DM), as risk factor for dementia of both vascular and Alzheimer's type, with T2DM patients predominantly presenting with AD as the most common cause of dementia (Sun and Alkon 2006; Whitmer 2007; Kodl and Seaquist 2008; Luchsinger 2008; Craft 2009; Roriz-Filho et al. 2009), although the higher mortality in T1DM makes such a distinction less clear. Neuropathological features including brain atrophy, white matter hyperintense and microvascular lesions with amyloid plaque burden, as well as increased amyloid and NFT in the hippocampus, all associate with cognitive deficits observed in diabetes patients (Sun and Alkon 2006; Roriz-Filho et al. 2009). Further, diabetic patients, especially the older age group, develop cerebrovascular disease with reduced blood flow, brain ischemia and stroke (Roriz-Filho et al. 2009). The extent of vascular changes is greater in T2DM than in T1DM due to the co-existence of multiple cardiovascular risk factors (i.e., dyslipidemia, hypertension). However, non-vascular mechanisms may also play an important role in the development of AD since such a risk persists in diabetic patients even when vascular factors are controlled for (Cole and Frautschy 2007). Prominently, insulin resistance (or insulin deficiency in T1DM) is now thought to underlie diabetes-associated cognitive decline and dementia (Luchsinger 2008; de la Monte 2009; Freude et al. 2009; Zhao and Townsend 2009). Supporting evidence for this mechanism comes from animal models of diabetes. Thus, rodent models of spontaneous and experimental diabetes show AD-like changes such as amyloidosis, tau hyperphosphorylation, neurite degeneration and neuronal loss (Li et al. 2007; Jolivalt et al. 2008; Kim et al. 2009a). The changes were more severe in the typ2 model and appear to be associated with insulin resistance and possibly hypercholesterolemia. It is to be noted that T2DM and a pre-diabetic condition are an increasing diagnosis due to unhealthy life-style as is a state of hypercholesterolemia under a broader but related cluster of diseases termed metabolic syndrome.
Dietary life-style, metabolic syndrome and AD
There is accumulating evidence for dietary life style negatively impacting upon brain function. In particular, consumption of high calorie ‘Western diet’ rich in unsaturated fatty acids and cholesterol is associated with the development of Alzheimer-like cognitive impairment (Martins et al. 2006; Pasinetti and Eberstein 2008). Several studies with animal models of AD have shown that such diets can cause increased amyloidosis, altered synaptic plasticity and behavior, essential features of Alzheimer pathology. In addition, high fat diet (Julien et al. 2010; Ma et al. 2009) and excessive sucrose intake (Cao et al. 2007) have been shown to enhance tau pathology in Tg mce. That hypercholesterolemia can cause tau hyperphosphorylation in the brain has been shown using apoE-deficient mice fed a high cholesterol diet (Rahman et al. 2005). However, there have been only limited studies examining the effects of diet on AD-like changes in non-transgenic animals. In a series of early studies Greenwood and Winocur (Winocur and Greenwood 2005) found that normal rats fed a high fat diet for 3 months had severely impaired cognitive ability. Later work by Wu et al (Wu et al. 2004) showed that the deleterious effects of saturated fat diet on cognitive function was related to altered synaptic plasticity and a down-regulation of brain-derived neurotrophic factor (BDNF). More recent studies have confirmed high fat diet-induced cognitive impairment tied to altered hippocampal morphology/plasticity in normal rats (Granholm et al. 2008; Stranahan et al. 2008). Using normal C57BL/6 mice, we showed that a high fat/high cholesterol diet induced loss of working memory correlated with striking neuroinflammatory changes and increased APP processing (Thirumangalakudi et al. 2008).
With regard to underlying mechanisms of diet-induced brain dysfunction, although it is possible that different dietary components including carbohydrates, triglycerides and cholesterol can have a direct influence on neuronal structure-function (with the caveat of BBB for certain lipids), it is more likely that the major effects are mediated indirectly via metabolic and vascular changes. It is now clear that long-term consumption of the Western diet can lead to the development of a pre-diabetes state that may progress into T2DM. Associated with this progressive metabolic disturbance is a clinical entity termed metabolic syndrome that in the human represents a clustering of cardiovascular risk factors including abdominal obesity, hypertension, low HDL, hyperglycemia and hypertriglyceridemia, all of which can be considered risk for sporadic AD (Milionis et al. 2008; Pasinetti and Eberstein 2008). Metabolic syndrome features co-exist and interact thereby increasing the risk of development of atherosclerotic complications as well. The scope of the syndrome has been expanded recently to include a novel component i.e., non-alcoholic fatty liver disease (NAFLD) and a new description suggests the existence of metabolic syndrome as a multifactorial endocrine disease and not a cluster of coincidental features (Bruce and Byrne 2009). Mechanistically, there are two important, interdependent processes that lie at the center of metabolic syndrome i.e., metabolic inflammation and insulin resistance.
Metabolic inflammation
Once identified as a lipid-storage disease, atherosclerosis is now recognized as a subacute inflammatory condition of the vessel wall characterized by monocyte/macrophage (and T cell) infiltration (Libby 2006; Hansson 2009). Oxidized lipids/lipoproteins participate in this process by inducing endothelium activation and expression of adhesion molecules as well as by transforming macrophages into foam cells that cause sclerotic plaque formation. Both obesity and prediabetic conditions are also characterized by low-grade inflammation (Rocha and Libby 2009). As with atherosclerosis, obesity was once considered a storage disease (-stored triglycerides in adipose tissue), but is now recognized as a low-grade inflammatory condition due to the accumulation of adipose tissue macrophages representing the main source of the circulating inflammatory mediators (Heilbronn and Campbell 2008; Bourlier and Bouloumie 2009). In both atherosclerosis and obesity/pre-diabetes, inflammatory signals interfere with insulin action and promote insulin resistance.
Insulin resistance
Along with metabolic inflammation, insulin resistance represents a central mechanism and a common pathological feature of components of the metabolic syndrome (Zeyda and Stulnig 2009). In fact, it is thought that metabolic inflammation comprising atherogenic and adipogenic inflammatory processes is a precursor to the development of insulin resistance in the main target organs i.e., adipose tissue, liver and muscle (Hotamisligil 2006; Rocha and Libby 2009). Over-consumption of lipids beyond what macrophages can normally handle can result in lipotoxicity and accumulation of the metabolites (oxidized LDL, advanced glycation end-products or AGEs, free fatty acids or FFA, cholesterol, and ceramide) within the adipose and non-adipose tissue compartments to induce chronic inflammation by promoting macrophage infiltration and activation (Lionetti et al. 2009; Prieur et al. 2009). Excessive secretion of proinflammatory cytokines then induces insulin resistance by interfering with insulin/IGF-IR signaling pathways in turn causing increased circulating FFA and reduced glucose uptake. Normally, insulin/IGF-IR signaling following ligand binding proceeds through Tyr phosphorylation of insulin receptor substrates (IRS) followed by activation of 2 major down-stream pathways: the phosphatidylinositol 3-kinase (PI3K)-Akt pathway largely responsible for insulin action on glucose uptake and suppression of gluconeogenesis and the MAPK pathway that regulates gene expression and, by interaction with PI3K, cell growth and differentiation. Phosphorylation of IRS on critical Ser sites, as opposed to Tyr, renders IRS inactive and prone to degradation resulting in a blockade of insulin signaling (Boura-Halfon and Zick 2009; Sun and Liu 2009). Ser phosphorylation is mediated by proinflammatory signaling kinases such as IKK, p38 MAPK and JNK that are activated by cytokines/chemokines such as TNFα, IL1β, MCP1, CRP etc. produced by activated macrophages. Another important mechanism whereby IRS is inactivated is via lipid metabolite-induced endoplasmic reticulum (ER) stress and JNK activation (Hotamisligil 2008; Zeyda and Stulnig 2009).
The two metabolic syndrome-associated processes (i.e., inflammation and insulin resistance) may also represent convergent mechanisms through which comorbid metabolic disorders promote the development of AD. The following sections will discuss further on this hypothesis.
Cerebrovascular dysfunction as a convergent mechanism between metabolic disorders and AD
There is evidence, both from human post-mortem analyses and animal models, for a significant vascular contribution to AD pathogenesis. The frequent vascular deposition of Aβ40 (vs. Aβ42 of senile plaques) in AD is a feature that is in common with cerebral amyloid angiopathy (CAA) (Kumar-Singh 2008; Thal et al. 2008; Bell and Zlokovic 2009). It has been suggested that AD and cerebrovascular disease may work synergistically to cause cognitive decline. In fact, mixed vascular and AD dementia is an increasingly prevalent diagnosis so that the line between Alzheimer's dementia and vascular dementia (VaD) is blurred. For many patients markers of vascular injury co-exist with traditional AD hallmarks (de la Torre 2009; Rocchi et al. 2009). The two-way pathological interaction is supported by the fact that vascular amyloid can cause injury and BBB dysfunction resulting in impaired Aβ clearance across the BBB (Bell and Zlokovic 2009). In turn, the presence of brain infarction and compromised vasoreactivity can influence the course of AD due to BBB dysfunction, cerebral hypoperfusion and ischemia resulting in increased CAA as well as parenchymal Aβ deposit (Zhu et al. 2007; Kumar-Singh 2008; Smith and Greenberg 2009)
Both atherosclerosis and diabetes are associated with cerebrovascular disease potentially representing a link to VaD and AD (Cechetto et al. 2008; Roriz-Filho et al. 2009; Rocchi et al. 2009). As noted above, a key mechanism by which hypercholesterolemia leads to the development of atherosclerotic lesions in large vessels including arteries is via the promotion of vascular inflammation. However, it is clear that elevated cholesterol can also cause a proinflammatory state in the microvasculature of different tissues including the brain long before the establishment of large vessel pathology (Ishikawa et al. 2004; Stokes 2006). There is evidence that diet-induced hypercholesterolemia in rodents causes the cerebral microvasculature to undergo oxidative stress and to assume a proinflammatory and prothrombogenic phenotype (Ishikawa et al. 2004). In a model of genetic hypercholesterolemia i.e., LDL receptor knockouts fed a Western diet, there occurs brain arteriole inflammation with increased levels of chemokines (Buga et al. 2006). Interestingly, these mice also display evidence of neuroinflammation (microglial activation) (Thirumangalakudi et al. 2008) and impaired cognitive performance (Mulder et al. 2004; Buga et al. 2006; Thirumangalakudi et al. 2008). It is of interest that a recent study by Franciosi et al (Franciosi et al. 2009) has demonstrated striking cerebrovascular pathology with characteristics of AD-like vascular defects in normal mice following a long-term (9 months) feeding of cholesterol-rich diet. Some features (i.e., thickening of basement membrane, presence of string and tortuous and loped vessels) resemble early microvascular changes seen in AD. Using APP/PS1 mouse model, it has been shown that cerebrovascular abnormalities including altered cerebral blood flow correlated with impaired cognition ensue in response to dietary cholesterol even before its effects on amyloid deposition (Hooijmans et al. 2009).
Even in the case of dietary fat, it is likely that excessive exposure to the triglycerides/fatty acids causes ‘lipotoxicity’ to cerebrovasculature (Takechi et al. 2009). In addition, cerebrovascular changes similar to that observed with hypercholesterolemia may occur under a diabetogenic condition elicited by the high fat diet. Diabetes interferes with both the barrier and transport functions of the cerebral microvessels (Huber 2008). A consequence of vascular abnormalities would be a compromised entry of circulating trophic factors (insulin, IGF-I, leptin etc) that normally support neuronal function and activity thereby exacerbating the neurodegenerative process (Neumann et al. 2008; Aleman and Torres-Aleman 2009). The mechanisms underlying vascular abnormalities may include low-grade inflammation and oxidative stress due to an upregulation of the receptor for AGEs i.e., RAGE (Yan et al. 2007). Excessive accumulation of AGEs is known to occur in aging, diabetes and other conditions of insulin resistance representing a key mediator of oxidative stress and inflammation. Interestingly, increased expression of RAGE is seen in regions of the brain affected by AD and its overexpression accelerates AD pathology and cognitive impairment (Takeuchi and Yamagishi 2008; Yan et al. 2009). A direct connection between diabetes-associated cerebrovascular inflammation and accelerated AD pathology with memory dysfunction has been recently demonstrated using a mouse model of AD with T2DM (Takeda et al. 2010). Interestingly, the early onset cognitive impairment was linked to increased vascular amyloid deposit rather than parenchymal load and, with altered brain insulin signaling.
Neuroinflammation in AD: the vascular connection
A large body of evidence indicates that neuroinflammation, primarily mediated by activated microglia, is functionally associated with a number of neurodegenerative diseases including AD (Akiyama et al. 2000; Heneka and O'Banion 2007; Eikelenboom et al. 2008). The evidence for an association between microglia-mediated inflammatory cascade and the progression, if not also pathogenesis, of AD is supported by early postmortem immunohistochemical studies revealing the presence of activated microglia in the periphery of amyloid plaques (Akiyama et al. 2000). Further, transgenic animals expressing mutant human APP develop plaque pathology accompanied with microglial activation that can be attenuated by anti-inflammatory treatments (Heneka and O'Banion 2007). Although such an approach in humans has been rather disappointing (Imbimbo 2009), inflammation in AD continues to be a subject of intense study as a potential treatment target through modulation of either detrimental (amyloidogenesis) or beneficial (amyloid clearance) roles of microglia/macrophages (Lucin and Wyss-Coray 2009). The past few years have seen changing views of the role of microglia in AD and other neurodegenerative diseases reflecting, as with their predecessors in the periphery (Auffray et al. 2009), a spectrum of different activation states (Colton 2009) that at the extreme ends fall into polarized M1 (proinflammatory) and M2 (anti-inflammatory/reparative) phenotypes, perhaps corresponding to their amyloidogenic vs. amyloid clearance roles in AD. On the other hand, there is also the suggestion that resident microglia may be ill equipped in the latter capacity perhaps due to age and advanced disease state (Njie et al. 2010; Hickman et al. 2008). Instead, it is proposed that peripherally derived monocytes may perform this function better thereby suggesting the potential therapeutic utility of enhanced recruitment of these cells in reducing amyloid pathology (Gate et al. 2010; Hickman and El Khoury 2010; Malm et al. 2010; Yong and Rivest 2009). While the idea of recruiting blood-borne monocytes to clear amyloid deposits is attractive, it seems that their effectiveness would be dependent on their appropriate phenotype (Town et al. 2008). Considering that the metabolic inflammation is characterized by M1-polarized tissue-associated (vascular, adipose, etc) and circulating monocytes (Rocha and Libby 2009), it seems that increased brain recruitment of these cells in metabolic disorders would be more harmful than beneficial. Therefore, it would be critical that any attempts at harnessing systemic immune system should take into consideration the ‘pre-existing condition’ of metabolic disorders.
A vascular component to inflammatory changes in AD is well recognized. Thus, as noted above, there is clear evidence for vascular deposition of Aβ40 (vs. Aβ42 of senile plaques) in association with CAA and increased neurovascular inflammation and permeability in AD and AD models. Microvessels isolated from AD brain were shown to have high levels of cytokines and chemokines (Grammas et al. 2006) suggesting a cerebrovascular contribution to neuroinflammatory processes in AD (Zlokovic 2005). Studies with a vasculotropic mutant APPTg mouse model show a direct relation between accumulation of vascular amyloid and neuroinflammation correlated with impaired cognitive performance (Xu et al. 2007). Since in familial AD and transgenic models, Aβ can trigger an inflammatory response by activating microglia (and microvascular endothelial cells), neuroinflammation represents a secondary consequence of AD-associated amyloidogenesis. The mechanisms underlying diet-induced neuroinflammation (Thirumangalakudi et al. 2008) however, are unclear, but as noted above, it is possible that systemic inflammation that characterizes high fat/cholesterol-induced atherogenic and diabetogenic changes may adversely affect cerebral microvasculature, compromising its barrier properties. A damaged or dysfunctional cerebrovasculature may trigger an activation of perivascular microglia in addition to promoting systemic macrophage invasion into the brain parenchyma. The recruitment of local immune cells (microglia) to the inflamed brain arterioles could then initiate a cascade of events leading to altered amyloid processing, neurodegeneration and synaptic/cognitive dysfunction [Fig 1]. Leaky barrier may also allow increased influx of blood-bourne cytokines, oxidized lipoproteins and cholesterol metabolites that would further contribute to the pathological sequelae. In fact, there is evidence for increased plasma and brain levels of 27-OH cholesterol in AD and it has been suggested that hypercholesterolemia-induced brain dysfunction may derive from an excessive influx of this oxysterol (Bjorkhem 2006). Both oxidized lipoproteins and oxysterols are known to be proinflammatory in atherosclerosis and may initiate a local inflammatory response in the brain at perivascular sites as well.
Fig 1.
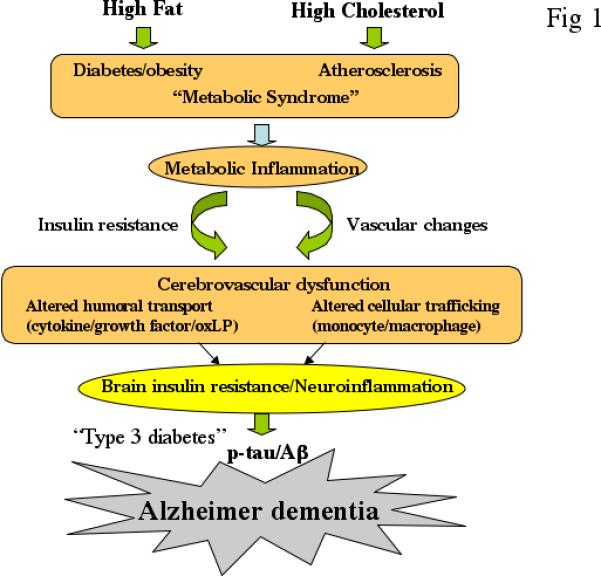
A model for dietary link to dementia via metabolic disorders. It is argued that the two key pathogenic processes associated with metabolic disorders i.e., inflammation and insulin resistance may re-emerge within the brain due to cerebrovascular dysfunction with increased BBB permeability allowing macrophage/cytokine entry and reduced transport of trophic factors. Potential signaling pathways that further lead to AD-like molecular and cognitive changes via increased Aβ and tau phosphorylation are illustrated in Fig 2.
Impaired brain insulin signaling in AD: ‘the insulin-resistant brain state’
As already noted, while cognitive capacities are often impaired in patients with diabetes, the links between diabetes and AD are mutual (Sun and Alkon 2006). Thus, AD patients often have hyperinsulinemia and hyperglycemia as compared with healthy controls and there is evidence of both peripheral and central insulin resistance in AD. There are also other striking similarities between the two diseases including impaired glucose and energy metabolism, elevated AGEs and oxidative stress, cellular injury (i.e., pancreatic β cells vs. neurons), amyloidogenesis (Aβ vs. islet amyloid polypeptide deposits) and impaired expression and activity of insulin degrading enzyme (IDE) (Neumann et al. 2008). Since IDE can degrade Aβ, this activity can potentially be compromised by excessive insulin, a competing substrate, resulting in increased Aβ. However, such a role of hyperinsulinemia presents an apparent paradox since insulin is considered an important neurotrophic factor. Both insulin and IGF-I play important homeostatic roles in the brain including glucose metabolism, energy balance and trophic support as well as maintenance of normal synaptic activity and cognitive function. In fact, cerebroventricular administration of insulin in rodents (Biessels et al. 1998) and its intranasal delivery in AD (Dhamoon et al. 2009) has been shown to improve cognition. Recent studies also show that insulin signaling protects synapses against pathogenic binding of Aβ oligomers (De Felice et al. 2009). The explanation for peripheral hyperinsulinemia being detrimental seems to lie in the reciprocal relation between peripheral and brain insulin levels. Thus, peripheral hyperinsulinemia is associated with an inhibition of brain insulin production resulting in a higher risk of AD (Luchsinger 2008). Also, it may interfere with amyloid clearance through BBB since the same insulin transporter is also involved in Aβ transport out of the brain (Sun and Alkon 2006). There is also evidence for an impaired transport into the brain of IGF-I (and perhaps, other trophic factors) due to cerebrovascular abnormalities (Neumann et al. 2008; Aleman and Torres-Aleman 2009) as might occur in metabolic disorders. It is likely that a diet-induced pre-diabetic (and atherogenic) condition induces similar cerebrovascular dysfunction thereby blocking insulin/IGF transport essentially creating an insulin/IGF-deficient environment in the brain. This would lead to impaired insulin/IGFR signaling. Although IGF-I can be produced endogenously in the brain, the levels of both insulin and IGF-I are found reduced in the AD brain (Neumann et al. 2008; de la Monte 2009). A generalized down-regulation of insulin/IGF-IR signaling including reduced receptor levels and signaling proteins is seen in postmortem AD brains (Moloney et al. 2010). These findings and the concept of AD-associated hypometabolism have led to the description of the so-called, ‘insulin-resistant brain state’ in the context of sporadic AD (Salkovic-Petrisic et al. 2009). There is also the suggestion that AD represents ‘Type 3 diabetes’ (de la Monte 2009). The mechanism whereby brain insulin resistance develops in AD is thought to involve Aβ oligomers interfering with neuronal insulin/IGFR signaling (Liao and Xu 2009). In contrast, it has been suggested that increased amyloidogenesis may follow diet-induced alterations in insulin receptor signaling (Pasinetti and Eberstein 2008). There may be an additional mechanism for the induction of insulin resistant brain state in this situation. Thus, it is likely that, as in systemic insulin resistance, increased expression of neuroinflammatory mediators may negatively interact with brain insulin/IGFR signaling to induce insulin resistance (Fig 2). Again, the mechanism may involve Ser phosphorylation of IRS1 by the two stress-activated protein kinases (SAPKs) i.e., JNK and p38 MAP kinase. The activation of the MAP kinase cascades and NFκB pathway down-stream of the cytokine and innate immune receptors (i.e., Toll-like receptors or TLRs and scavenger receptors) in the macrophage/microglial compartment, on the other hand, would provide the signal for increased expression of the mediators that would target the vulnerable neurons.
Fig 2.
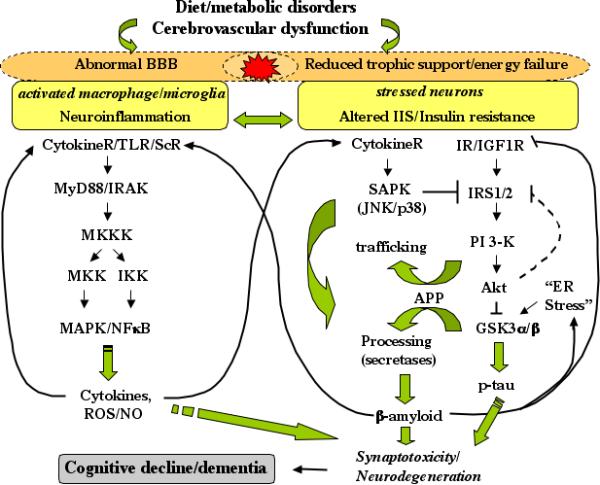
Hypothetical scheme depicting altered brain insulin signaling and neuroinflammatory cascade leading to AD-like cognitive decline in metabolic disorders. It is likely that proinflammatory mediators released by activated immune cells interfere with neuronal IIS thereby exacerbating their insulin resistant state. Reduced cerebral blood flow and energy failure due to dysfunctional cerebrovasculature would also contribute to neuronal metabolic defects. Impaired IIS will lead to an activation of GSK3 and hyperphosphorylation of tau implicated in synaptic dysfunction and neurodegeneration. Altered APP processing would cause increased Aβ leading to neuronal/synaptic loss or dysfunction. It is also known that Aβ can induce neuronal insulin resistance by interfering with IR/IGF1R signaling thereby promoting a vicious cycle. In addition, cytokines and ROS can directly damage neurons and synapses with the outcome of AD-type cognitive decline. BBB, blood brain barrier; IIS, insulin/IGF1R signaling; IR, insulin receptor; NO, nitric oxide; ROS, reactive oxygen species; SAPK, stress-activated protein kinase; ScR, scavenger receptor; TLR, toll-like receptors.
The consequences of dysregulated insulin signaling may also include altered APP trafficking (PI3K-dependent) and hyperphosphorylated tau due to GSK3 activation. Normally, insulin inactivates GSK3 via PI3K-Akt-mediated phosphorylation of the enzyme isoforms. There is now substantial evidence for a key role of GSK3 in AD through multiple mechanisms including tau phosphorylation, Aβ production and inflammation (Balaraman et al. 2006; Hooper et al. 2008). Its inhibition reduces Aβ while its conditional inactivation reverses AD-like phenotype in mouse models correlated with normal tau phosphorylation (Engel et al. 2006). In a dual-hit hypothesis of LOAD, GSK3 takes a central place linking Aβ and tau (Small and Duff 2008).
The model proposed (Fig 2) depicts the possibility of a direct neuro/synaptotoxicity, not necessarily involving Aβ, exerted by proinflammatory cytokines and reactive oxygen and nitrogen species (ROS/NO). While oxidative stress-mediated neurotoxicity is obvious, there is evidence for the roles of specific cytokines in hippocampal neurodegeneration and cognitive loss. Thus, for example, sustained expression of IL-1β in the hippocampus has been shown to impair long-term contextual and spatial memory in transgenic mice (Moore et al. 2009). Also, there is much interest in targeting TNFα as a therapy for AD because of its evident role in synaptic dysfunction (Tobinick 2009).
Metabolism-based treatment options for AD
The above discussion points to several potential targets of therapeutic intervention along the pathway of metabolic syndrome to AD including lipid metabolism, inflammation and insulin signaling/resistance. Obviously, the statin drugs continue to be investigated with the caveat that the efficacy does not simply relate to their cholesterol lowering effects (Fonseca et al. 2010). There is great interest in the use of omega 3 fatty acids due to their multiple beneficial effects including anti-inflammatory and insulin sensitizing roles (Cole and Frautschy 2010). Similarly, PPARγ agonists (thiazolidines, TZDs) used for treating diabetes based on their potent insulin sensitizer roles are expected to benefit AD as well (Jiang et al. 2008). PPAR agonists also represent a better anti-inflammatory strategy than the use of controversial NSAIDs and have added benefit of reducing vascular oxidative stress (Hamel et al. 2008). Of particular interest, another nuclear receptor, liver x receptor (LXR), is also receiving increasing attention due to its multiple beneficial roles including cholesterol homeostasis, antiinflammation and insulin sensitization (Hong and Tontonoz 2008). With respect to AD, LXR activation has an additional desirable effect of reducing amyloidogenic APP processing, and there are recent reports demonstrating the efficacies of synthetic LXR agonists in reducing amyloid pathology, neuroinflammation and memory deficit in mouse models of AD (Fitz et al. 2010; Riddell et al. 2007; Zelcer et al. 2007).
Continued efforts at defining the convergent mechanisms and mediators underlying metabolic disorders and AD should further promote development and devising of therapeutic strategies that are commonly applicable to treat both AD and metabolic disorders. However, the approach may stumble on tissue-specific differences in the actions of certain drugs as noted recently for the antidiabetic drug, metformin (Chen et al. 2009). Thus, the drug was found to have an undesired effect of increasing the generation of Aβ, although in combined use, it enhanced insulin's effect in reducing Aβ. With respect to targeting insulin/IGFR signaling per se, it is to be noted that although the strategy of boosting the brain levels of IGF-1 (and insulin) in AD seems logical (Aleman and Torres-Aleman 2009), an apparently contradictory view has also emerged that in fact, suggests a neuroprotective role of reduced IGF signaling against amyloid proteotoxicity, perhaps in accordance with the ‘paradoxical’ role of IIS in aging and longevity (Cohen and Dillin 2008; Freude et al. 2009). Thus, Cohen et al (Cohen et al. 2009) show that when transgenic mice expressing mutant human APP/PS1 are made partially deficient in IGF1R by crossing with long-lived, IGF1R heterozygous mice, they are protected against the development of Alzheimer's symptoms including synaptic loss, neuroinflammation and behavioral impairment correlated with a conversion of the toxic amyloid oligomers into less toxic fibrillar aggregates. The authors suggest a feedback down-regulation or desensitization of brain IGF1R to reconcile the reduced AD pathology observed in response to IGF-1 administration in other studies. Obviously, the significance of altered brain insulin/IGF1R signaling and insulin resistance in AD requires further clarification in terms of multiple alternative signaling pathways (including threshold/gating effects) down-stream of insulin/IGF receptors (Shineman et al. 2009).
Conclusion
Despite continued (-but abating) debate on the epidemiological and clinicopathological relevance of systemic metabolic diseases to AD, there have been intense research efforts at defining convergent pathogenic processes using a repertoire of experimental models. It seems that cerebrovascular abnormalities commonly associated with metabolic disorders represent an important link that would further support the emerging vascular hypothesis of AD. While identification of shared risk factors including diet, lifestyle and environmental toxins (–not discussed here) has emphasized the importance and feasibility of preventive strategies against the development of both cardiometabolic and cognitive conditions, uncovering of shared mechanisms underlying these co-morbid disease processes point to an opportunity to develop/screen common treatments. As discussed in this review, some of the key disease mechanisms including inflammation and insulin resistance occur both in the periphery and in the brain suggesting common mediators (i.e., cytokines, pro-oxidants) and/or deficiency in homeostatic regulators (i.e., insulin, IGF, HDL, antioxidants). There are indications that targeting such mechanisms and mediators is therapeutically feasible in AD. This is evident in trials using some of the drugs approved for treating atherosclerosis and diabetes including statins and TZDs. It is expected that dissection of the convergent mechanisms underlying metabolic disorders and AD would further promote the development of refined therapeutic strategies that are effective in either AD or systemic disorders. Finally, it is important to note that insulin provides a direct negative feedback to hypothalamic nuclei that control whole body energy and glucose homeostasis and that dietary fat is known to induce hypothalamic inflammation resulting in insulin/leptin resistance (Koch et al. 2008; Milanski et al. 2009). Therefore, CNS insulin resistance (and neuroinflammation) may be a common denominator of metabolic disorders and cognitive dysfunctions the targeting of which seems logical in both these conditions.
Acknowledgment
This work was supported by NIH grants, R01NS051575 and R21NS063183.
Abbreviations
- AD
Alzheimer's disease
- Aβ
beta-amyloid
- APP
amyloid precursor protein
- BBB
blood brain barrier
- CAA
cerebral amyloid angiopathy
- GSK3
glycogen synthase kinase 3
- IIS
insulin/IGF1R signaling
- IR
insulin receptor
- IRS
insulin receptor substrate
- LDLR
low density lipoprotein receptor
- PI 3K
phosphatidyl inositiol 3 kinase
- RAGE
receptor for advanced gycation end products
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- VaD
vascular dementia
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman A, Torres-Aleman I. Circulating insulin-like growth factor I and cognitive function: neuromodulation throughout the lifespan. Prog Neurobiol. 2009;89:256–265. doi: 10.1016/j.pneurobio.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Audoy-Remus J, Richard JF, Soulet D, Zhou H, Kubes P, Vallieres L. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci. 2008;28:10187–10199. doi: 10.1523/JNEUROSCI.3510-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- Balaraman Y, Limaye AR, Levey AI, Srinivasan S. Glycogen synthase kinase 3beta and Alzheimer's disease: pathophysiological and therapeutic significance. Cell Mol Life Sci. 2006;63:1226–1235. doi: 10.1007/s00018-005-5597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res. 1998;800:125–135. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260:493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296:E581–591. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- Bourlier V, Bouloumie A. Role of macrophage tissue infiltration in obesity and insulin resistance. Diabetes Metab. 2009;35:251–260. doi: 10.1016/j.diabet.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- Bruce KD, Byrne CD. The metabolic syndrome: common origins of a multifactorial disorder. Postgrad Med J. 2009;85:614–621. doi: 10.1136/pgmj.2008.078014. [DOI] [PubMed] [Google Scholar]
- Buga GM, Frank JS, Mottino GA, Hendizadeh M, Hakhamian A, Tillisch JH, Reddy ST, Navab M, Anantharamaiah GM, Ignarro LJ, Fogelman AM. D-4F decreases brain arteriole inflammation and improves cognitive performance in LDL receptor-null mice on a Western diet. J Lipid Res. 2006;47:2148–2160. doi: 10.1194/jlr.M600214-JLR200. [DOI] [PubMed] [Google Scholar]
- Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2007;282:36275–36282. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer's disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139–1146. doi: 10.1016/S0140-6736(04)15900-X. [DOI] [PubMed] [Google Scholar]
- Cechetto DF, Hachinski V, Whitehead SN. Vascular risk factors and Alzheimer's disease. Expert Rev Neurother. 2008;8:743–750. doi: 10.1586/14737175.8.5.743. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9:759–767. doi: 10.1038/nrn2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GM, Frautschy SA. DHA may prevent age-related dementia. J Nutr. 2010;140:869–874. doi: 10.3945/jn.109.113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer's Disease. Exp Gerontol. 2007;42:10–21. doi: 10.1016/j.exger.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Insulin resistance and Alzheimer's disease. BMB Rep. 2009;42:475–481. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC. Cerebrovascular and cardiovascular pathology in Alzheimer's disease. Int Rev Neurobiol. 2009;84:35–48. doi: 10.1016/S0074-7742(09)00403-6. [DOI] [PubMed] [Google Scholar]
- Dhamoon MS, Noble JM, Craft S. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2009;72:292–293. doi: 10.1212/01.wnl.0000344246.91081.2c. author reply 293-294. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Veerhuis R, Familian A, Hoozemans JJ, van Gool WA, Rozemuller AJ. Neuroinflammation in plaque and vascular beta-amyloid disorders: clinical and therapeutic implications. Neurodegener Dis. 2008;5:190–193. doi: 10.1159/000113699. [DOI] [PubMed] [Google Scholar]
- Engel T, Hernandez F, Avila J, Lucas JJ. Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca AC, Resende R, Oliveira CR, Pereira CM. Cholesterol and statins in Alzheimer's disease: Current controversies. Exp Neurol. 2010;223:282–293. doi: 10.1016/j.expneurol.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Franciosi S, Gama Sosa MA, English DF, Oler E, Oung T, Janssen WG, De Gasperi R, Schmeidler J, Dickstein DL, Schmitz C, Gandy S, Hof PR, Buxbaum JD, Elder GA. Novel cerebrovascular pathology in mice fed a high cholesterol diet. Mol Neurodegener. 2009;4:42. doi: 10.1186/1750-1326-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer's disease: from model organisms to human disease. Curr Alzheimer Res. 2009;6:213–223. doi: 10.2174/156720509788486527. [DOI] [PubMed] [Google Scholar]
- Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer's disease: the blood-borne identity. J Neural Transm. 2010;117:961–970. doi: 10.1007/s00702-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammas P, Samany PG, Thirumangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer's disease microvessels: implications for disease pathogenesis. J Alzheimers Dis. 2006;9:51–58. doi: 10.3233/jad-2006-9105. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Bimonte-Nelson HA, Moore AB, Nelson ME, Freeman LR, Sambamurti K. Effects of a saturated fat and high cholesterol diet on memory and hippocampal morphology in the middle-aged rat. J Alzheimers Dis. 2008;14:133–145. doi: 10.3233/jad-2008-14202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E, Nicolakakis N, Aboulkassim T, Ongali B, Tong XK. Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer's disease. Exp Physiol. 2008;93:116–120. doi: 10.1113/expphysiol.2007.038729. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Inflammatory mechanisms in atherosclerosis. J Thromb Haemost. 2009;7(Suppl 1):328–331. doi: 10.1111/j.1538-7836.2009.03416.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14:1225–1230. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:168–173. doi: 10.2174/187152710791011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. 2008;18:461–467. doi: 10.1016/j.gde.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooijmans CR, Van der Zee CE, Dederen PJ, Brouwer KM, Reijmer YD, van Groen T, Broersen LM, Lutjohann D, Heerschap A, Kiliaan AJ. DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiol Dis. 2009;33:482–498. doi: 10.1016/j.nbd.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond) 2008;32(Suppl 7):S52–54. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JD. Diabetes, cognitive function, and the blood-brain barrier. Curr Pharm Des. 2008;14:1594–1600. doi: 10.2174/138161208784705441. [DOI] [PubMed] [Google Scholar]
- Imbimbo BP. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer's disease. Expert Opin Investig Drugs. 2009;18:1147–1168. doi: 10.1517/13543780903066780. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–244. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Heneka M, Landreth GE. The role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in Alzheimer's disease: therapeutic implications. CNS Drugs. 2008;22:1–14. doi: 10.2165/00023210-200822010-00001. [DOI] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer's disease and correction by insulin. J Neurosci Res. 2008;86:3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009a;150:5294–5301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009b;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch L, Wunderlich FT, Seibler J, Konner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Bruning JC. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest. 2008;118:2132–2147. doi: 10.1172/JCI31073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodl CT, Seaquist ER. Cognitive dysfunction and diabetes mellitus. Endocr Rev. 2008;29:494–511. doi: 10.1210/er.2007-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh S. Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008;7(Suppl 1):67–82. doi: 10.1111/j.1601-183X.2007.00380.x. [DOI] [PubMed] [Google Scholar]
- Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J Nutr Health Aging. 2002;6:315–319. [PubMed] [Google Scholar]
- Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- Liao FF, Xu H. Insulin signaling in sporadic Alzheimer's disease. Sci Signal. 2009;2:pe36. doi: 10.1126/scisignal.274pe36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- Lionetti L, Mollica MP, Lombardi A, Cavaliere G, Gifuni G, Barletta A. From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutr Metab Cardiovasc Dis. 2009;19:146–152. doi: 10.1016/j.numecd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer's disease: an epidemiological perspective. Eur J Pharmacol. 2008;585:119–129. doi: 10.1016/j.ejphar.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, Chen PP, Hudspeth B, Chen C, Zhao Y, Vinters HV, Frautschy SA, Cole GM. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm T, Koistinaho M, Muona A, Magga J, Koistinaho J. The role and therapeutic potential of monocytic cells in Alzheimer's disease. Glia. 2010;58:889–900. doi: 10.1002/glia.20973. [DOI] [PubMed] [Google Scholar]
- Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, Nolan D, Gandy SE, Martins RN. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer's disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–736. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- McAllister AK, van de Water J. Breaking boundaries in neural-immune interactions. Neuron. 2009;64:9–12. doi: 10.1016/j.neuron.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merched A, Xia Y, Visvikis S, Serot JM, Siest G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer's disease. Neurobiol Aging. 2000;21:27–30. doi: 10.1016/s0197-4580(99)00103-7. [DOI] [PubMed] [Google Scholar]
- Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DM, Anhe G, Amaral ME, Takahashi HK, Curi R, Oliveira HC, Carvalheira JB, Bordin S, Saad MJ, Velloso LA. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29:359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milionis HJ, Florentin M, Giannopoulos S. Metabolic syndrome and Alzheimer's disease: a link to a vascular hypothesis? CNS Spectr. 2008;13:606–613. doi: 10.1017/s1092852900016886. [DOI] [PubMed] [Google Scholar]
- Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 31:224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Moore AH, Wu M, Shaftel SS, Graham KA, O'Banion MK. Sustained expression of interleukin-1beta in mouse hippocampus impairs spatial memory. Neuroscience. 2009;164:1484–1495. doi: 10.1016/j.neuroscience.2009.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- Mulder M, Jansen PJ, Janssen BJ, van de Berg WD, van der Boom H, Havekes LM, de Kloet RE, Ramaekers FC, Blokland A. Low-density lipoprotein receptor-knockout mice display impaired spatial memory associated with a decreased synaptic density in the hippocampus. Neurobiol Dis. 2004;16:212–219. doi: 10.1016/j.nbd.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer's disease: molecular links & clinical implications. Curr Alzheimer Res. 2008;5:438–447. doi: 10.2174/156720508785908919. [DOI] [PubMed] [Google Scholar]
- Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinetti GM, Eberstein JA. Metabolic syndrome and the role of dietary lifestyles in Alzheimer's disease. J Neurochem. 2008;106:1503–1514. doi: 10.1111/j.1471-4159.2008.05454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieur X, Roszer T, Ricote M. Lipotoxicity in macrophages: evidence from diseases associated with the metabolic syndrome. Biochim Biophys Acta. 2009;1801:327–337. doi: 10.1016/j.bbalip.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- Rahman A, Akterin S, Flores-Morales A, Crisby M, Kivipelto M, Schultzberg M, Cedazo-Minguez A. High cholesterol diet induces tau hyperphosphorylation in apolipoprotein E deficient mice. FEBS Lett. 2005;579:6411–6416. doi: 10.1016/j.febslet.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, Reinhart PH, Jacobsen JS. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Rocchi A, Orsucci D, Tognoni G, Ceravolo R, Siciliano G. The role of vascular factors in late-onset sporadic Alzheimer's disease. Genetic and molecular aspects. Curr Alzheimer Res. 2009;6:224–237. doi: 10.2174/156720509788486644. [DOI] [PubMed] [Google Scholar]
- Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409. doi: 10.1038/nrcardio.2009.55. [DOI] [PubMed] [Google Scholar]
- Roriz-Filho JS, Sa-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, Moriguti JC, Roriz-Cruz M. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta. 2009;1792:432–443. doi: 10.1016/j.bbadis.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- Salkovic-Petrisic M, Osmanovic J, Grunblatt E, Riederer P, Hoyer S. Modeling Sporadic Alzheimer's Disease: The Insulin Resistant Brain State Generates Multiple Long-Term Morphobiological Abnormalities Inclusive Hyperphosphorylated Tau Protein and Amyloid-beta. A Synthesis. J Alzheimers Dis. 2009;18:729–750. doi: 10.3233/JAD-2009-1184. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Granholm AC, Kindy MS, Bhat NR, Greig NH, Lahiri DK, Mintzer JE. Cholesterol and Alzheimer's disease: clinical and experimental models suggest interactions of different genetic, dietary and environmental risk factors. Curr Drug Targets. 2004;5:517–528. doi: 10.2174/1389450043345335. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Shineman DW, Dain AS, Kim ML, Lee VM. Constitutively active Akt inhibits trafficking of amyloid precursor protein and amyloid precursor protein metabolites through feedback inhibition of phosphoinositide 3-kinase. Biochemistry. 2009;48:3787–3794. doi: 10.1021/bi802070j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009;40:2601–2606. doi: 10.1161/STROKEAHA.108.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DL, Kuo YM, Roher A, Martin T, Lukas RJ. Alterations of Alzheimer's disease in the cholesterol-fed rabbit, including vascular inflammation. Preliminary observations. Ann N Y Acad Sci. 2000;903:335–344. doi: 10.1111/j.1749-6632.2000.tb06384.x. [DOI] [PubMed] [Google Scholar]
- Stokes KY. Microvascular responses to hypercholesterolemia: the interactions between innate and adaptive immune responses. Antioxid Redox Signal. 2006;8:1141–1151. doi: 10.1089/ars.2006.8.1141. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK, Alkon DL. Links between Alzheimer's disease and diabetes. Drugs Today (Barc) 2006;42:481–489. doi: 10.1358/dot.2006.42.7.973588. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Liu F. Phosphorylation of IRS proteins Yin-Yang regulation of insulin signaling. Vitam Horm. 2009;80:351–387. doi: 10.1016/S0083-6729(08)00613-4. [DOI] [PubMed] [Google Scholar]
- Takechi R, Galloway S, Pallebage-Gamarallage MM, Lam V, Mamo JC. Dietary fats, cerebrovasculature integrity and Alzheimer's disease risk. Prog Lipid Res. 2009;49:159–170. doi: 10.1016/j.plipres.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi M, Yamagishi S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer's disease. Curr Pharm Des. 2008;14:973–978. doi: 10.2174/138161208784139693. [DOI] [PubMed] [Google Scholar]
- Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, Kindy MS, Bhat NR. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem. 2008;106:475–485. doi: 10.1111/j.1471-4159.2008.05415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick E. Tumour necrosis factor modulation for treatment of Alzheimer's disease: rationale and current evidence. CNS Drugs. 2009;23:713–725. doi: 10.2165/11310810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es MA, van den Berg LH. Alzheimer's disease beyond APOE. Nat Genet. 2009;41:1047–1048. doi: 10.1038/ng1009-1047. [DOI] [PubMed] [Google Scholar]
- Whitmer RA. Type 2 diabetes and risk of cognitive impairment and dementia. Curr Neurol Neurosci Rep. 2007;7:373–380. doi: 10.1007/s11910-007-0058-7. [DOI] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26(Suppl 1):46–49. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Wollmer MA. Cholesterol-related genes in Alzheimer's disease. Biochim Biophys Acta. 2010;1801:762–773. doi: 10.1016/j.bbalip.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- Wolozin B, Manger J, Bryant R, Cordy J, Green RC, McKee A. Re-assessing the relationship between cholesterol, statins and Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:63–70. doi: 10.1111/j.1600-0404.2006.00687.x. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–1707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- Xu F, Grande AM, Robinson JK, Previti ML, Vasek M, Davis J, Van Nostrand WE. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid beta-protein precursor transgenic mice. Neuroscience. 2007;146:98–107. doi: 10.1016/j.neuroscience.2007.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Bierhaus A, Nawroth PP, Stern DM. RAGE and Alzheimer's disease: a progression factor for amyloid-beta-induced cellular perturbation? J Alzheimers Dis. 2009;16:833–843. doi: 10.3233/JAD-2009-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SF, D'Agati V, Schmidt AM, Ramasamy R. Receptor for Advanced Glycation Endproducts (RAGE): a formidable force in the pathogenesis of the cardiovascular complications of diabetes & aging. Curr Mol Med. 2007;7:699–710. [PubMed] [Google Scholar]
- Yong VW, Rivest S. Taking advantage of the systemic immune system to cure brain diseases. Neuron. 2009;64:55–60. doi: 10.1016/j.neuron.2009.09.035. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc Natl Acad Sci U S A. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyda M, Stulnig TM. Obesity, inflammation, and insulin resistance--a mini-review. Gerontology. 2009;55:379–386. doi: 10.1159/000212758. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer's disease. Biochim Biophys Acta. 2009;1792:482–496. doi: 10.1016/j.bbadis.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Zhu X, Smith MA, Honda K, Aliev G, Moreira PI, Nunomura A, Casadesus G, Harris PL, Siedlak SL, Perry G. Vascular oxidative stress in Alzheimer disease. J Neurol Sci. 2007;257:240–246. doi: 10.1016/j.jns.2007.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]