

Abstract
Many chromatin-associated proteins contain two sequence motifs rich in phenylalanine/tyrosine residues of unknown function. These so-called FYRN and FYRC motifs are also found in transforming growth factor beta regulator 1 (TBRG1)/nuclear interactor of ARF and MDM2 (NIAM), a growth inhibitory protein that also plays a role in maintaining chromosomal stability. We have solved the structure of a fragment of TBRG1, which encompasses both of these motifs. The FYRN and FYRC regions each form part of a single folded module (the FYR domain), which adopts a novel α + β fold. Proteins such as the histone H3K4 methyltransferases trithorax and mixed lineage leukemia (MLL), in which the FYRN and FYRC regions are separated by hundreds of amino acids, are expected to contain FYR domains with a large insertion between two of the strands of the β-sheet.
Keywords: p14ARF, chromatin, taspase1, proteolysis
Introduction
In recent years, it has become clear that chromatin structure plays a central role in regulating processes, such as transcription, DNA repair, and DNA replication. Consequently, proteins that modify or remodel chromatin are currently subject to intense interest. Many of these proteins are modular and contain domains that reoccur in numerous nuclear proteins.1 The FYRN and FYRC sequence motifs are two poorly characterized phenylalanine/tyrosine-rich regions of around 50 and 100 amino acids, respectively, that are found in a variety of chromatin-associated proteins [Fig. 1(A)].1–3 They are particularly common in histone H3K4 methyltransferases most notably in a family of proteins that includes human mixed linage leukemia (MLL) and the Drosophila melanogaster protein trithorax. Both of these enzymes play a key role in the epigentic regulation of gene expression during development,9 and the gene coding for MLL is frequently rearranged in infant and secondary therapy-related acute leukemias.10 They are also found in transforming growth factor beta regulator 1 (TBRG1), a growth inhibitory protein induced in cells undergoing arrest in response to DNA damage and transforming growth factor (TGF)-β1.11 As TBRG1 has been shown to bind to both the tumor suppressor p14ARF and MDM2, a key regulator of p53, it is also known as nuclear interactor of ARF and MDM2 (NIAM).12 In most proteins, the FYRN and FYRC regions are closely juxtaposed, however, in MLL and its homologues they are far distant and it is not clear if each motif corresponds to an independent protein module or if both motifs are components of a single domain. To be fully active, MLL must be proteolytically processed by taspase1, which cleaves the protein between the FYRN and FYRC regions.13,14 The N-terminal and C-terminal fragments remain associated after proteolysis apparently as a result of an interaction between the FYRN and FYRC regions. How proteolytic processing regulates the activity of MLL is not known. Intriguingly, the FYRN and FYRC motifs of a second family of histone H3K4 methyltransferases, represented by MLL2 and MLL4 in humans and TRR in Drosophila melanogaster, are closely juxtaposed.
Figure 1.
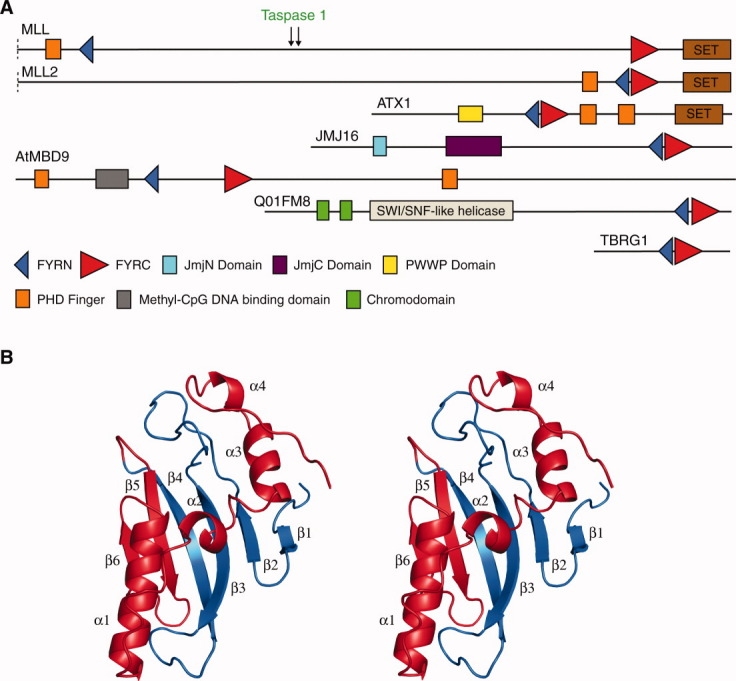
A: Domain architectures of selected proteins that contain FYRN and FYRC motifs. For convenience only the C-terminal regions of MLL and MLL2 are shown. The taspase1 cleavage sites in MLL are indicated with arrows. ATX1, JMJ16 and AtMBD9 are all proteins from Arabidopsis thaliana. ATX1 is a histone H3K4 methylase4 and JMJ16 is a Jumonji C (JmjC) domain containing histone H3K4 demethylase.5 AtMBD9 is a methyl-CpG binding domain (MBD) containing protein involved in plant development.6 Q01FM8 is a chromodomain helicase from the alga Ostreococcus tauri.7 The domains shown are those identified by the SMART database.8 B: Crystal structure of the FYR domain of TBRG1 shown as ribbon model in stereoview. Secondary structure elements are labeled. The FYRN and FYRC regions are colored in blue and red, respectively.
Although FYRN and FYRC motifs are found in proteins that play important roles in the regulation of cell growth and differentiation, little is known about their structure and function. In this study, we report the crystal structure of a fragment of TBRG1 that contains these motifs and show that each forms part of a single protein module, which adopts a novel α + β fold. The structure provides insights into how these motifs mediate the heterodimerization of the products of the proteolytic processing of MLL and allows the identification of potential ligand binding sites.
Results and Discussion
The FYRN and FYRC regions of TBRG1 are components of a single domain FYR with a novel α + β fold
Attempts were made to express and purify the FYRN/FYRC regions of several of the proteins in which they are closely spaced. A fragment of TBRG1 (residues 179–324), proved the most amenable to study as it could be easily expressed in E. coli, was simple to purify and readily crystallized. Chemical denaturation of this protein by GdmCl shows a cooperative unfolding profile typical of a two-state transition when monitored both by intrinsic tryptophan fluorescence and by circular dichroism [Fig. 2(A)]. Thermal denaturation of the protein monitored by differential scanning calorimetry (DSC) also revealed a single unfolding transition with a midpoint of 41.8°C [Fig. 2(B)]. This data indicates that in TBRG1 the FYRN and FYRC regions are not separate independently folded domains but are components of a distinct protein module. Crystals of the protein were grown by hanging-drop vapor diffusion, and its structure was determined to 1.6 Å resolution using multiple wavelength anomalous dispersion phasing and a selenomethionine-substituted derivative (Table I). The FYRN and FYRC motifs both form part of a single FYR domain that adopts an α + β fold consisting of a six-stranded antiparallel β-sheet followed by four consecutive α-helices [Fig. 1(B)]. The FYRN region corresponds to β-strands 1–4 and their connecting loops, whereas the FYRC motif maps to β-strand 5, β-strand 6 and helices α1 to α4. The two central β-strands are twisted and much longer than those at the periphery of the β-sheet (strands 1 and 6). Strands 2 and 3 are connected through the longest loop in the protein, which is stabilized by a network of hydrogen bonds. A short turn connects β-strand 6 to helix α1, and a longer loop places the short helix α2 in an L-shaped conformation with respect to α1. This motif is followed by two α-helices displaying a similar L-shaped conformation (α3-loop-α4). A search of the protein data bank using DALI15 revealed no significant similarity to other structures.
Figure 2.
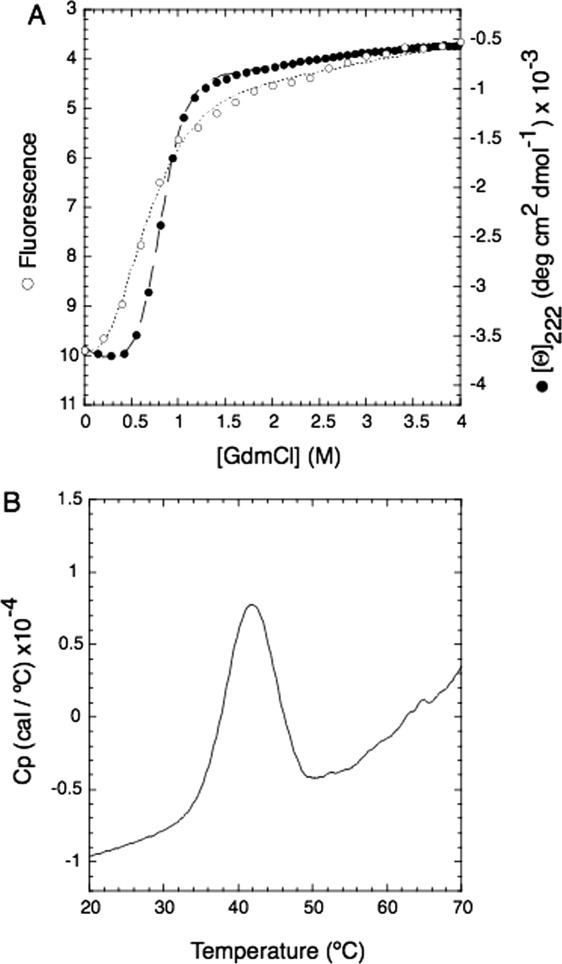
Thermodynamic stability of the FYR domain. A: Denaturation curves of a fragment of TBRG1 that encompasses the FYRN and FYRC motifs fitted to a two-state model following the intrinsic fluorescence of tryptophan (left: y-axis; white circles, dotted line) and molar ellipticity at 222 nm (right: y-axis; black circles, dashed line) as a function of [GdmCl]. B: Thermal denaturation of the same protein monitored by DSC. The apparent melting temperature (Tm) is 41.8°C.
Tabel I.
Summary of Data Collection and Refinement Statistics of the FYR Domain of TBRG1
| Native | SeMet | |||
|---|---|---|---|---|
| Data collection | ||||
| Space group | P3121 | P3121 | ||
| Cell dimensions | ||||
| a, b, c (Å) | 53.867, 53.867, 91.297 | 53.976, 53.976, 91.297 | ||
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | ||
| Molecules/AU | 1 | 1 | ||
| Peak | Inflection | Remote | ||
| Wavelength (Å) | 0.9796 | 0.9797 | 0.9798 | 0.9763 |
| Resolution (Å) | 32.6–1.6 | 41.6–2.0 | 32.6–2.0 | 32.6–2.0 |
| Rsym or Rmergea | 0.079 (0.422) | 0.068 (0.163) | 0.054 (0.134) | 0.059 (0.160) |
| I/σI | 18.8 (5.7) | 23.7 (12.3) | 20.0 (10.0) | 17.4 (8.3) |
| Completeness (%) | 99.8 (98.9) | 98.7 (97.9) | 98.9 (98.0) | 98.5 (97.6) |
| Redundancy | 10.3 (10.4) | 10.5 (10.9) | 5.3 (5.5) | 5.3 (5.5) |
| Refinement | ||||
| Resolution (Å) | 23.3–1.6 | |||
| No. reflections | 20876 | |||
| Rworkb/Rfreec | 0.178, 0.202 | |||
| No. atoms | ||||
| Protein | 1118 | |||
| Ligand/ion | 6 (2 Glycerol) | |||
| Water | 160 | |||
| B-factors (Å2) | ||||
| Protein | 19.64 | |||
| Ligand/ion | 28.09 | |||
| Water | 29.24 | |||
| R.M.S.D.d | ||||
| Bond lengths (Å) | 0.012 | |||
| Bond angles (°) | 1.391 | |||
| Ramachandrane | 90.0%, 0.0% | |||
Values in parentheses refer to the last resolution shell.
Rsym = ΣhΣi|I(h,i)−I(h)|/ΣhΣi I(h,i) where I(h,i) are symmetry related intensities and I(h) is the mean intensity of the reflection with unique index h.
Rcryst (%) = Σ|Fo − Fc|/ΣFo, where, Fo is the observed structure factor amplitude, and Fc is the structure factor calculated from the model.
Rfree (%) is calculated in the same manner as Rcryst using 5% of all reflections excluded from refinement stages using high-resolution data.
R.M.S.D., root-mean-square deviation.
Percentage of residues in the “most favored region” of the Ramachandran plot and percentage of outliers (PROCHECK).
Role of conserved residues
A structure-based sequence alignment of selected FYRN/FYRC motifs is shown in Figure 3(A). Most of the conserved tyrosine and phenylalanine residues, after which these motifs are named, are involved in interactions that stabilize the fold. The side chains of Y216, Y234, F247, and F294, for example, all form part of the main hydrophobic core of the protein that comprises residues in the helices, the sheet and some of the loop regions [Fig. 3(B)]. The other residues that make up this core are also conserved or are replaced by other hydrophobic amino acids. Y317 and F322 are in an extended loop region at the C-terminus of the protein and are involved in hydrophobic interactions that help to position the C-terminus of the domain close to the N-terminus, a feature often seen in protein modules [Fig. 3(B)]. The side chains of most of the conserved tyrosines form hydrogen bonds that stabilize the fold. The hydroxyl of Y234 in strand 4 hydrogen bonds to the carbonyl of A223 and the amide of M225, which are in the turn between strands 3 and 4. Other residues are involved in long-range hydrogen bonds that link the two motifs. The hydroxyl of Y222, which is at the end of β-strand 3, for example, hydrogen bonds to the Nɛ group of H268 in α1 [Fig. 3(D)]. Although H268 is replaced by a tryptophan in most other FYRC motif containing proteins, it is likely that similar interactions are formed with the Nɛ group of this amino acid. The hydroxyl of Y216 forms a hydrogen bond with the carbonyl group of F294, which is at the C-terminus of helix 2. Interestingly, the carbonyl groups of the two preceding residues in this helix form hydrogen bonds with the side chain of a highly conserved arginine (R220) in β-strand 3, suggesting that the correct positioning of this helix is critical to the stability of the fold [Fig. 3(D)]. Analysis of the conservation of solvent-exposed residues using the ConSurf server16 reveals two potentially functionally important regions [Fig. 3 (C,D)]. One of these consists of a pocket on the protein surface lined by Y212 and F247, which also contains a highly conserved serine (S263). The second group of conserved solvent-exposed residues includes Y222 and R220. Two tryptophan residues, corresponding to A223 and H268 in TBRG1, that are conserved in FYRN/FYRC motifs from chromatin-associated proteins are also predicted to be located in this region of the protein, which would result in a different shape complementarity of this surface patch and hence potentially different molecular recognition features of the FYR domain.
Figure 3.

Sequence conservation in FYR domains. A: Structure-based alignment of the sequences of selected FYR domains. The large insertions in MLL and related proteins are indicated in parentheses. Residues are colored based on degree of sequence conservation. B: Ribbon representation of the TBRG1 FYR domain showing the position of conserved phenylalanine and tyrosine residues in the hydrophobic core of the domain in green. C: Surface representation of the TBRG1 FYR domain highlighting conserved solvent-exposed residues. Highly conserved residues are colored green. Residues equivalent to the tryptophan residues conserved in chromatin-associated FYR domains are colored yellow. D: Stereo view of the two conserved surfaced patches. Hydrogen bonds mediated via the side chains of R220 and Y222 are highlighted with a black dashed line. The color-coding is the same as in panel (C).
Implications for the proteolytic processing of MLL
Based on the degree to which structurally important residues are conserved, in particular those that participate in long-range interactions that link the FYRN/FYRC regions, it is likely that in other proteins these motifs adopt a similar fold to that seen in TBRG1. For proteins such as MLL, in which the FYRN/FYRC motifs are separated by hundreds of amino acids, the structure is expected to contain a large insertion between strands 4 and 5 of the sheet. This would explain why, when MLL is cleaved between the FYRN/FYRC motifs, the products of the reaction remain associated, as during this process the protein essentially is cut within an exposed loop of a folded domain. Why the FYRN/FYRC motifs of MLL and related proteins, but not those of the MLL2/MLL4 family of histone methyltransferases, have a protease cleavable insertion between them is not clear. Proteolytic processing is required for the correct nuclear sublocalization of MLL, although the mechanism by which this is achieved has not been determined. It seems unlikely that the cleavage of this loop would directly affect the structure or function of the fold. It has been suggested that in the uncleaved form of MLL the FYRN and FYRC regions are held in a conformation that prevents them from associating and proteolysis allows them to come together to form a functional unit.13 This hypothesis is consistent with the structural data presented here, as both the FYRN and FYRC regions are integral to the fold and the potential binding sites identified from sequence conservation are composed of residues from either side of the cleavage site. It is, however, possible that the protease targets a regulatory module located within the insertion between the FYRN and FYRC regions and their role in proteolytic processing is only to serve as a means of ensuring that the resulting fragments do not dissociate. It will only be possible to distinguish between these possibilities once the function of these motifs in MLL and related proteins has been established.
FYRN and FYRC motifs are found in association with modules that create or recognize histone modifications in proteins from a wide range of eukaryotes, and it is likely that in these proteins they have a conserved role related to some aspect of chromatin biology. Based on the structural data reported here, a binding rather than a catalytic function seems more likely. The nature of a potential ligand, however, remains unclear, as the FYR domain does not resemble any of the protein modules known to recognize potential binding partners such as modified histones. TBRG1-like proteins, in contrast, are restricted to metazoans and appear to have evolved in parallel with the p53/MDM2 pathway.17,18 As discussed above, the sequences of TBRG1-like FYRN and FYRC motifs are distinct from those in other proteins. Although TBRG1 is located in the nucleus, it has not so far been shown to be associated with chromatin. While it is possible that in TBRG1 these motifs have a similar role to those in other proteins and help to link the antiproliferative pathways in which it is involved with chromatin, it is also possible that they may have acquired an alternative function. Even though FYRN and FYRC motifs are found in an array of biologically and medically important proteins, there is still, clearly, much to be learnt about their function, and the data reported here will provide a structural basis for these efforts.
Materials and Methods
Protein expression and purification
The DNA coding for residues 179–324 of human TBRG1 was amplified by PCR from a human cDNA library (Clontech) and cloned into a modified pRSETA (Invitrogen) expression vector containing a hexahistidine-tagged lipoyl domain and a TEV cleavage site. Protein was expressed in E. coli C41(DE3) and purified by Ni-affinity chromatography followed by TEV protease cleavage. This was followed by a second Ni-affinity chromatography step to remove the cleaved fusion partner and gel filtration.
CD and fluorescence spectroscopy
CD measurements were performed using a Jasco J-815 spectropolarimeter. FYR domain (20 mM) was incubated with different concentrations of guanidinium hydrochloride [GdmCl] for 4 h in 20 mM Tris pH 7.5, 150 mM NaCl, and 1 mM DTT, while following the amplitude of the negative band at 222 nm as a function of denaturant. Fluorescence measurements were recorded on a Cary Eclipse fluorescence spectrophotometer equipped with a Hamilton Microlab titrator controlled by laboratory software. The excitation and emission wavelengths were 296 and 330 nm, respectively. Slit widths for both excitation and emission were kept at 7 nm, with a photomultiplier voltage of 600 V. Samples (1.4 mL) containing 5 μM FYR domain in 20 mM Tris pH 7.5, 150 mM NaCl and 1 mM DTT were thermostatted at 25 ± 0.1°C with constant stirring. Refolding was carried out by injecting 30 μL into an equal volume of 6 M GdmCl solution containing an equimolar concentration of FYR domain. The time between injections was 6 min.
Differential scanning calorimetry
Calorimetric measurements were performed by using a VP-DSC microcalorimeter (MicroCal), equipped with an AutoSampler. Scans were obtained at a protein concentration of 180 μM, in buffer containing 50 mM potassium phosphate pH 7.2, 150 mM NaCl and 1 mM DTT. A scan rate of 50°C/h was used over a temperature range from 10–110°C. The reversibility of the transition was checked by cooling and reheating the same sample. Results from the DSC measurements were analyzed with the MicroCal Origin™ program.
Crystallization and structure determination
The FYR domain was crystallized at 3.4 mg/mL by sitting-drop vapor diffusion in 1.0 M K/Na tartrate and 100 mM HEPES, pH 7.5 at 17°C. Crystals used for phasing were grown using selenomethionine-substituted FYR domain at the same concentration and conditions as for the native proteins. The crystals were flash-frozen in liquid nitrogen after the addition of glycerol to 20%. The FYR domain crystals belonged to the spacegroup P3121 with one molecule in the asymmetric unit. Cell constants and crystallographic data are summarized in Table I. Datasets were collected at beamline I02 of the Diamond Light Source (native and seleno-methionine). All data were indexed and integrated with MOSFLM and further processed using the CCP4 package.19 Initially, five selenomethionine sites were found with SHELXD20 using the peak dataset (Table I). The coordinates of these sites were imported into SHARP21 and phases were calculated using the inflection and remote datasets. The model was built manually with the program MAIN and refined using CNS22 and PHENIX.23 The coordinates and structure factors have been deposited in the protein data bank (http://www.pdb.org) with the accession code 2wzo.
Acknowledgments
The authors thank H. Tidow and A. Andreeva for careful reading of this manuscript.
References
- 1.Doerks T, Copley RR, Schultz J, Ponting CP, Bork P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002;12:47–56. doi: 10.1101/gr.203201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balciunas D, Ronne H. Evidence of domain swapping within the jumonji family of transcription factors. Trends Biochem Sci. 2000;25:274–276. doi: 10.1016/s0968-0004(00)01593-0. [DOI] [PubMed] [Google Scholar]
- 3.Prasad R, Zhadanov AB, Sedkov Y, Bullrich F, Druck T, Rallapalli R, Yano T, Alder H, Croce CM, Huebner K, Mazo A, Canaani E. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene. 1997;15:549–560. doi: 10.1038/sj.onc.1201211. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez-Venegas R, Pien S, Sadder M, Witmer X, Grossniklaus U, Avramova Z. ATX-1, an Arabidopsis homolog of trithorax, activates flower homeotic genes. Curr Biol. 2003;13:627–637. doi: 10.1016/s0960-9822(03)00243-4. [DOI] [PubMed] [Google Scholar]
- 5.Lu F, Li G, Cui X, Liu C, Wang XJ, Cao X. Comparative analysis of JmjC domain-containing proteins reveals the potential histone demethylases in Arabidopsis and rice. J Integr Plant Biol. 2008;50:886–896. doi: 10.1111/j.1744-7909.2008.00692.x. [DOI] [PubMed] [Google Scholar]
- 6.Peng M, Cui Y, Bi YM, Rothstein SJ. AtMBD9: a protein with a methyl-CpG-binding domain regulates flowering time and shoot branching in Arabidopsis. Plant J. 2006;46:282–296. doi: 10.1111/j.1365-313X.2006.02691.x. [DOI] [PubMed] [Google Scholar]
- 7.Derelle E, Ferraz C, Rombauts S, Rouze P, Worden AZ, Robbens S, Partensky F, Degroeve S, Echeynie S, Cooke R, Saeys Y, Wuyts J, Jabbari K, Bowler C, Panaud O, Piegu B, Ball SG, Ral JP, Bouget FY, Piganeau G, de Baets B, Picard A, Delseny M, Demaille J, van de Peer Y, Moreau H. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci USA. 2006;103:11647–11652. doi: 10.1073/pnas.0604795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenney K, Shilatifard A. A COMPASS in the voyage of defining the role of trithorax/MLL-containing complexes: linking leukemogensis to covalent modifications of chromatin. J Cell Biochem. 2005;95:429–436. doi: 10.1002/jcb.20421. [DOI] [PubMed] [Google Scholar]
- 10.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 11.Babalola GO, Schultz RM. Modulation of gene expression in the preimplantation mouse embryo by TGF-alpha and TGF-beta. Mol Reprod Dev. 1995;41:133–139. doi: 10.1002/mrd.1080410203. [DOI] [PubMed] [Google Scholar]
- 12.Tompkins VS, Hagen J, Frazier AA, Lushnikova T, Fitzgerald MP, di Tommaso A, Ladeveze V, Domann FE, Eischen CM, Quelle DE. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J Biol Chem. 2007;282:1322–1333. doi: 10.1074/jbc.M609612200. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeda S, Chen DY, Westergard TD, Fisher JK, Rubens JA, Sasagawa S, Kan JT, Korsmeyer SJ, Cheng EH, Hsieh JJ. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 2006;20:2397–2409. doi: 10.1101/gad.1449406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 16.Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- 17.Belyi VA, Levine AJ. One billion years of p53/p63/p73 evolution. Proc Natl Acad Sci USA. 2009;106:17609–17610. doi: 10.1073/pnas.0910634106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane DP, Cheok CF, Brown C, Madhumalar A, Ghadessy FJ, Verma C. Mdm2 and p53 are highly conserved from placozoans to man. Cell Cycle. 2010;9:540–547. doi: 10.4161/cc.9.3.10516. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project Number, 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Sheldrick GM. Phase annealing in SHELX-90; direct methods for larger structures. Acta Crystallogr. 1990;A46:467–473. [Google Scholar]
- 21.Fortelle EL, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacememt and multiwavelength anomalous diffraction methods. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 22.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]