

Abstract
Signal transduction and activator of transcription 3 (Stat3) is activated by cytokines and growth factors in lung cancers and regulates expression of genes implicated in cell growth, survival, and transformation. Previously, we found that mice with a deletion of the G protein-coupled receptor, family C, group 5, member a (Gprc5a) gene develop lung tumors indicating that Gprc5a is a tumor suppressor. Herein, we show that epithelial cells from Gprc5a knockout mouse lung (Gprc5a−/− cells) survive better in vitro in medium deprived of exogenous growth factors and form more colonies in semi-solid medium than their counterparts from wildtype mice (Gprc5a+/+ cells). Stat3 Tyrosine 705 phosphorylation and expression of several Stat3-regulated anti-apoptotic genes were higher in Gprc5a−/− than in Gprc5a+/+ cells. Both cell types secreted Leukemia inhibitory factor (Lif), however, whereas Stat3 activation was persistent in Gprc5a−/− cells it was transient in Gprc5a+/+ cells. Lung adenocarcinoma cells isolated from Gprc5a−/− mice also exhibited autocrine Lif-mediated Stat3 activation. The level of Socs3, the endogenous Stat3 inhibitory protein, was higher in Gprc5a+/+ than in Gprc5a−/− cells and expression of the tumor suppressor stabilized Socs3. Inhibition of Stat3 signaling in Gprc5a−/− normal and cancer cells by the Jak2 inhibitor AG490 or by a dominant negative Stat3(Y705F) increased starvation-induced apoptosis and inhibited colony formation. These results demonstrate that persistent Stat3 activation is important for the survival and transformation of Gprc5a−/− lung cells and suggest that the tumor suppressive effects of Gprc5a are mediated, at least in part, by inhibition of Stat3 signaling via Socs3 stabilization.
Keywords: Stat3, Gprc5a, lung cancer, Lif, apoptosis
Introduction
Because lung cancer is the leading cause of cancer death (1) it is important to advance the understanding of cellular and molecular aberrations associated with lung tumorigenesis to improve diagnosis, prevention and therapy of this disease. Various genetic and epigenetic changes have been implicated in lung cancer including activation of oncogenes by mutations or amplification, inactivation of tumor suppressor genes by loss of heterozygosity, and by transcription silencing via promoter hypermethylation (2).
It has also been shown that Signal Transducer and Activator of Transcription 3 (STAT3) is frequently activated in lung cancer and functions as an oncogene (3, 4). STAT3 signaling is triggered by cytokines [(e.g., interleukine 6 (IL-6)] and growth factors that regulate cell proliferation, differentiation, survival, invasion, inflammation and immunity (5). IL-6 and related cytokines bind to specific cell surface receptors that become associated through their cytoplasmic domain with tyrosine kinases such as the Janus kinase (JAK) family kinases (6), which phosphorylate specific tyrosine residues in STAT proteins (e.g. Y705 in STAT3), leading to homo- or heterodimerization with other STAT proteins then translocation into the nucleus to regulate gene transcription (5, 7). The JAK/STAT3 signaling pathway plays important roles in carcinogenesis because many Stat3 target genes including cyclin D1, Bcl-XL, Mcl1, Hspa1a and Cryab are involved in cell proliferation and survival (8–10). Therefore, it has been targeted for cancer therapy (4, 11). For example, STAT3 activation can be suppressed by JAK inhibitors like the tyrphostin AG490, which induce apoptosis in cancer cells and inhibit tumor growth (12, 13).
G protein-coupled receptor, family C, group 5, member A (GPRC5A) is a retinoic acid-induced gene that is primarily expressed in human and mouse lung epithelial cells (14, 15). .GPRC5A expression was suppressed in the majority of human NSCLC cell lines (14) and tumor tissues from human NSCLC patients (15). Our recent finding that deletion of the Gprc5a gene in mice leads to the development of spontaneous lung tumors strongly indicated that Gprc5a functions as a lung-specific tumor suppressor in mice (15).
Herein, we report that normal lung epithelial cells from Gprc5a−/− mice exhibit persistent Stat3 activation by autocrine Lif. Stabilization of the endogenous Stat3 inhibitory protein Socs3 by Gprc5a appears to be a possible mechanism for the differential activation of Stat3 in Gprc5a+/+ and Gprc5a−/− cells. This is the first report on activation of Jak/Stat3 signaling by loss of a GPCR rather than by activation of GPCR as reported previously (16, 17).
Materials and Methods
Cell lines and culture conditions
Epithelial cells were derived from normal tracheas of three weeks old Gprc5a−/− and Gprc5a+/+ mice (C57Bl/6 × 129sv) and from a spontaneous lung adenocarcinoma from a 24 months old Gprc5a−/− male mouse. Briefly, tracheas and tumor tissue were dissected from mice, cut into small fragments, and incubated in a tissue dissociating solution ACCUMAX™ (Innovative Cell Technologies) then transferred to PRIMARIA tissue culture dishes (BD Biosciences) and incubated in AmnioMAX-C100 medium (Invitrogen). The epithelial cells were then detached by trypsinization and sub-cultured and grown in keratinocyte-serum-free medium (K-SFM) (Invitrogen). The cell lines were karyotyped by G banding in the Institutional Molecular Cytogenetics Facility (MD Anderson Cancer Center) and found to be of mouse origin. The normal epithelial cells (Gprc5a+/+ and Gprc5a−/− cells) were cultured in K-SFM supplemented with epidermal growth factor (EGF, 5 ng/ml) and bovine pituitary extract (BPE, 50 µg/ml) (Invitrogen) and the tumor cells (MDA959) were cultured in Dulbecco’s modification of Eagle’s medium (DMEM) and Ham’s F12 medium (1:1) supplemented with 10% fetal bovine serum. The MDA959 cells were tumorigenic (1-cm diameter tumor developed 46 days after injecting 4×106 cells suspended in 1:1 Matrigel subcutaneously) in syngeneic mice, whereas the Gprc5a−/− cells were non-tumorigenic under the same conditions. Human embryonic kidney cell line HEK293T from ATCC was cultured in the same medium with serum as the MDA959 cells.
Reagents and antibodies
AG490 was from EMD Chemicals. Antibodies against pStat3(Y705), Stat3, pErk1/2, Erk1/2 Bcl-XL and Socs3 were from Cell Signaling Technology. Mcl-1 antibody was from Epitomics. Hspa1a and Cryab antibodies were from Assay Designs. Flag antibody to detect expression of flag-tagged dominant negative Stat3(Y705F) was from Sigma. Neutralizing antibodies against Lif, Il-6, Osm (Oncostatin M) and normal goat IgG were from R&D Systems. ESGRO® (LIF) was from Millipore. EGF was from Invitrogen.
Plasmid construction
Plasmid pRc/CMV-Stat3(Y705F) generated in Dr. James E. Darnell, Jr’s laboratory (The Rockefeller University) (18, 19) was from Addgene (plasmid 8709). The plasmid pRc/CMV-Stat3(Y705F) was digested using SacI and the released Stat3(Y705F)-Flag cDNA was then subcloned into pIRES2-EGFP as an intermediate step before releasing Stat3(Y705F)-Flag cDNA by digestion of pIRES2-EGFP-Stat3(Y705F)-Flag using BamHI and subcloning it into pIREShyg3 to generate pIREShyg3-Stat3(Y705F)-Flag expression vector. The coding sequence of GPRC5A was obtained from cDNA of 292G lung tumor cells and a Myc tag was added to the C-terminus using PCR. The Myc-tagged GPRC5A cDNA was then subcloned into pcDNA3.1(+) to generate the pcDNA3.1(+)-CPRC5A-Myc plasmid. SOCS3 cDNA released from pCMV6-Entry-SOCS3 (Origene) using BamHI and XhoI was subcloned into pCMV-3Tag-8 (Stratagene) to generate the SOCS3-Flag expression plasmid. SOCS3 cDNA with C-Terminal HA tag added by PCR was subcloned into pIREShyg3 to generate pIREShyg3-SOCS3-HA plasmid.
Transfection and stable transfected cells
All transfections were done with Fugene 6 transfection reagent (Roche). Gprc5a−/− and MDA959 cells were transfected with pIREShyg3 or pIREShyg3-Stat3(Y705F)-Flag expression vector. After 48 hours, stably transfected cells were selected by using hygromycin from Clontech. The Gprc5a−/− cells were also transfected with pIREShyg3-SOCS3-HA plasmid. The HEK293T cells were co-transfected transiently with the GPRC5A and SOCS3 expression plasmids mentioned above.
Immunoblotting
Cells were washed with PBS then lysed to extract total proteins using cell lysis buffer (Cell Signaling Technology) and subjected to western immunoblotting as described before (15). The signal strengths were quantitated by densitometric analysis using Quantity One software.
Enzyme-linked immunosorbent assay
Conditioned media from 24 hr cell cultures were analyzed by Lif Quantikine ELISA Kit from R&D Systems.
RNA extraction and real-time quantitative PCR
Total RNA was extracted from Gprc5a+/+ and Gprc5a−/− cells using TRI Reagent from Molecular Research Center and analyzed by Quantitative real-time PCR reaction (QPCR) using Applied Biosystems 7500 Fast Real-Time PCR System. The primers were from Applied Biosystems. Mouse Actin was used as an internal control gene. The expression data was normalized to actin and analyzed using the 7500 Fast System Software.
Anchorage-independent growth assay
Cells were suspended in 0.2 ml of Matrigel (Collaborative Biomedical Products, Becton Dickinson Labware) diluted 1:1 (vol/vol) with growth medium. Colony formation was evaluated after two weeks as described before (15).
Apoptosis assay
Apoptosis was induced by starving cells in K-SFM medium without supplements for 48 hours and measured by using FITC-Annexin V Apoptosis Detection Kit (BD Biosciences) using the manufacturer’s instructions.
Statistical analyses
All analyses were performed in triplicates and the significance of differences etween groups was calculated using the student’s t test. P < 0.05 was considered to be statistically significant.
Results
Decreased starvation-induced apoptosis and increased anchorage-independent growth in Gprc5a−/− cells compared with Gprc5a+/+ cells
Gprc5a−/− and Gprc5a+/+ cells showed the expected differential expression of the Gprc5a mRNA (Fig. 1A, upper) and protein (Fig. 1A, lower). The Gprc5a−/− cells were more resistant than Gprc5a+/+ cells to apoptosis induced by starvation (incubation in K-SFM without EGF and BPE) for 48 hours (Fig. 1B, upper). Starvation induced 22.9% cell death in Gprc5a+/+ cells compared with only 9.1% in Gprc5a−/− cells (Fig. 1B, lower) indicating that loss of Gprc5a resulted in increased cell survival. Next, we compared the ability of Gprc5a+/+ and Gprc5a−/− cells to form colonies in semi-solid medium. Although both cell types failed to form colonies in agar or agarose (data not shown), Gprc5a−/− cells formed many more colonies than Gprc5a+/+ cells when cultured in suspension in Matrigel (Fig. 1C) suggesting that the knockout cells expressed a more transformed phenotype (20).
Figure 1.

Properties of cultured Gprc5a+/+ and Gprc5a−/− cells. A, Cells were cultured in K-SFM with EGF and BPE for 24 hours and extracted for mRNA analysis by QPCR (upper) and for protein analysis by western blotting (lower). B, Cells were starved for 48 hours then harvested and subjected to analysis of apoptosis. Upper shows results of one such assay and lower shows the mean ± SD of three independent experiments. C, Cells were suspended in Matrigel and cultured for two weeks. Upper shows photomicrographs of cultures at high magnification and colonies in three wells (each cell line) were counted and the data are presented in bar graph as mean ± SD colonies/well (lower). *, P < 0.05.
EGF-independent autocrine activation of Stat3 in Gprc5a−/− cells
STAT3 is activated in the majority of lung cancers and has been implicated in cell survival and transformation (21, 22). Therefore, we asked whether Stat3 activation is different between Gprc5a+/+ and Gprc5a−/− cells. We found that the tyrosine 705 phosphorylated Stat3 [pStat3(Y705)], which is the activated form, was higher in Gprc5a−/− cells compared with Gprc5a+/+ cells when these cells were cultured in K-SFM supplemented with EGF and BPE (Fig. 2A, left). Because EGFR signaling was reported to activate Stat3 (23–25), we examined the effect of EGF on Stat3 activation in these cells. We found that EGF signaling was activated similarly in these cells as indicated by the transient increase in pErk1/2 level. In contrast, pStat3 level, which was higher in Gprc5a−/− cells than in Gprc5a+/+ cells before EGF treatment, decreased 30 min after EGF addition and began to increase slightly after 3 hours (Fig. 2A, middle) suggesting that EGFR activation does not mediate Stat3 phosphorylation in either of these cells. The surprising decrease in pStat3 early after EGF treatment of the Gprc5a−/− cells was reversed after longer incubation (> 3 hours) and remained high up to 12 hours (Fig. 2A, right). A similar response pattern but with a substantially lower level of pStat3 was observed in Gprc5a+/+ cells. Because in this experiment, we replaced the “old” EGF-free cell growth medium with fresh EGF-supplemented medium, we surmised that the transient decrease in pStat3 level is due to removal of autocrine factor(s) from the conditioned medium (“old”). Indeed, we found that Stat3 was activated in Gprc5a−/− cells cultured in medium without EGF for 24 hours but the level of pStat3 decreased sharply in parallel cultures one hour after replacing culture medium conditioned for the preceding 23 hours with fresh medium containing no EGF (Fig. 2B, left). We then considered two possible explanations for the different level of pStat3 between Gprc5a+/+ and Gprc5a−/− cells: 1) Gprc5a−/− cells generate more autocrine Stat3 activator(s) than Gprc5a+/+ cells do and 2) both cell types produce autocrine factor(s) but differ in the response to such factor(s). We found that 24-hours conditioned media from either Gprc5a+/+ or Gprc5a−/− cells activated Stat3 in Gprc5a−/− cells, whereas the response of Gprc5a+/+ cells to these media was very low (Fig. 2B, right). The increased pStat3 level in Gprc5a−/− cells was associated with increased expression of several Stat3-regulated cell survival genes including at both the mRNA (Fig. 2C) and protein (Fig. 2D) levels. These results suggest that the difference in Stat3 activation between Gprc5a+/+ and Gprc5a−/− cells is due to their different response to some autocrine Stat3 activator(s).
Figure 2.

EGF-independent Stat3 activation in Gprc5a−/− cells. A, left, Gprc5a+/+ and Gprc5a−/− cells were incubated in K-SFM with BPE and EGF (5 ng/ml) for 24 hours and then extracted and analyzed by immunoblotting. Middle, Gprc5a+/+ and Gprc5a−/− cells were starved overnight in K-SFM then treated with EGF (10 ng/ml) in K-SFM for up to 3 hours, then harvested and subjected to immunoblotting. Right, cells were starved overnight in K-SFM, treated with EGF (10 ng/ml) for up to 12 hours, and analyzed by immunoblotting. This treatment was done by replacing the basal culture medium with EGF containing medium at different times and harvesting the cells at the same time. The 0 hour point represents cells cultured in medium without EGF for 12 hours. The cells were harvested and subjected to immunoblotting. B, left, Gprc5a+/+ and Gprc5a−/− cells were cultured in K-SFM and harvested after 24 hours. In parallel cultures, the cells were cultured for 23 hours and then their “old” medium was replaced with fresh medium and the cells were incubated in this medium for one hour. The cells were harvested and analyzed by immunoblotting. Right, Gprc5a+/+ and Gprc5a−/− cells were incubated in medium conditioned for 24 hours by other cultures of either Gprc5a+/+ or Gprc5a−/− cells as indicated. The cells were harvested after one hour and analyzed by immunoblotting. C, Gprc5a+/+ and Gprc5a−/− cells were starved for 48 hours and extracted for mRNA analysis by QPCR for Stat3 regulated genes. *, P < 0.05. D, cells treated as in C were analyzed by western blotting for Stat3 regulated proteins. Actin was used to control for loading.
Persistent activation of Stat3 in Gprc5a−/− cells is mediated by autocrine Lif
Il-6 and related cytokines are known to activate Stat3 by a Jak-dependent pathway. Therefore, we decided to study the effect of AG490 [alpha-cyano-(3,4-dihydroxy)-N-benzylcinnamide] (12), one of the most studied Janus kinase 2 (Jak2) inhibitors that blocks Stat3 activation (13). The Gprc5a−/− cells were treated with 30 µM AG490 for 24 hours based on many reports in which this inhibitor was used at concentrations between 25 and 100 µM for 16 to 24 hours to block Jak2 and stat3 in vitro. We found that AG490 completely blocked the tyrosine (Y705) phosphorylation of Stat3 indicating that Stat3 was activated by the Jak/Stat3 pathway (Fig. 3A). We then examined the effects of neutralizing antibodies against the Jak/Stat3 activators Il-6, Lif, and Osm on Stat3 activation in Gprc5a−/− cells. Antibody against Lif suppressed Stat3 activation, whereas antibodies against Il-6 and Osm failed to do so (Fig. 3B, upper), indicating that Lif might be the autocrine activator of Stat3 in Gprc5a−/− cells. Because conditioned medium from Gprc5a+/+ cells also activated Stat3 in Gprc5a−/− cells (Fig. 2B, right) we next asked whether Lif was mediating this effect. We found that Lif antibody was able to suppress the ability of conditioned medium from Gprc5a+/+ and Gprc5a−/− cells to activate Stat3 in Gprc5a−/− cells indicating that Lif is the major autocrine Stat3 activator in both cells (Fig. 3B, lower). We found that Gprc5a−/− cells released more Lif into their conditioned medium than Gprc5a+/+ cells (Fig. 3C). However, since conditioned media from both Gprc5a+/+ and Gprc5a−/− cells activated Stat3 in Gprc5a−/− cells to a similar level (Fig. 2B, right), Lif level was not the limiting factor for the different Stat3 activation between the two cell types. To further assess the differential response to Lif, we treated these cells with exogenous Lif and found that Stat3 activation was transient in the Gprc5a+/+ cells, peaking around 30 min and declining rapidly by 60 min (Fig. 3D). In contrast, Stat3 activation in Gprc5a−/− cells was persistent; after peaking at 30 min it remained activated for at least 90 min (Fig. 3D). These results suggest that Gprc5a loss leads to a prolonged response to Lif as indicated by persistent Stat3 activation that was also reported to occur in various tumor cells (26).
Figure 3.

Identification of Lif as an autocrine inducer of persistent Stat3 activation in Gprc5a−/− cells. A, Gprc5a−/− cells were treated with AG490 (30 µM) or DMSO (0.1% V:V) in K-SFM for 24 hours. The cells were then harvested and analyzed by immunoblotting. B, upper, Gprc5a−/− cells were cultured in K-SFM with neutralizing antibodies against mouse Il-6, Lif, and Osm or with normal goat IgG (all at 30 µg/ml) for 24 hours. The cells were then analyzed for Stat3 activation by immunoblotting. Lower, media were conditioned for 24 hours by Gprc5a+/+ and Gprc5a−/− cell cultures then collected and centrifuged. The supernatants were treated for 1 hour with Lif neutralizing antibody or with normal goat IgG (both at 30 µg/ml). These media were used to treat Gprc5a−/− cells that had been starved for one hour by incubation in fresh K-SFM. After 1 hour of incubation with the treated media, the cells were analyzed for Stat3 activation. C, Gprc5a+/+ and Gprc5a−/− cells were cultured in fresh K-SFM at 2 × 105 per 35-mm diameter well and their 24 hours conditioned media were collected and analyzed by ELISA for secreted Lif. D, upper, Gprc5a+/+ and Gprc5a−/− cells were treated with exogenous Lif (1000 units/ml) in K-SFM. After the indicated times cells were harvested and analyzed for Stat3 activation by immunoblotting. Lower, Graphic representation of the ratios of pStat3 to Stat3 calculated from the intensities of western blotting bands on X-Ray films acquired using Quantity One software.
Autocrine Lif mediates Stat3 activation in MDA959 lung tumor cells
Next we investigated whether Stat3 was also activated in MDA959 tumor cells. We found that MDA959 cells had a constitutively active Stat3, which was completely inhibited by AG490 (Supplementary Fig. S1A), implicating the Jak/Stat3 signaling pathway in this activation. MDA959 cells were found to secrete Lif into their medium (Supplementary Fig. S1B) and Lif antibody decreased the level of pStat3 induced by conditioned medium from MDA959 cells (Supplementary Fig. S1C), suggesting that autocrine Lif activated Stat3 also in these lung tumor cells.
Implication of reduced level of Socs3 in Stat3 activation in Gprc5a−/− cells
Because Socs3 is an endogenous Stat3-inducible inhibitor of Stat3, we asked whether it is involved in the differential response of Gprc5a+/+ and Gprc5a−/− cells to Lif. We found that the level of Socs3 mRNA was slightly higher, whereas the protein level was lower in the knockout cells suggesting that it was regulated at the post-transcriptional level (Fig. 4A). To determine whether the decreased Socs3 protein in Gprc5a−/− cells was caused by increased proteasome-mediated degradation, we treated both Gprc5a+/+ and Gprc5a−/− cells with the proteasome inhibitor MG132 and found that it did not increase the Socs3 protein level in either cell line (Fig. 4B), suggesting the Socs3 protein is not regulated by proteasome mediated degradation in these cells. Over-expression of exogenous human SOCS3 in the knockout cells suppressed Stat3 activation by autocrine as well as exogenous Lif (Fig. 4C). These results indicate that the lower Socs3 level in the knockout cells compared to the Gprc5a+/+ cells may be responsible for the persistent versus transient activation of Stat3 in the two cell lines. Using HEK 293T cells, which do not express endogenous GPRC5A (unpublished results), co-transfected with Myc-tagged GPRC5A and Flag-tagged SOCS3 or with the SOCS3 expression vector only, we found that the level of SOCS3 was higher in the presence of GPRC5A and its rate of degradation was lower in the presence of GPRC5A (Fig. 4D). These findings suggest that GPRC5A can stabilize SOCS3.
Figure 4.
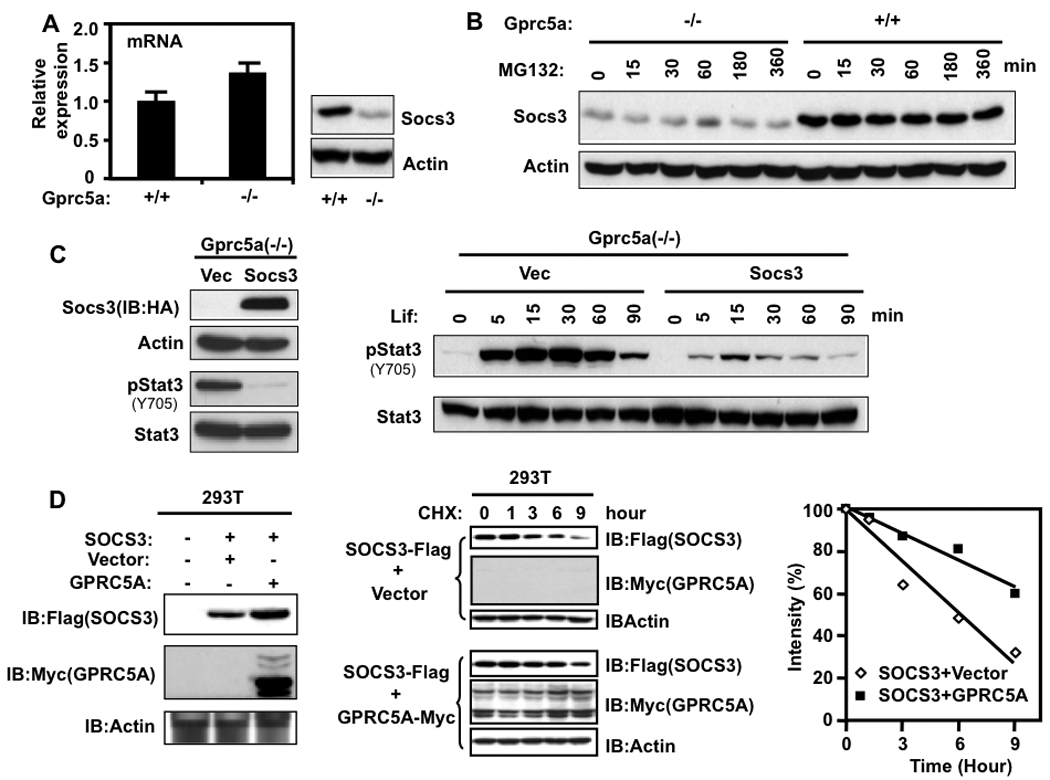
Involvement of Socs3 protein in the persistent activation of Stat3 in Gprc5a−/− cells. A, Gprc5a+/+ and Gprc5a−/− cells were starved for 48 hours and then their total mRNA was extracted and analyzed for Socs3 mRNA by QPCR (left) and their total protein was analyzed for Socs3 protein by immunoblotting (right). B, cells were treated with 10 µM MG132 for different times and analyzed by immunoblotting. C, left, Gprc5a−/− cells transfected with vector or SOCS3-HA were starved for 48 hours then extracted for protein analysis by immunoblotting. Right, Gprc5a−/− cells transfected with vector or SOCS3-HA were treated with exogenous Lif (1000 units/ml) in K-SFM. After the indicated times, cells were harvested and analyzed for Stat3 activation by immunoblotting. D, left, HEK293T cells transfected with the indicated vectors were cultured for 48 hours and then analyzed by immunoblotting. Middle, HEK293T cells transfected with the indicated vectors for 48 hours were treated with 20 µM cycloheximide for different times and then the cells were analyzed by immunoblotting. Right, Densitometry results for the western blotting bands in the middle panel were plotted.
Inhibition of Jak/Stat3 signaling in Gprc5a−/− cells and MDA959 lung tumor cells partially reverses the transformed phenotype
Stat3(Y705F) is a specific dominant negative inhibitor of Stat3 (18, 27). Stable transfection of Stat3(Y705F) expression vector decreased Stat3 tyrosine phosphorylation in Gprc5a−/− cells compared to control vector (Fig. 5A, three top panels). Stat3(Y705F) expression suppressed several antiapoptotic Stat3-regulated genes at the protein (Fig. 5A, lower 5 panels) and mRNA levels (Fig. 5B). The abrogation of Stat3 activation in Stat3(Y705F) transfected Gprc5a−/− cells increased starvation-induced apoptosis (Fig. 6A) and decreased the number of colonies in semi-solid medium (Fig. 6B) relative to control vector. Similar effects were observed after transfection of Stat3(Y705F) into tumorigenic MDA959 cells (Supplementary Fig. S2). These data suggest that Stat3 activation is important for the transformed phenotypes of Gprc5a−/− cells and MDA959 tumor cells.
Figure 5.
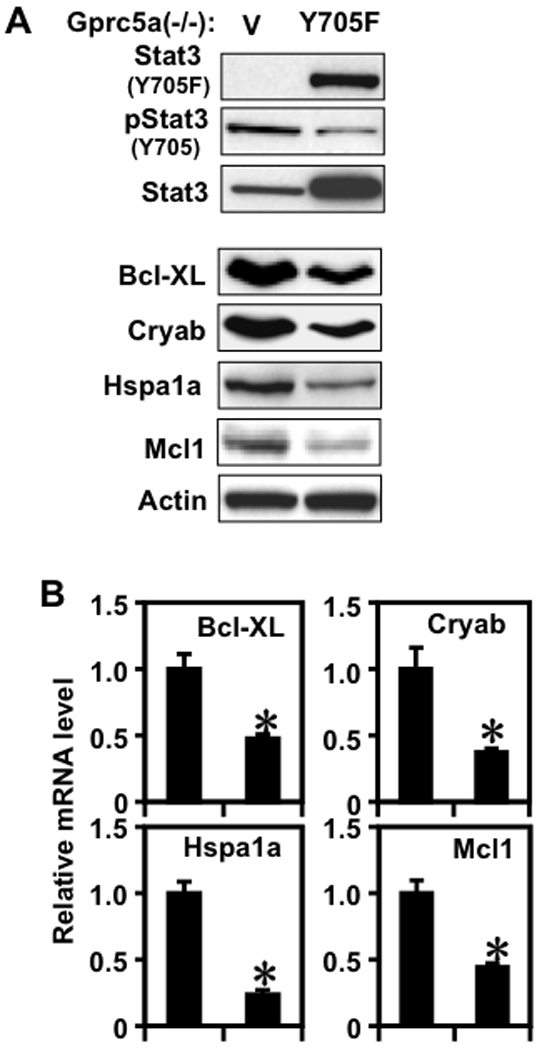
Dominant negative Stat3 inhibits Stat3 activation and the expression of anti-apoptotic genes. Gprc5a−/− cells transfected with vector or Stat3(Y705F) were starved for 48 hours then extracted for protein analysis by immunoblotting (A) and for mRNA analysis by QPCR (B) for Stat3 activation and regulated genes.
Figure 6.
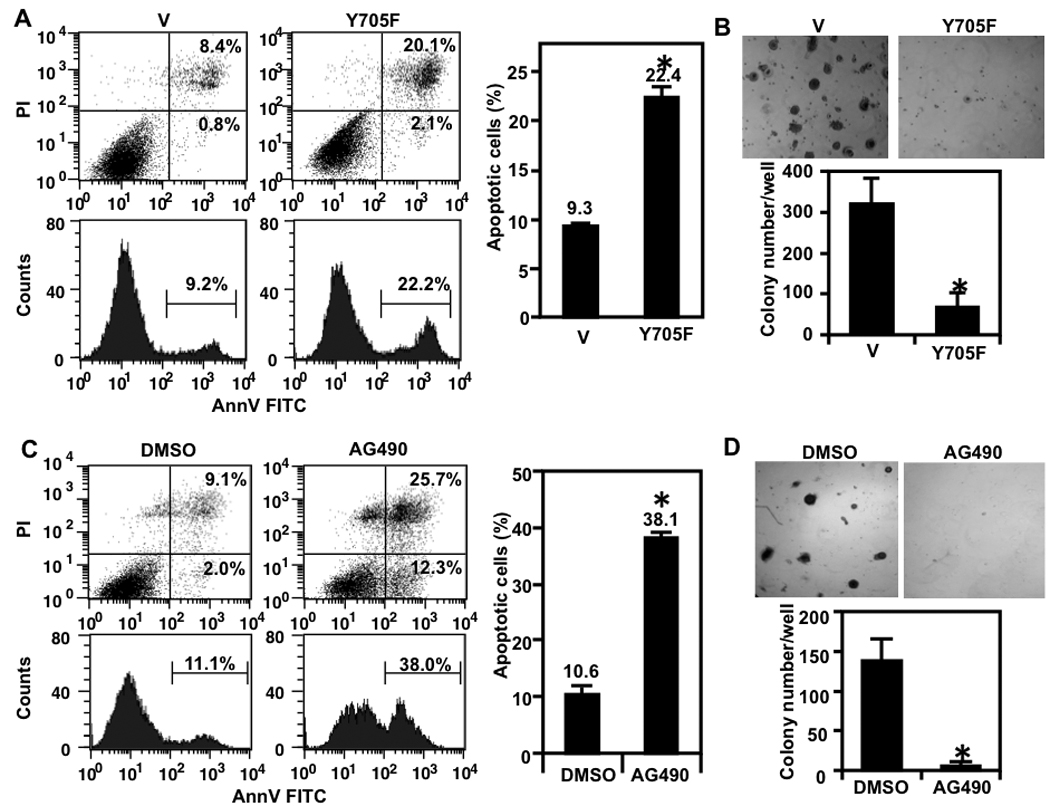
Dominant negative Stat3 and AG490 increase stress-induced apoptosis and inhibit colony formation in Gprc5a−/− cells. A. Gprc5a−/− cells transfected with vector or Stat3(Y705F) were starved for 48 hours then harvested and subjected to analysis of apoptosis. Left shows results of one assay and right shows the mean ± SD of three independent experiments. B, Gprc5a−/− cells transfected with vector or Stat3(Y705F) were suspended in Matrigel and analyzed for colony formation after two weeks. Photomicrographs of cultures at high magnification are shown (upper) and the colonies in three wells were counted and the data are presented in bar graph as mean ± SD colonies/well (lower). C, Gprc5a−/− cells were treated with AG490 (30 µM) or DMSO in K-SFM for 48 hours then harvested and subjected to analysis of apoptosis. Left shows the flow cytometry results of one assay and right shows the mean ± SD of three independent experiments. D, Gprc5a−/− cells were suspended in Matrigel with AG490 (30 µM) or DMSO and analyzed for colony formation after two weeks. Photomicrographs of cultures at high magnification are shown (upper) and the colonies in three wells (each treatment) were counted and the data are presented in bar graph as mean ± SD colonies/well (lower). *, P < 0.05.
Having demonstrated that AG490 can suppress Stat3 activation in both Gprc5a−/− (Fig. 3A) and MDA959 cells (Supplementary Fig. S1A), we examined the effects of this small molecule on cell survival and anchorage-independent growth and found that treatment with AG490 increased starvation-induced apoptosis by about 3 fold and dramatically decreased colony formation in Matrigel in Gprc5a−/− cells (Fig. 6C and D, respectively) and in MDA959 cells (Supplementary Fig. S2). These results indicate that inhibition of Stat3 activation by AG490 can also suppress the expression of the transformed phenotype of these cells.
Discussion
We explored potential mechanism(s) of action of the orphan G protein coupled receptor Gprc5a, which functions as a tumor suppressor in mouse lung in vivo (15). Epithelial cell lines isolated from tracheas of wild type mice and knockout mice as well as from a spontaneous lung adenocarcinoma from an adult Gprc5a−/− mouse (MDA959) exhibited different characteristics. Cells from normal trachea of a Gprc5a−/− mouse exhibited resistance to starvation-induced apoptosis and ability to form colonies in semi-solid medium, both of which are features associated with cell transformation (28). In contrast, Gprc5a+/+ cells were more sensitive to starvation induced apoptosis than the Gprc5a−/− cells and failed to grow in semi-solid medium. In these two properties, the Gprc5a−/− cells were comparable to the MDA959 adenocarcinoma cells. However, the MDA959 cells are tumorigenic, whereas the Gprc5a−/− cells are nontumorigenic. Thus, our findings demonstrate that the Gprc5a−/− cells exhibit properties associated with the transformed cell phenotype but not the tumorigenic phenotype and therefore, they may be considered premalignant cells (20, 28). It is important to note that the acquisition of the malignant (tumorigenic) phenotype by cultured epithelial cells may require multiple genetic and epigenetic changes because human bronchial epithelial cells immortalized by overexpression of the human telomerase reverse transcriptase, transfection of CDK4 to bypass the p16/RB pathway, silencing the p53 pathway and introduction of mutant K-RASV12 or mutant EGFR, were able to form colonies in semi-solid medium but were still non-tumorigenic (29).
The relative resistance of Gprc5a−/− cells to starvation-induced apoptosis compared to Gprc5a+/+ cells suggests increased self-sufficiency in growth signals, enhanced ability to evade apoptosis or both (28). Because Stat3 activation plays important roles in both mouse and human lung cancer development (23, 30), we examined it in our cells and found that Stat3 signaling pathway was activated in Gprc5a−/− cells to a higher level than in Gprc5a+/+ cells. This activation was independent of exogenous EGF but depended on factor(s) released by both Gprc5a+/+ and Gprc5a−/− cells. The increased expression of antiapoptotic Stat3 target genes including Bcl-XL, Cryab, Hspa1a, and Mcl1 in the Gprc5a−/− cells is a likely mechanism for their relative resistance to apoptosis.
Further studies using the Jak/Stat3 inhibitor AG490 demonstrated that Jak activity is required for Stat3 activation and identified Lif, a member of the Il-6 family cytokines, as the autocrine mediator of Stat3 activation in the Gprc5a−/− cells. Previous reports have shown that human carcinoma cell lines including lung cancer produce Lif (31) and that Lif functions as an autocrine or paracrine growth factor (32). Our studies have shown that while both Gprc5a+/+ and Gprc5a−/− cells produce and release Lif, their response to Lif was different insofar as Lif induced a persistent Stat3 activation in the Gprc5a−/− cells but only a transient activation of Stat3 in the Gprc5a+/+ cells. This difference in Stat3 activation may explain the higher levels of the anti-apoptotic genes and proteins and the partial resistance of the cells to starvation. We also found that Stat3 was constitutively activated by autocrine Lif in MDA959 lung tumor cells suggesting that Lif mediated Stat3 activation happened in both premalignant and malignant cells.
The mechanism by which Stat3 is activated persistently in Gprc5a−/− cells but only transiently in Gprc5a+/+ cells may be related to the difference in the levels of the Socs3 protein in these cells. The higher level of Socs3 in the wildtype cells appears to be due to its stabilization by Gprc5a. Therefore, the absence of Gprc5a in the knockout cells leads to a decrease in Socs3 level. This is a new mechanism for Socs3 regulation leading to Stat3 activation. Previous reports highlighted the role of promoter methylation as a mechanism for SOCS3 suppression in human lung cancers (33).
The importance of Stat3 activation for expression of the transformed phenotype in Gprc5a−/− cells and in MDA959 cells was demonstrated by the finding that blocking Stat3 activation by dominant negative Stat3(Y705F) or by AG490 increased the sensitivity of both cell lines to starvation-induced apoptosis and decreased their colony forming potential. Interestingly, transgenic mice overexpressing constitutively activated Stat3 in alveolar type II epithelial cells were reported to develop spontaneous lung adenocarcinomas (30).
The relevance of our findings with mouse cells to human lung cancer is indicated by our previous findings that GPRC5A expression in various lung cancers (NSCLC both adenocarcinomas and squamous cell carcinomas) is much lower than in normal lung tissues (15). Others reported that STAT3 is persistently activated by chronic stimulation of JAK by cytokines in a variety of human tumors including lung tumors (4, 34). Further, STAT3 target genes have been proposed as biomarkers for chronic obstructive pulmonary disease (COPD) and lung adenocarcinoma diagnosis and prognosis (35).
In conclusion, based on our findings we propose that Gprc5a can suppress Stat3 activation by stabilizing Socs3 and that the loss of its expression leads to persistent Stat3 activation induced by autocrine Lif in normal Gprc5a−/− cells and in MDA959 cells. This effect is important for the growth of these cells in vitro and may also be important for the development of lung cancer in the Gprc5a−/− mice in vivo. For example, it would be interesting to cross our Gprc5a−/− mice with lung specific Stat3−/− mice (36) or with Lif−/− mice (37) and determine whether the double knockout (Gprc5a−/−Stat3−/−) or (Gprc5a−/−Lif−/−) mice develop less lung tumors compared to Gprc5a−/− mice.
Supplementary Material
Acknowledgements
This study was supported by the Samuel Waxman Cancer Research Foundation and by the Cancer Center Support Grant P30 CA16672 (the Flow Cytometry and the DNA Analysis Cores). We thank Dr. James E. Darnell, Jr. for the dominant negative Stat3 plasmid.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Meuwissen R, Berns A. Mouse models for human lung cancer. Genes Dev. 2005;19:643–664. doi: 10.1101/gad.1284505. [DOI] [PubMed] [Google Scholar]
- 3.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 4.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 5.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeh TC, Pellegrini S. The Janus kinase family of protein tyrosine kinases and their role in signaling. Cell Mol Life Sci. 1999;55:1523–1534. doi: 10.1007/s000180050392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 8.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Sekkai D, Gruel G, Herry M, et al. Microarray analysis of LIF/Stat3 transcriptional targets in embryonic stem cells. Stem Cells. 2005;23:1634–1642. doi: 10.1634/stemcells.2005-0182. [DOI] [PubMed] [Google Scholar]
- 11.Costantino L, Barlocco D. STAT 3 as a target for cancer drug discovery. Curr Med Chem. 2008;15:834–843. doi: 10.2174/092986708783955464. [DOI] [PubMed] [Google Scholar]
- 12.Meydan N, Grunberger T, Dadi H, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 13.Levitzki A. Tyrosine kinases as targets for cancer therapy. Eur J Cancer. 2002;38 Suppl 5:S11–S18. doi: 10.1016/s0959-8049(02)80598-6. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Y, Lotan R. Molecular cloning and characterization of a novel retinoic acid-inducible gene that encodes a putative G protein-coupled receptor. J Biol Chem. 1998;273:35008–35015. doi: 10.1074/jbc.273.52.35008. [DOI] [PubMed] [Google Scholar]
- 15.Tao Q, Fujimoto J, Men T, et al. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst. 2007;99:1668–1682. doi: 10.1093/jnci/djm208. [DOI] [PubMed] [Google Scholar]
- 16.Burger M, Hartmann T, Burger JA, Schraufstatter I. KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene. 2005;24:2067–2075. doi: 10.1038/sj.onc.1208442. [DOI] [PubMed] [Google Scholar]
- 17.Ho MK, Su Y, Yeung WW, Wong YH. Regulation of transcription factors by heterotrimeric G proteins. Curr Mol Pharmacol. 2009;2:19–31. doi: 10.2174/1874467210902010019. [DOI] [PubMed] [Google Scholar]
- 18.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18:2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen Z, Darnell JE., Jr Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–2067. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tucker RW, Sanford KK, Handleman SL, Jones GM. Colony morphology and growth in agarose as tests for spontaneous neoplastic transformation in vitro. Cancer Res. 1977;37:1571–1579. [PubMed] [Google Scholar]
- 21.Song L, Turkson J, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22:4150–4165. doi: 10.1038/sj.onc.1206479. [DOI] [PubMed] [Google Scholar]
- 22.Gao J, McConnell MJ, Yu B, et al. MUC1 is a downstream target of STAT3 and regulates lung cancer cell survival and invasion. Int J Oncol. 2009;35:337–345. [PMC free article] [PubMed] [Google Scholar]
- 23.Gao SP, Mark KG, Leslie K, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 25.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H, Herrmann A, Deng JH, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakajima K, Yamanaka Y, Nakae K, et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 1996;15:3651–3658. [PMC free article] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 29.Sato M, Vaughan MB, Girard L, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–2128. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Du H, Qin Y, Roberts J, Cummings OW, Yan C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007;67:8494–8503. doi: 10.1158/0008-5472.CAN-07-0647. [DOI] [PubMed] [Google Scholar]
- 31.Kamohara H, Sakamoto K, Ishiko T, et al. Human carcinoma cell lines produce biologically active leukemia inhibitory factor (LIF) Res Commun Mol Pathol Pharmacol. 1994;85:131–140. [PubMed] [Google Scholar]
- 32.Kellokumpu-Lehtinen P, Talpaz M, Harris D, Van Q, Kurzrock R, Estrov Z. Leukemia-inhibitory factor stimulates breast, kidney and prostate cancer cell proliferation by paracrine and autocrine pathways. Int J Cancer. 1996;66:515–519. doi: 10.1002/(SICI)1097-0215(19960516)66:4<515::AID-IJC15>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 33.He B, You L, Uematsu K, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100:14133–14138. doi: 10.1073/pnas.2232790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qu P, Roberts J, Li Y, et al. Stat3 downstream genes serve as biomarkers in human lung carcinomas and chronic obstructive pulmonary disease. Lung Cancer. 2009;63:341–347. doi: 10.1016/j.lungcan.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hokuto I, Ikegami M, Yoshida M, et al. Stat-3 is required for pulmonary homeostasis during hyperoxia. J Clin Invest. 2004;113:28–37. doi: 10.1172/JCI200419491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Escary JL, Perreau J, Duménil D, Ezine S, Brûlet P. Leukaemia inhibitory factor is necessary for maintenance of haematopoietic stem cells and thymocyte stimulation. Nature. 1993;363:361–364. doi: 10.1038/363361a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.