

Abstract
BACKGROUND AND PURPOSE
Oxidative stress [i.e. increased levels of reactive oxygen species (ROS)] has been suggested as a pathomechanism of different diseases, although the disease-relevant sources of ROS remain to be identified. One of these sources may be NADPH oxidases. However, due to increasing concerns about the specificity of the compounds commonly used as NADPH oxidase inhibitors, data obtained with these compounds may have to be re-interpreted.
EXPERIMENTAL APPROACH
We compared the pharmacological profiles of the commonly used NADPH oxidase inhibitors, diphenylene iodonium (DPI), apocynin and 4-(2-amino-ethyl)-benzolsulphonyl-fluoride (AEBSF), as well as the novel triazolo pyrimidine VAS3947. We used several assays for detecting cellular and tissue ROS, as none of them is specific and artefact free.
KEY RESULTS
DPI abolished NADPH oxidase-mediated ROS formation, but also inhibited other flavo-enzymes such as NO synthase (NOS) and xanthine oxidase (XOD). Apocynin interfered with ROS detection and varied considerably in efficacy and potency, as did AEBSF. Conversely, the novel NADPH oxidase inhibitor, VAS3947, consistently inhibited NADPH oxidase activity in low micromolar concentrations, and interfered neither with ROS detection nor with XOD or eNOS activities. VAS3947 attenuated ROS formation in aortas of spontaneously hypertensive rats (SHRs), where NOS or XOD inhibitors were without effect.
CONCLUSIONS AND IMPLICATIONS
Our data suggest that triazolo pyrimidines such as VAS3947 are specific NADPH oxidase inhibitors, while DPI and apocynin can no longer be recommended. Based on the effects of VAS3947, NADPH oxidases appear to be a major source of ROS in aortas of SHRs.
Keywords: hypertension, NADPH oxidase, NOX, oxidative stress, reactive oxygen species
Introduction
Growing evidence suggests that an overproduction of reactive oxygen species (ROS), such as superoxide, hydrogen peroxide, hydroxyl radicals and peroxynitrite, contributes to the pathogenesis of cardiovascular diseases (Stocker and Keaney, 2004). With respect to the relevant enzymatic sources of these ROS, the family of NADPH oxidases has received considerable attention (Griendling et al., 2000; Cave et al., 2006).
NADPH oxidases are comprised of membrane proteins (i.e. the catalytic flavin–heme protein); NOX, of which five isoforms exist (NOX1–5); and the non-catalytic 22 kDa binding protein, p22phox (Lassegue and Clempus, 2003). Other components of the NADPH oxidase complex can include a cytosolic organizer (p47phox or NOXO1), an activator (p67phox or NOXA1) and other proteins (p40phox and Rac). These cytosolic subunits vary with the different NOX isoforms (Opitz et al., 2007). Upon phosphorylation, cytosolic subunits translocate to the NOX-containing membrane resulting in ROS formation. However, NOX4 and NOX5 may be independent of cytosolic subunits and constitutively active (Brandes and Kreuzer, 2005; Bedard and Krause, 2007; Lambeth et al., 2007; Opitz et al., 2007) or regulated by intracellular free calcium (Bedard and Krause, 2007) respectively.
The only known function of NADPH oxidases is to generate ROS. This feature makes NADPH oxidases distinct from other sources of ROS such as xanthine oxidase (XOD) or uncoupled NOS as these enzymes produce ROS only after conversion to a dysfunctional state. This is triggered by ROS derived from a primary source. NADPH oxidases may be this primary source of ROS, initiating oxidative stress and converting other enzymes into an ROS-producing state (McNally et al., 2003) to eventually induce cardiovascular pathophysiology. To test this hypothesis, both genetic and pharmacological approaches have been pursued.
According to studies using genetically modified mice, NOX1 plays a role in the pathophysiology of hypertension (Matsuno et al., 2005; Gavazzi et al., 2006) and in neointima formation in mice (Lee et al., 2009). However, pharmacological evidence – and thus the potential of translating these findings into therapeutic applications – has not yet been provided. This is mainly due to the lack of specific NADPH oxidase inhibitors. In particular, there are concerns about the specificity of compounds currently used as NADPH oxidase inhibitors. For example, one of the most frequently applied compounds, apocynin, may also act as an antioxidant (Heumuller et al., 2008) and a Rho kinase inhibitor (Schluter et al., 2008). Other compounds that have been widely used include 4-(2-aminoethyl)-benzenesulphonyl fluoride (AEBSF) (Bayraktutan, 2005), which blocks p47phox translocation to the membrane but also inhibits serine proteases (Diatchuk et al., 1997), as well as diphenylene iodonium (DPI). DPI also inhibits other flavin-containing enzymes such as NOS and XOD (O'Donnell et al., 1993), as well as cholinesterases and the internal calcium pump (Tazzeo et al., 2009).
We here directly compared the specificity and efficacy of the most commonly investigated NADPH oxidase inhibitors in three different cell lines, and isolated enzyme preparations using several assays for ROS detection. We also analysed the compounds in situ in vascular tissue sections of spontaneously hypertensive rats (SHRs). We are aware that many, if not all, of the ROS assays have limitations with respect to specificity and artefacts (Dikalov et al., 2007). Therefore, we followed current recommendations and used several of the most frequently used ROS detection systems in parallel to ensure that our data are not only representative, but also comparable to previous papers.
We included the triazolo pyrimidine VAS3947, a novel derivative of VAS2870 with higher solubility. VAS3947 has been developed by in silico optimization of VAS2870 (Tegtmeier et al., 2005), which was recently shown to inhibit NADPH oxidase activity in primary rat vascular smooth muscle cells (VSMCs) (ten Freyhaus et al., 2006), and human umbilical vein endothelial cells (HUVECs) (Stielow et al., 2006). VAS2870 also inhibited wound margin H2O2 production without obvious toxicity in zebrafish larvae (Niethammer et al., 2009), as well as platelet-derived growth factor (PDGF) BB-stimulated vasculogenesis of embryonic stem cell-derived endothelial cells (Lange et al., 2009). We here show that, among the tested compounds, only VAS3947 was an apparently specific inhibitor of NADPH oxidases, which did not interfere with commonly used ROS assays. Using this novel inhibitor, we also provide pharmacological evidence that NADPH oxidases are indeed a relevant source of ROS in the SHR model of hypertension. Having established the specificity, future experiments are justified to determine the mechanism of action, the isoform specificity and the in vivo actions of this novel NADPH oxidase inhibitor compound class, which is beyond the scope of this study.
Methods
RNA isolation and RT-PCR
Total RNA was isolated from the human CaCo-2 and HL60, and the rat A7r5 cell lines using the RNeasy Kit (Qiagen, Hilden, Germany) and treated with DNase I (Invitrogen, Karlsruhe, Germany) according to the manufacturers' protocols. Total RNA was then reverse transcribed by Superscript III using the protocol for random hexamer primers (Invitrogen). Thereafter, each reaction was treated with RNase H (Invitrogen) for 20 min at 37°C before PCR was performed [94°C 5 min, 94°C 1 min–60°C 1 min–72°C 30 s (35×), 72°C 10 min] using the following specific primers: human NOX1 (5′-tctctccagcctatctcatg-3′, 5′-ctcattcatgctctcctctg-3′), NOX2 (5′-tcctccaccaaaaccatccg-3′, 5′-aaaaccgcaccaacctctcac-3′), human NOX3 (5′-ctgccctgacagatgtatttc-3′, 5′-gtcagtattttcgtcccagtg-3′), human NOX4 (5′-tctggctctccatgaatgtc-3′, 5′-agaagttgagggcattcacc-3′), human NOX5 (5′-gtgcatcatggaagtcaacc-3′, 5′-ccaaaagtatctcagagccc-3′), or rat NOX1 (5′-cctgctcattttgcaaccac-3′, 5′-catgagaaccaaagccacag-3′), rat NOX2 (5′-gacagacttcggacagtttg-3′, 5′-actctagcttggatacctgg-3′) and rat NOX4 (5′-gtgtttgagcagagcttctg-3′, 5′-gtgaagagaagctttctggg-3′). Purified PCR fragments were subcloned in pCR2.1 TOPO (Invitrogen) and validated by sequencing (GENterprise Gesellschaft für Genanalyse und Biotechnologie mbH, Mainz, Germany).
Cell culture
A7r5 cells (rat, smooth muscle embryonic aorta, ATCC-No. CRL 1444) were cultured in Dulbecco's modified Eagle's medium (Sigma, Deisenhofen, Germany) supplemented with 0.1% glucose, 10% heat-inactivated calf serum, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 2 mM glutamine. CaCo-2 cells (human, adenocarcinoma, colon, ATCC-No. HTB 37) were cultured with medium of the same composition as for A7r5 cells, but additionally supplemented with 1% non-essential amino acids. Cells were cultured at 37°C under an atmosphere of 6% CO2 until they reached 70–80% confluence. Cells were washed with phosphate-buffered saline (PBS) buffer (2.7 mM KCl, 1.5 mM KH2PO4, 137 mM NaCl, 8 mM Na2HPO4, pH 7.3) and detached using a solution of trypsin (0.05%) and EDTA (0.02%) in PBS buffer. Subsequently, cells were counted and resuspended in reaction buffer [140 mM NaCl, 5 mM KCl, 0.8 mM MgCl2× 2H2O, 1.8 mM CaCl2× 2H2O, 1 mM Na2HPO4, 25 mM HEPES, 0,1% (w/v) glucose, complete EDTA-free protease inhibitor cocktail; pH 7.3] to a concentration of 2 × 106 cells·mL−1.
HL-60 cells (human, promyeloblast, ATCC-No. CCL 240) were cultured in RPMI-1640 medium (PAA Laboratories GmbH, Pasching, Austria) supplemented with 5% fetal calf serum, penicillin (100 U·mL−1), streptomycin (100 µg·mL−1) and glutamine (2 mM). Cell suspensions (5 × 105 cells·mL−1) were incubated with 1.25% DMSO for 6 days to induce differentiation into granulocyte-like cells. Differentiated cells were centrifuged at 300× g, washed with Hanks's balanced salt solution (HBSS, PAA Laboratories GmbH) and resuspended in HBSS to a final concentration of 4.4 × 106 cells·mL−1.
NADPH oxidase activity assay in cell homogenates
In CaCo-2 and A7r5 whole-cell homogenates (WCHs), NADPH oxidase-catalysed ROS formation was measured using lucigenin-enhanced chemiluminescence according to the protocol of Li and Shah (2001) with slight modifications. A7r5 and CaCo-2 cells were suspended in reaction buffer (2 × 106 cells·mL−1) and then underwent three freeze (−20°C) thaw cycles to produce WCH. We routinely confirmed complete lysis of the cells under the microscope. The chemiluminescent probe lucigenin was added to WCH to a final concentration of 5 µM. Then, 100 µL aliquots of WCH (2 × 105 cells equivalent) were transferred to individual wells of an opaque white 96-well plate. Inhibitors were directly added to the wells in the specified concentrations, and the reaction mixture was incubated for 30 min at 37°C in the dark. Subsequently, the reaction was started by the addition of 100 µM NADPH, and chemiluminescence was recorded every 3 min over a period of 9 min (CaCo-2) or 21 min (A7r5) in a luminescence reader (Fluoroscan Ascent FL; Thermo Labsystems, Vantaa, Finland). All measurements were performed in duplicate and repeated at least three times. Results are expressed as area under the curve (AUC), normalized as either a percentage of the NADPH-induced signal in untreated WCH or as a percentage of the superoxide dismutase (SOD) (250 U·mL−1)/catalase (CAT) (1000 U·mL−1) inhibitable control signal.
Whole-cell NADPH oxidase activity assay
During the oxidative burst stimulated by phorbol myristate acetate (PMA) in HL-60 cells, NADPH oxidase activity was measured using the superoxide-specific chemiluminescent probe L012. L012 was added to DMSO-differentiated HL-60 cell suspensions to a final concentration of 100 µM. At this concentration, L012 is reportedly devoid of redox cycling properties (Sohn et al., 1999). Then, 100 µL aliquots of cell suspension (4.4 × 105 cells) were transferred to individual wells of a 96-well plate. Inhibitors were directly added to the wells at the specified concentrations. Following a 30 min incubation at 37°C in the dark, the oxidative burst was initiated by the addition of 100 nM PMA, and the chemiluminescence signal was recorded every 5 min over a period of 60 min in the luminescence reader mentioned above. All measurements were performed in duplicate and repeated at least three times. Results were expressed as AUC normalized as a percentage of the signal generated by untreated PMA-stimulated HL-60 cells.
A second series of experiments was performed in which we used cytochrome c reduction as an alternative measure of superoxide production. Cytochrome c was added to the DMSO-differentiated HL-60 cell suspension to a final concentration of 100 µM. Then, 100 µL aliquots (4.4 × 105 cells) were transferred to individual wells of a 96-well plate. Following the direct addition of inhibitors, cells were incubated for 30 min at 37°C in the dark. Subsequently, the oxidative burst was initiated by the addition of PMA (final concentration 100 nM) and the cells incubated for 120 min at 37°C. Absorbance was then measured at 540 nm (isosbestic point of cytochrome c) and 550 nm (SpectraMax 340; Molecular Devices, Sunnyvale, CA, USA). Each experiment was performed in duplicate and repeated at least three times. Signals were calculated by normalization of the signals obtained at 540 nm. The blank signal (HBSS buffer, cytochrome c, PMA) was subtracted and all values expressed relative to the control signal (cells, cytochrome c, PMA).
Additional cytochrome c assays were performed using opsonized zymosan (OPZ)-treated HL-60 cells instead of PMA treated. OPZ was prepared from zymosan A from Saccharomyces cerevisiae (Vejrazka et al., 2005). Briefly, 5 mg·mL−1 zymosan A was incubated with normal human serum for 30 min at 37°C. Finally, zymosan was washed and resuspended in HBSS; 500 µg·mL−1 OPZ was used for stimulation of oxidative burst.
In situ tissue ROS assay
All animal care and experimental procedures complied with the recommendations of the Federation of European Laboratory Animals Science Association, and were approved by the local ethics committee. Rats (SHR and WKY) were bred in the animal facilities of the Rudolf Buchheim Institute, Giessen, and fed standard rodent chow and water ad libitum. Thoracic aortas of 12- to 14-month-old SHRs (n = 6) and WKY (n = 6) were embedded in Tissue Tek O.C.T. Compound (Sakura Finetek, Torrance, CA, USA). Unfixed frozen cross sections (5 µm) were incubated with dihydroethidium (DHE) (5 µM; Invitrogen, Darmstadt, Germany) in a light-protected moist chamber at 37°C for 30 min. Before incubation with DHE, serial sections were treated with either l-NAME (100 µM), pegylated SOD (PEG-SOD) (250 U·mL−1), oxypurinol (100 µM) or VAS3947 (10 µM) for 30 min at 37°C. Images were obtained with a DM 6000 B fluorescence microscope (Leica, Wetzlar, Germany) using the same imaging settings for each sample. For semi-quantitative analysis of ROS production, total fluorescence intensity was analysed with the FW4000 software (Leica) using three to six images from three sections per aortic ring for each experimental condition.
Xanthine/XOD activity assay
Xanthine (final concentration: 500 µM) and cytochrome c (final concentration: 100 µM) were dissolved in HBSS, and 100 µL aliquots of this solution were transferred to individual wells of a 96-well plate. After addition of the inhibitors, the mixtures were allowed to equilibrate for 20 min. The reaction was started by the addition of 100 µL XOD (final concentration: 20 mU·mL−1), and absorbance at 540 and 550 nm was recorded 10 min after the start of the reaction (SpectraMax 340; Molecular Devices). Superoxide production was calculated by normalization of the signals obtained at 540 nm. The blank signal (without xanthine) was subtracted.
A L012-based xanthine/XOD assay was performed with the same substances and concentrations as mentioned above using 100 µM L012 instead of cytochrome c. After an equilibration time of 20 min, the reaction was started with XOD (20 mU·mL−1), and the chemiluminescence was recorded for 20 min in a Fluoroscan FL microplate reader. Signals were calculated as AUC and related to the xanthine/XOD-derived control signal.
Endothelial NOS (eNOS) activity assay
eNOS activity was determined in homogenates of Sf9 cells (derived from Spodoptera frugiperda) transfected with a recombinant baculoviral vector expressing human eNOS (Schmidt et al., 1996; Frey et al., 1999). A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline was performed according to the method of Knipp and Vasak (2000) with slight modifications. Briefly, 90 µL reaction mixture pH 7.5 (100 mM Tris–HCl, 0.05 µM calmodulin, 1 mM CaCl2, 1 µM flavin adenine dinucleotide, 1 µM flavin mononucleotide, 250 µM CHAPSO, 1 mM l-arginine, 5 µM tetrahydrobiopterin, 1 mM NADPH, 2 mM reduced glutathione) was incubated with the specified concentration of NADPH oxidase inhibitors (or the respective solvents) and 5 µL of eNOS cell homogenate for 60 min at 37°C. The reaction was stopped by the addition of 300 µL colour-developing reagent (COLDER), consisting of one part solution A (80 mM diacetylmonoxime, 2 mM thiosemicarbazide) and three parts solution B [3 M H3PO4, 6 M H2SO4, 2 mM NH4Fe(SO4)2]. The mixture was heated for 15 min at 95°C, cooled to room temperature and centrifuged for 5 min at 16 000× g. Then, 300 µL of each sample was transferred to a 96-well microtiter plate, and the absorption at 540 nm was recorded (SpectraMax 340; Molecular Devices). A citrulline calibration curve was performed by mixing 100 µL of 0, 3.125, 6.25, 12.5, 25, 50, 100, 200 or 400 µM citrulline in 100 mM Tris buffer containing 2 mM reduced glutathione with 300 µL COLDER solution, and subsequently treated as described above. Enzyme activity was calculated using the citrulline calibration curve. The effects of NADPH oxidase inhibitors were expressed as a percentage of the respective control solutions. Signal inhibition by l-NAME (1 mM) demonstrated specificity of the assay for NOS. Standards were measured in duplicate, while all other measurements were performed in triplicate and repeated at least twice.
Data analysis
Data analysis was performed using Prism 4.0 for Macintosh (GraphPad Software, San Diego, CA, USA). IC50 values of concentration-dependent inhibition curves were calculated with a non-linear regression analysis using an algorithm for sigmoidal dose–response with variable slopes. Results are expressed as mean ± SEM. Statistical differences between the means were analysed by one-way anova followed by Bonferroni's multiple range test. A value of P < 0.05 was considered to be significant.
Materials
N,N-dimethyl-9,9–biacridinium dinitrate (lucigenin), cytochrome c from horse heart, NG-nitro-l-arginine methyl ester (l-NAME), oxypurinol, SOD from bovine erythrocytes, CAT from bovine liver, PMA, DPI chloride (dissolved in DMSO; 0.3–10 µM) were purchased from Sigma (Deisenhofen, Germany); 8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4-(2H,3H) dione (L012) from Wako (Osaka, Japan); XOD from cow's milk and complete EDTA-free protease inhibitor cocktail from Roche (Mannheim, Germany); 4′-hydroxy-3′-methoxyacetophenone (apocynin) from Calbiochem (Darmstadt, Germany; in 40% ethanol, 3–1000 µM); AEBSF from Merck (Darmstadt, Germany; in HBSS, 3–1000 µM); 3-benzyl-7-(2-oxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS3947) from Vasopharm GmbH (Würzburg, Germany; in DMSO). Synthesis and analytical data for VAS3947 have been described by Tegtmeier et al. (2005). The structure of VAS3947 is shown in Figure 1. Solvent controls were performed for all assays and did not have any significant effect, if not stated otherwise.
Figure 1.
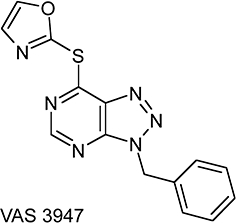
Chemical structure of VAS3947 [3-benzyl-7-(2-oxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine]. VAS3947 was characterized by NMR and mass spectrometry [1H nmr (DMSO-D6): δ 5.90 (s, 2H, CH2), 7.3–7.4 (m, 5H, Ph), 7.6 (s, 1H, Ar), 8.55 (s, 1H, Ar), 8.95 (s, 1H, H-5). ms: (+APCI) m/z 311 [M+H]+).
Results
CaCo-2, HL-60 and A7r5 cells collectively express all known NOX subunits
First, we wanted to establish cell models that physiologically expressed different NOX isoforms. Therefore, we analysed the NOX mRNA expression patterns using non-quantitative RT-PCR (Figure 2A). In contrast to previous reports, our batch of CaCo-2 cells not only expressed NOX1 mRNA, the predominant isoform in colon (Rokutan et al., 2006), but also mRNAs of NOX2, NOX4 and NOX5. As reported, HL-60 cells mainly expressed NOX2 mRNA (Teufelhofer et al., 2003), the main isoform in human neutrophils (Babior et al., 2002). However, we also detected NOX5 in these cells. Finally, A7r5 cells contained not only NOX4 mRNA, the predominant isoform in VSMCs (Wingler et al., 2001), but also NOX3, previously thought to be confined to the inner ear (Banfi et al., 2004) and NOX1 (Suh et al., 1999). Thus, none of the cell lines was specific for any NOX isoform, and they expressed different NOX isoform patterns. Jointly, the chosen cell lines expressed all NOX isoforms relevant to the cardiovascular system under basal conditions (i.e. NOX1, NOX2, NOX4 and human NOX5).
Figure 2.
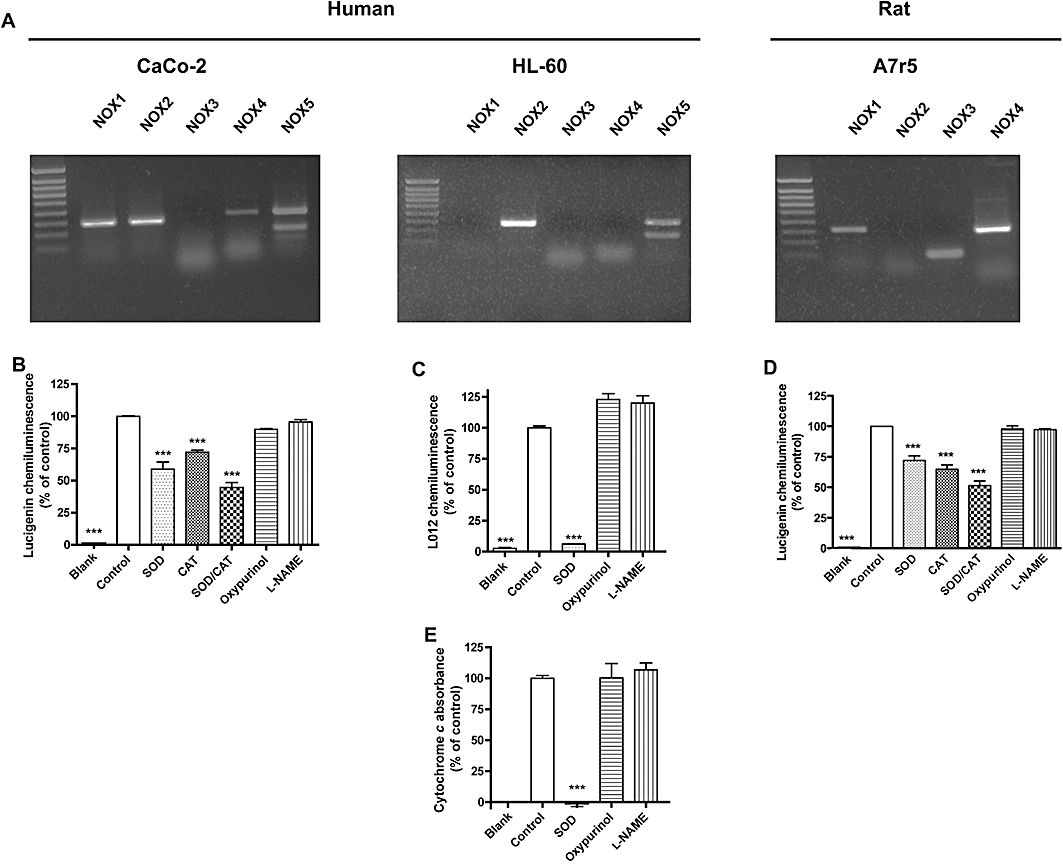
NOX expression and NADPH oxidase activity in three different cell lines. (A) Reverse transcriptase PCR of the five human (NOX1–5) and four rodent (NOX1–4) NOX isoform mRNAs in the human colon epithelial cell line CaCo-2, the human leucoblast cell line HL-60 and the rat aortic smooth muscle cell line A7r5. All cell lines expressed more than one NOX subunit. For measurement of NADPH oxidase activity, CaCo-2 homogenates + substrate NADPH (B), PMA-treated HL-60 cells (C and E) and A7r5 homogenates + substrate NADPH (D) were incubated in the absence or presence of SOD (250 U·mL−1), CAT (1000 U·mL−1), oxypurinol (100 µM) or l-NAME (100 µM). ROS formation was measured by lucigenin (B and D), or L012 (B) chemiluminescence, or by cytochrome c reduction (E). In none of the cells, detectable ROS formation originated from NOS (inhibited by l-NAME) or XOD (inhibited by oxypurinol). Only in HL-60 cells, the assay signals were fully inhibited by SOD and thus due to superoxide. In CaCo-2 and A7r5 cell homogenates, even the combination of SOD and CAT only partially attenuated the assay signal, suggesting that only a fraction was due to superoxide and/or H2O2. In later experiments, only the SOD/CAT-inhibited signal component was considered as a measure of superoxide/H2O2 formation in CaCo-2 and A7r5 cell homogenates. Data labelled ‘blank’ represent signals in the absence of cells or homogenate. Values represent means ± SEM of n≥ 6 observations; ***P < 0.001, significantly different from control values.
Cellular ROS formation is independent of XOD and NOS
We next characterized the likely enzymatic source and nature of ROS generated in our chosen cell models. The NADPH-dependent lucigenin (5 µM) chemiluminescence in homogenates represents a frequently used cell-free NADPH oxidase assay. At that low concentration, lucigenin is claimed to be free of relevant redox cycling and artefactual signals (Munzel et al., 2002). Using this assay, we found that both SOD and CAT attenuated the signal in CaCo-2 and A7r5 cell homogenates (Figure 2B,D), and we observed an additive effect when combining SOD and CAT. Inhibition of the lucigenin signals by CAT (Rost et al., 1998; Janiszewski et al., 2002), in the absence and presence of SOD, suggested that lucigenin is also sensitive to H2O2 (Rost et al., 1998). Both species, superoxide and H2O2, may originate from NADPH oxidases (Cai, 2005). Therefore, the SOD/CAT inhibitable fraction of the CaCo-2 and A7r5 signal was utilized as basal ROS formation. Nevertheless, the inhibition by SOD and CAT was not complete. Although we made sure that all cells were lysed, some cell organelles or intracellular vesicles may stay intact under our lysis protocol. NADPH oxidases and ROS can be highly localized, and SOD and CAT may not enter these organelles, which may explain these observations.
In PMA-stimulated HL-60 cells, the oxidative burst was measured by L012-mediated chemiluminescence (Figure 2C) (Daiber et al., 2004) and cytochrome c reduction (Figure 2E). SOD completely abolished the PMA-stimulated signal in both assays, confirming that superoxide is the only detectable ROS released from PMA-stimulated HL-60 cells. Consequently, the entire L012 signal was taken as PMA-stimulated ROS formation in HL-60 cells.
To determine the enzymatic source of ROS, ROS formation was re-assessed in the absence or presence of the NOS inhibitor, l-NAME, and the XOD inhibitor, oxypurinol. Both compounds failed to attenuate the ROS signals in any of the assays, suggesting that neither XOD nor uncoupled NOS are relevant sources of ROS in our cell models under the conditions used.
Antioxidant effects
NOX inhibitors may also reduce ROS signals by direct antioxidant effects (i.e. radical scavenging). Therefore, we performed a xanthine/XOD cytochrome c reduction assay, which generates superoxide independent of NADPH oxidase activity. As shown in Figure 3A, DPI and oxypurinol, but none of the other compounds, potently attenuated the XOD signal, suggesting a direct inhibition of XOD. This is consistent with DPI being a flavoprotein inhibitor (Lange et al., 2009) as both NOX and XOD are flavoproteins. Interestingly, when using L012 to detect ROS from xanthine/XOD, apocynin potently interfered with the signal (Figure 3B). This confirms recent reports that apocynin directly scavenges reaction products of H2O2, which are detected by L012, but not by cytochrome c (Heumuller et al., 2008). Surprisingly, AEBSF increased the L012 signal more than eightfold, suggesting a previously unrecognized interaction between L012 and AEBSF. This may be particularly problematic when AEBSF is used in homogenizing buffers as a serine protease inhibitor (Kobayashi et al., 2007; Baumer et al., 2008). VAS3947 (30 µM) was the only compound that did not interfere with xanthine/XOD-derived L012 signals, suggesting this compound is free of antioxidant or scavenging effects relevant to ROS detection.
Figure 3.
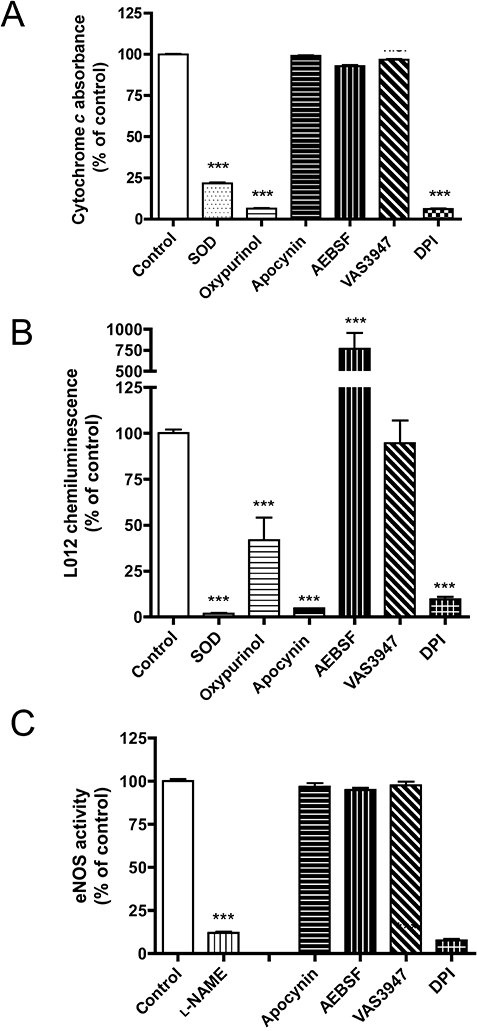
Effect of the NADPH oxidase inhibitors apocynin (1 mM), AEBSF (1 mM), DPI (10 µM) and VAS3947 on the activity of the flavo-protein XOD (30 µM VAS3947), and the flavo-haem protein, eNOS (10 µM VAS3947). XOD activity was assayed by cytochrome c reduction (A) or L012 chemiluminescence (B); eNOS activity was determined by l-arginine to l-citrulline conversion using recombinantly expressed eNOS (C). SOD (250 U·mL−1) and oxypurinol (1 mM) served as positive controls for XOD activity and l-NAME (1 mM) for eNOS activity. While apocynin did not scavenge superoxide in the cytochrome c reduction assay, it interfered with the L012 chemiluminescence assay. DPI inhibited both XOD and eNOS, and was thus no longer considered for NADPH oxidase enzyme assays in the present study. Values represent means ± SEM of n≥ 5 experiments. ***P < 0.001, significantly different from control values.
Inhibition of non-NOX flavoproteins
In addition to XOD, eNOS was used as a flavoprotein control to test the specificities of NADPH oxidase inhibitors. eNOS, like the catalytic NOX subunit, binds NADPH and contains both a flavin and haem cofactor. As expected, DPI (10 µM) also abolished eNOS activity (Figure 3C), while apocynin (1 mM), AEBSF (1 mM) and VAS3947 (10 µM) had no effect. As DPI inhibited both XOD and eNOS activity in concentrations similar to those needed for NADPH oxidase inhibition, it was no longer considered for further NADPH oxidase enzyme assays in the present study. We also tested higher concentrations (up to 100 µM) of VAS3947, and neither found an effect on XOD nor on eNOS activity (not shown).
Concentration dependence and efficacy
We next investigated the concentration dependence and efficacy of the remaining compounds, apocynin, AEBSF and VAS3947. In CaCo-2 cell homogenates, VAS3947 completely blocked NADPH-dependent ROS production with an IC50 of 12 µM, whereas apocynin and AEBSF caused only modest inhibition (Figure 4A). In contrast, the L012 chemiluminescence signal from HL-60 cells was inhibited by all three compounds (Figure 4B). The IC50 values for apocynin and VAS3947 were 97 and 2 µM respectively. Surprisingly, AEBSF exerted a biphasic response with concentrations <300 µM increasing the signal (EC50 = 30 µM) and higher concentrations being inhibitory (IC50 = 0.42 mM). Considering the increased signal in the L012 xanthine/XOD assay in the presence of AEBSF (Figure 3B), a direct interaction between AEBSF and L012 is likely. Because apocynin exerted non-specific effects in the L012 xanthine/XOD assay, we hypothesized that the inhibition seen in this HL-60 cell assay may not necessarily be due to inhibition of NADPH oxidases. Indeed, when using cytochrome c as a detection reagent in HL-60 cells (Figure 4B), apocynin had only a minor effect, while VAS3947 still completely inhibited with a similar IC50 value (1 µM) as observed in the L012 assay. In contrast to the L012 assay, AEBSF inhibited the cytochrome c reduction signal in a monophasic manner and had a high IC50 value of 0.3 mM similar to the second inhibitory phase with L012 (0.42 mM; see above).
Figure 4.
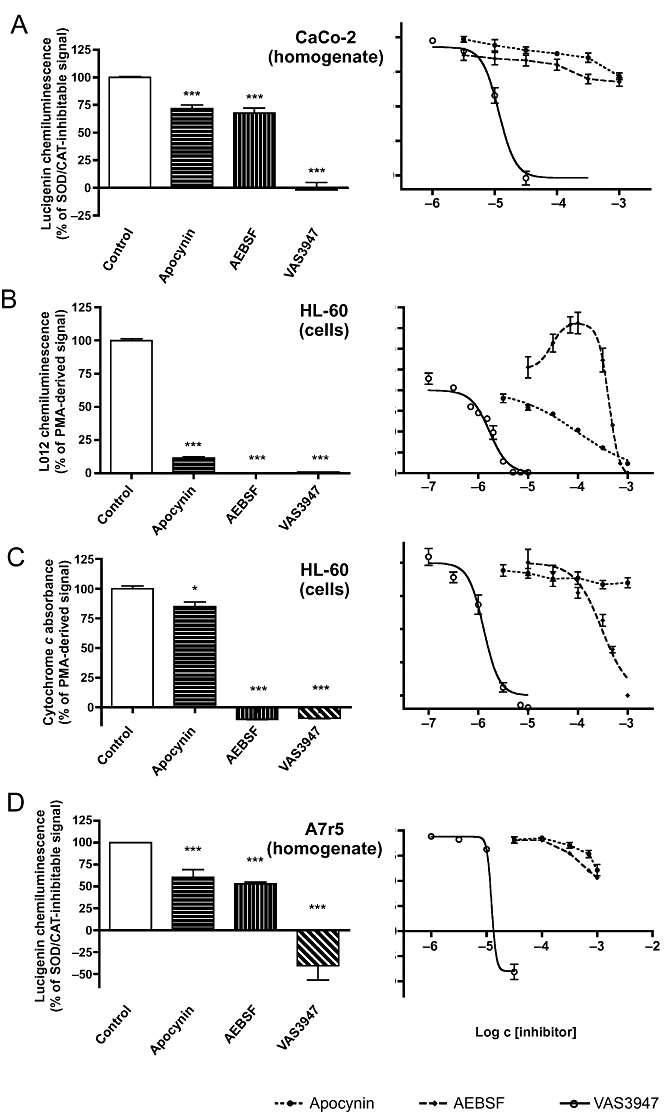
Maximal inhibition and potency of apocynin, AEBSF and VAS3947 to inhibit NADPH oxidase activity. Activity was measured in CaCo-2 (A) or A7r5 (D) cell homogenates as NADPH-induced and SOD/CAT-inhibitable lucigenin-derived chemiluminescence, or in DMSO-differentiated HL-60 cells as measured by L012-derived chemiluminescence (B) or cytochrome c reduction (C). The left panels depict maximal inhibition; the right panels concentration–response curves. Concentrations used for maximal inhibition were in CaCo-2 cell homogenates 1 mM apocynin, 1 mM AEBSF and 30 µM VAS3947 (A); in HL-60 cell-mediated L012 chemiluminescence 1 mM apocynin, 100 µM AEBSF and 10 µM VAS3947 (B); in HL-60 cell-mediated cytochrome c reduction 1 mM apocynin, 1 mM AEBSF and 10 µM VAS3947 (C); and in A7r5 cell homogenates 1 mM apocynin, 1 mM AEBSF and 30 µM VAS3947 (D). IC50 values for VAS3947 were 12 µM (A), 2 µM (B), 1 µM (C) and 13 µM (D). Apocynin had a potency in the micromolar range (97 µM) only in the HL-60/L012 assay (B). AEBSF had the lowest potency (420 µM in B, 304 µM in C; 1.2 mM in D), and in the HL-60/L012 assay it even enhanced signal intensity at low micromolar concentrations (EC50 = 30 µM). Values represent means ± SEM of n≥ 5 experiments. *P < 0.05, ***P < 0.001, significantly different from control values.
We hypothesized that apocynin was insufficiently metabolized by PMA-stimulated HL-60 cells and thus did not inhibit cytochrome c reduction mediated by these cells. Previous observations suggest that apocynin may be a pro-drug requiring activation by myeloperoxidases (Heumuller et al., 2008). Therefore, HL-60 cells were stimulated with OPZ, which, in contrast to PMA, activates not only NADPH oxidases, but also myeloperoxidase. Indeed, under these conditions, apocynin attenuated cytochrome c reduction (data not shown). VAS3947 and AEBSF were as effective in OPZ as in PMA-treated HL-60 cells (data not shown). In summary, VAS3947 was the only compound to effectively and consistently inhibit the oxidative burst signal in HL-60 cells independent of the stimulus and the read-out reagent.
Results obtained with homogenates of the third cell line, A7r5, were similar to those obtained with CaCo-2 cell homogenates when using the same lucigenin assay. Apocynin and AEBSF partially, and VAS3947 fully attenuated the NADPH-stimulated signal (Figure 4D). The IC50 values calculated from the concentration–response curves suggested millimolar potency for apocynin (1.9 mM) and AEBSF (1.2 mM), and micromolar potency for VAS3947 (13 µM). Thus, VAS3947 also inhibits NADPH oxidase in a vascular cell line mainly expressing NOX4 and NOX3, whereas apocynin and AEBSF were only weak inhibitors in this model with IC50 values approximately 100 times that for VAS3947.
NADPH oxidases are the main source of ROS in the SHR aorta
Having established that apocynin and AEBSF have considerably different IC50 values and are ineffective in some NOX model systems, whereas VAS3947 consistently inhibited NADPH oxidase activity in all three cell lines with similar IC50 values, we considered VAS3947 as the most reliable NADPH inhibitor. VAS3947 was therefore used as a pharmacological tool to investigate the source of ROS in aortas of aged SHRs (Figure 5), an established model of hypertension and cardiovascular oxidative stress. The in situ DHE stain is not specific for any ROS and rather summarizes superoxide-, peroxide- and peroxynitrite-mediated oxidative chemistry. This DHE stain of SHR aortas was significantly higher throughout the aortic wall compared to the signal in aortas from WKY control rats. Furthermore, it was blocked by PEG-SOD and was resistant to both l-NAME and oxypurinol. This suggested that superoxide was a major product leading to DHE oxidation, and that it was neither derived from eNOS nor XOD. Importantly, VAS3947 effectively inhibited DHE-detectable ROS formation in SHR aortas, suggesting that NADPH oxidases are a relevant enzymatic source of ROS throughout the aortic wall of aged SHRs.
Figure 5.
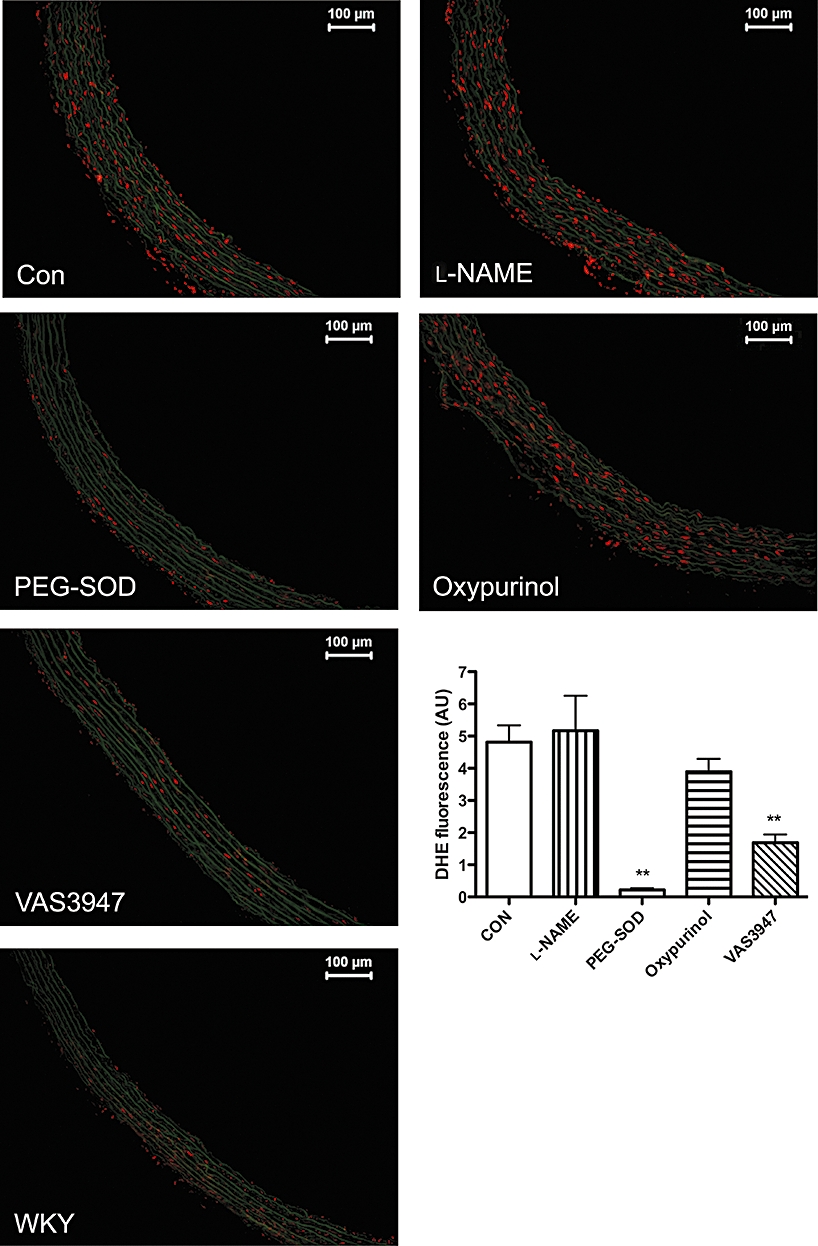
Effect of the NADPH oxidase inhibitors on ROS formation in aortic sections from 12- to 14-month-old SHRs as determined by in situ DHE tissue stain. DHE signals are enhanced in SHR versus WKY rat aorta, and correlate with enhanced levels of NOX1, 2 and 4. Sections were incubated either in the absence (Con) or presence of l-NAME (100 µM), PEG-SOD (250 U·mL−1), oxypurinol (100 µM) or VAS3947 (10 µM). Signal intensity was quantified and expressed as mean ± SEM of three experiments (n = 6). **P < 0.01, significantly different from control (CON).
Discussion
In the present study, we established the comparative pharmacology of the most frequently used and novel NADPH oxidase inhibitors, applying some of the most commonly used ROS assays. Of all inhibitors investigated, the novel triazolo pyrimidine, VAS3947, was the only compound that neither interfered with the flavoprotein, XOD, or the flavo-haem protein, eNOS, had no significant antioxidant activity, nor interfered with any of the ROS detection assays. Rather, VAS3947 effectively inhibited NADPH oxidase activity in three cellular models expressing different patterns of all known NOX isoforms. Therefore, at least in vitro, triazolo pyrimidines are new pharmacological tools for inhibiting NADPH oxidases. Applying this novel tool, we were able to establish that NADPH oxidases are a major source of ROS in aortas of aged hypertensive rats.
Based on genetic and other data, NADPH oxidases potentially play a major role in the development and progression of cardiovascular diseases (Williams and Griendling, 2007). This would suggest that it might be beneficial to therapeutically target NADPH oxidases. Apart from genetic mouse models, pharmacological agents are essential to provide proof-of-principle in non-mouse species and to serve as experimental therapeutics. The first triazolo pyrimidine described as an NADPH oxidase inhibitor was VAS2870, stemming from a systematic compound screen in HL-60 cells. VAS2870 inhibits NADPH oxidase activity in oxLDL-exposed HUVEC (Stielow et al., 2006) and PDGF-stimulated primary rat aortic VSMC (ten Freyhaus et al., 2006). VAS2870 also inhibits the stimulation of vasculogenesis of mouse embryonic stem cells upon treatment with PDGF-BB (Lange et al., 2009), and it inhibits wound margin H2O2 production without obvious toxicity in zebrafish larvae (Niethammer et al., 2009). In addition, VAS2870 does not interact with ROS in an antioxidant manner nor does it interfere with XOD (ten Freyhaus et al., 2006). However, a major disadvantage of VAS2870 is its poor solubility. Therefore, a closely related derivative, VAS3947, was developed with an approximately four times better solubility (data not shown). Apart from the solubility, VAS3947 has strikingly similar properties compared to VAS2870. For example, the IC50 values for NADPH oxidase activity of PMA-stimulated HL-60 cells, PMA-stimulated whole blood and freshly isolated human lymphocytes stimulated with PMA are essentially the same for both compounds (unpublished data). VAS3947 (this study) and VAS2870 (not shown) both effectively inhibit ROS production in aortas of SHRs as assessed by in situ DHE staining. Taken together, our evidence shows that: (i) the potential to inhibit NADPH oxidases is a class effect of triazolo pyrimidines; and (ii) this compound class inhibits NADPH oxidase activity in a variety of cell types and tissues of phagocytic, as well as non-phagocytic, origin with similar efficacy.
Our emphasis was to characterize the compounds in the most commonly used assays. We have therefore used alternative ways to characterize NADPH oxidase inhibitors: PMA-stimulated ROS release from HL-60 cells and NADPH-stimulated ROS production in cell homogenates. For detection, we used cytochrome c, L012 and lucigenin. We are aware that these assays do have drawbacks. Lucigenin-enhanced chemiluminescence has been widely used and is reported to be reasonably specific for superoxide. The lack of complete inhibition of the lucigenin signal by SOD that we observed was also reported by others (Rost et al., 1998). This indicates that it is not completely specific for superoxide. Lucigenin can penetrate cells and is prone to redox cycling. However, when 5 µM is used, the amount of artefact is probably insignificant (Dikalov et al., 2007). Compared to lucigenin, L012 is more sensitive for the detection of ROS (Daiber et al., 2004). H2O2 does not significantly contribute to L012-enhanced chemiluminescence. However, in addition to reacting with superoxide, L012 also detects peroxynitrite (Daiber et al., 2004). In contrast to lucigenin, L012 only detects extracellular ROS, as does cytochrome c, which considered as a ‘gold standard’ for detection of superoxide released in large amounts, such as during the respiratory burst of neutrophils. A disadvantage is its poor sensitivity (Dikalov et al., 2007).
In comparison to VAS3947, DPI is a less suitable pharmacological tool to validate the involvement of NADPH oxidase because of its effects on other flavo-enzymes, such as XOD and eNOS, as well as on cholinesterases and the internal calcium pump (Tazzeo et al., 2009). In our hands, apocynin had only minor efficacy and potency. This may in part be due to the fact that apocynin is a pro-drug requiring metabolic activation by myeloperoxidases. Furthermore, in PMA-stimulated HL-60 cells, the inhibition of the L012 signal by apocynin was caused by interference with the assay, either due to antioxidative effects or a direct interaction with L012. Thus, inhibitory effects of apocynin in the L012 assay may not necessarily reflect inhibition of NADPH oxidase activity (Heumuller et al., 2008). Interestingly, upon oxidation of apocynin during its activation, the resulting apocynin radical becomes a potent pro-oxidant with high capacity to oxidize thiols and NADPH. The depletion of NADPH potentially reduces NADPH oxidase activity (Castor et al., 2010). AEBSF was a similarly weak NADPH oxidase inhibitor in CaCo-2 and A7r5 cell homogenates, possibly indicating an only minor role for p47phox-dependent NADPH oxidases in these cells. Indeed, NOXO1 and/or NOXA1, homologues of p47phox and p67phox, may be of more relevance in CaCo-2 cells and not affected by AEBSF (Banfi et al., 2003; Takeya et al., 2003). In NADPH-stimulated homogenates from A7r5 cells, NOX4 may be the main source of ROS and independent of cytosolic subunits. While AEBSF attenuated the oxidative burst of HL-60 cells at high concentrations in the L012 and the cytochrome c assay, it may not be advisable to use this compound as an NADPH oxidase inhibitor because it obviously interferes with L012 and has off-target effects such as serine protease inhibition (Diatchuk et al., 1997).
The mechanism of action of triazolo pyrimidines such as VAS3947 and VAS2870 is unclear. In human leucocytes, VAS2870 does not inhibit translocation of p47phox to the membrane (ten Freyhaus et al., 2006), but may still interfere with oxidase assembly once the translocation has occurred. This assumption is supported by experiments showing that VAS2870 inhibits NOX2 activity in a cell-free system when added before (ten Freyhaus et al., 2006), but not after complex formation and stimulation of the oxidase (unpublished observations). The consistent potency and efficacy of VAS3947 in different cell and tissue models indicate that VAS3947, in contrast to apocynin, is not a pro-drug.
VAS3947 effectively suppressed the DHE signal in aortas of aged SHRs, suggesting that NADPH oxidases are indeed a major source of ROS in this disease model and that these enzymes can be pharmacologically targeted. However, the pharmacokinetic profile of VAS3947 and the in vivo efficacy, as well as long-term effects of NADPH oxidase inhibition, are unknown. In this context, peptide-based NADPH oxidase inhibitors are more advanced. The NOX2 homologous fusion peptide, gp91ds-tat (Rey et al., 2001), which interferes with cytosolic subunit translocation and activation of NOX2, inhibits angiotensin II-induced aortic superoxide production and hypertension in vivo (Rey et al., 2001). In addition, adenoviral expression of this peptide sequence in blood vessel adventitia had a beneficial effect, preventing neointimal hyperplasia (Dourron et al., 2005). Because our focus was on small synthetic molecules, and not on peptides, we did not include gp91ds-tat in our study. Nevertheless, NOX1-, NOX3-, NOX4- and NOX5-specific tat-peptide analogues may become useful pharmacological tools in the future.
In conclusion, apocynin, AEBSF and DPI appear to lack specificity as pharmacological agents when validating the involvement of NADPH oxidases in any physiological or pathological function. In contrast, triazolo pyrimidines such as VAS3947 are specific for NADPH oxidases versus NOS and XOD, and do not interfere with commonly used ROS assay systems. However, we cannot exclude the possibility that VAS3947, in addition to inhibiting NADPH oxidases, also interferes with alternative sources of ROS that we have not yet investigated, such as the mitochondrial electron chain. Future studies will be required to clarify the precise mechanism of action and in vivo efficacy of triazolo pyrimidines, and – once several derivatives become available – their isoform selectivity. For in vivo experiments, derivatives with higher solubility are essential. Importantly, by applying VAS3947 to the SHR model, we here have provided validated pharmacological evidence that NADPH oxidases are a relevant source of ROS in hypertension.
Acknowledgments
This work was funded by grants from the Deutsche Forschungsgemeinschaft, SFB547/C7 (H.H.H.W.S.), the Bayerische Forschungsstiftung (K.W. and H.H.H.W.S.) and the National Health and Medical Research Council of Australia (H.H.H.W.S.).
Glossary
Abbreviations
- AEBSF
(4-2-amino-ethyl)-benzolsulphonyl-fluoride hydrochloride
- AUC
area under the curve
- CAT
catalase
- COLDER
colour-developing reagent
- DHE
dihydroethidium
- DPI
diphenylene iodonium
- eNOS
endothelial NOS
- HBSS
Hanks's balanced salt solution
- HUVEC
human umbilical vein endothelial cell
- l-NAME
NG-nitro-l-arginine methyl ester
- NOX
NADPH oxidase
- OPZ
opsonized zymosan
- oxLDL
oxidized LDL
- PBS
phosphate-buffered saline
- PMA
phorbol myristate acetate
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rat
- SOD
superoxide dismutase
- VSMC
vascular smooth muscle cell
- WCH
whole-cell homogenate
- WKY
Wistar Kyoto (rats)
- XOD
xanthine oxidase
Conflict of interest
H.H.H.W.S. declares that he holds shares in Vasopharm GmBH, which develops NADPH oxidase inhibitors pharmaceutically. H.H.H.W.S., K.W. and P.S. are inventors of a patent on VAS3947, which is owned by Vasopharm GmbH. P.S. is employed by Vasopharm GmbH. K.W. is a former employee of Vasopharm GmbH.
Supplemental material
References
- Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- Baumer AT, Ten Freyhaus H, Sauer H, Wartenberg M, Kappert K, Schnabel P, et al. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. J Biol Chem. 2008;283:7864–7876. doi: 10.1074/jbc.M704997200. [DOI] [PubMed] [Google Scholar]
- Bayraktutan U. Coronary microvascular endothelial cell growth regulates expression of the gene encoding p22-phox. Free Radic Biol Med. 2005;39:1342–1352. doi: 10.1016/j.freeradbiomed.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res. 2005;96:818–822. doi: 10.1161/01.RES.0000163631.07205.fb. [DOI] [PubMed] [Google Scholar]
- Castor LR, Locatelli KA, Ximenes VF. Pro-oxidant activity of apocynin radical. Free Radic Biol Med. 2010;48:1636–1643. doi: 10.1016/j.freeradbiomed.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- Daiber A, August M, Baldus S, Wendt M, Oelze M, Sydow K, et al. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free Radic Biol Med. 2004;36:101–111. doi: 10.1016/j.freeradbiomed.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem. 1997;272:13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourron HM, Jacobson GM, Park JL, Liu J, Reddy DJ, Scheel ML, et al. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am J Physiol Heart Circ Physiol. 2005;288:H946–H953. doi: 10.1152/ajpheart.00413.2004. [DOI] [PubMed] [Google Scholar]
- Frey A, Schneider-Rasp S, Marienfeld U, Yu JC, Paul M, Poller W, et al. Biochemical and functional characterization of nitric oxide synthase III gene transfer using a replication-deficient adenoviral vector. Biochem Pharmacol. 1999;58:1155–1166. doi: 10.1016/s0006-2952(99)00196-3. [DOI] [PubMed] [Google Scholar]
- ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, et al. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Janiszewski M, Souza HP, Liu X, Pedro MA, Zweier JL, Laurindo FR. Overestimation of NADH-driven vascular oxidase activity due to lucigenin artifacts. Free Radic Biol Med. 2002;32:446–453. doi: 10.1016/s0891-5849(01)00828-0. [DOI] [PubMed] [Google Scholar]
- Knipp M, Vasak M. A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal Biochem. 2000;286:257–264. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Ohura I, Kawakita K, Yokota N, Fujiwara M, Shimamoto K, et al. Calcium-dependent protein kinases regulate the production of reactive oxygen species by potato NADPH oxidase. Plant Cell. 2007;19:1065–1080. doi: 10.1105/tpc.106.048884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S, Heger J, Euler G, Wartenberg M, Piper HM, Sauer H. Platelet-derived growth factor BB stimulates vasculogenesis of embryonic stem cell-derived endothelial cells by calcium-mediated generation of reactive oxygen species. Cardiovasc Res. 2009;81:159–168. doi: 10.1093/cvr/cvn258. [DOI] [PubMed] [Google Scholar]
- Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- Lee MY, Martin AS, Mehta PK, Dikalova AE, Garrido AM, Datla SR, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Shah AM. Differential NADPH- versus NADH-dependent superoxide production by phagocyte-type endothelial cell NADPH oxidase. Cardiovasc Res. 2001;52:477–486. doi: 10.1016/s0008-6363(01)00407-2. [DOI] [PubMed] [Google Scholar]
- McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, et al. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285:H2290–H2297. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- Munzel T, Afanas'ev IB, Kleschyov AL, Harrison DG. Detection of superoxide in vascular tissue. Arterioscler Thromb Vasc Biol. 2002;22:1761–1768. doi: 10.1161/01.atv.0000034022.11764.ec. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J. 1993;290(Pt 1):41–49. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz N, Drummond GR, Selemidis S, Meurer S, Schmidt HH. The ‘A's and ‘O's of NADPH oxidase regulation: a commentary on ‘Subcellular Localization and Function of Alternatively Spliced Noxo1 Isoforms’. Free Radic Biol Med. 2007;42:175–179. doi: 10.1016/j.freeradbiomed.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(–) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- Rokutan K, Kawahara T, Kuwano Y, Tominaga K, Sekiyama A, Teshima-Kondo S. NADPH oxidases in the gastrointestinal tract: a potential role of Nox1 in innate immune response and carcinogenesis. Antioxid Redox Signal. 2006;8:1573–1582. doi: 10.1089/ars.2006.8.1573. [DOI] [PubMed] [Google Scholar]
- Rost M, Karge E, Klinger W. What do we measure with luminol-, lucigenin- and penicillin-amplified chemiluminescence? 1. Investigations with hydrogen peroxide and sodium hypochlorite. J Biolumin Chemilumin. 1998;13:355–363. doi: 10.1002/(SICI)1099-1271(199811/12)13:6<355::AID-BIO502>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Schluter T, Steinbach AC, Steffen A, Rettig R, Grisk O. Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res. 2008;80:271–279. doi: 10.1093/cvr/cvn185. [DOI] [PubMed] [Google Scholar]
- Schmidt HH, Hofmann H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. No NO from NO synthase. Proc Natl Acad Sci U S A. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn HY, Gloe T, Keller M, Schoenafinger K, Pohl U. Sensitive superoxide detection in vascular cells by the new chemiluminescence dye L-012. J Vasc Res. 1999;36:456–464. doi: 10.1159/000025688. [DOI] [PubMed] [Google Scholar]
- Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HH, et al. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem Biophys Res Commun. 2006;344:200–205. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, et al. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- Tazzeo T, Worek F, Janssen L. The NADPH oxidase inhibitor diphenyleneiodonium is also a potent inhibitor of cholinesterases and the internal Ca(2+) pump. Br J Pharmacol. 2009;158:790–796. doi: 10.1111/j.1476-5381.2009.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegtmeier F, Walter U, Schinzel R, Wingler K, Scheurer P, Schmidt H. Compounds containing a N-heteroaryl moiety linked to fused ring moieties for the inhibition of NAD(P)H oxidases and platelet activation. 2005 European Patent 1 598 354 A1. [Google Scholar]
- Teufelhofer O, Weiss R-M, Parzefall W, Schulte-Hermann R, Micksche M, Berger W, et al. Promyelocytic HL60 cells express NADPH oxidase and are excellent targets in a rapid spectrophotometric microplate assay for extracellular superoxide. Toxicol Sci. 2003;76:376–383. doi: 10.1093/toxsci/kfg234. [DOI] [PubMed] [Google Scholar]
- Vejrazka M, Micek R, Stipek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta. 2005;1722:143–147. doi: 10.1016/j.bbagen.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Williams HC, Griendling KK. NADPH oxidase inhibitors: new antihypertensive agents? J Cardiovasc Pharmacol. 2007;50:9–16. doi: 10.1097/FJC.0b013e318063e820. [DOI] [PubMed] [Google Scholar]
- Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med. 2001;31:1456–1464. doi: 10.1016/s0891-5849(01)00727-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.