

Why do some tumors grow and spread rapidly, while others remain localized or grow slowly? Perhaps some tumors never make it past one transformed cell? Recent advances in genomics, proteomics, and other “-omics” have provided a plethora of data on molecular aberrations in malignant cells, but in most cases these data are hard to make sense of and so are currently clustered as “molecular signatures”. Through clinical experience, these “signatures” can be associated with the aggressiveness of the malignant disease and serve as guidelines for prognosis and selection of the form of treatment of the disease, but progress is likely to be slow until new concepts are generated from these data. Thus, understanding of the pathways that signal biological outcomes appears a critical priority. The report by Oka et al. in this issue of Cancer Biology & Therapy on DNA damage signaling draws attention to one such pathway, also known as the DNA Damage Response (DDR).1
In normal mammalian cells, DNA damage activates DNA repair genes, and this is signaled by ATM (Ataxia Telangiectasia Mutated), a kinase that activates another kinase, Chk2 (check point kinase 2), which can promote intra-S and G2 phase cell cycle arrest, as well as apoptosis. A parallel pathway consisting of ATR, an ATM related kinase, and Chk1 has similar outcomes, though there are some differences, as ATR is reported to have an essential role during DNA replication, and senses its blockage.2-5 These pathways converge on the p53 tumor suppressor gene “the guardian of the genome”,6 which can activate molecules such as p21Waf1,7 that arrest cell cycle progression to allow DNA damage to be repaired (Fig. 1).8 Alternatively, if the situation seems hopeless, p53 nudges the cell to self-destruction by enhancing the expression of pro-apoptotic molecules such as Bax or Bim.9,10 In either case, the cells do not self-renew so the mutations, some of which may cause self-generation of growth signals or survival under adverse conditions, are “nipped in the bud”. Thus, the DNA damage signaling proteins interact upstream with DNA repair proteins, and downstream with regulators of cell cycle progression and cell survival. The upstream components include those that promote increased DNA accessibility in the chromatin and localization of DNA repair complexes. A specific example is the phosphorylation by ATM/ATR of a variant of nucleosome core histone H2A known as H2AX, and its phosphorylated form (γH2AX) is a useful marker of DDR,5 as it is recognized by a specific antibody in tissue sections as discrete nuclear foci, where DNA repair is presumed to take place.
Figure 1.
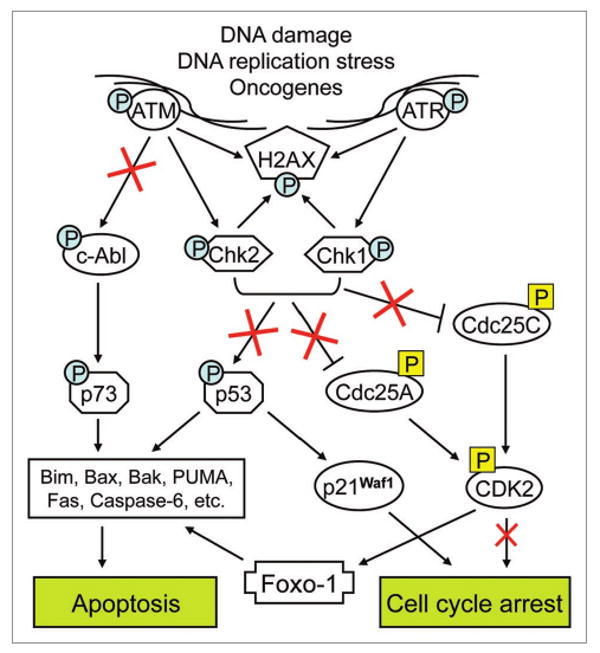
DNA Damage Response (DDR) pathways in normal and malignant cells. In normal cells, different types of genotoxic stress (e.g., DNA double-strand breaks) or DNA replication stress elicit DDR as part of the self-defense system. This involves the activation (via phosphorylation) of “sensor” kinases, ATM and ATR, which in turn activate Chk2 and Chk1 checkpoint kinases, respectively, all leading to phosphorylation of histone H2AX at the sites of DNA damage. These events invoke multiple downstream pathways which, depending on the extent of DNA damage, induce either cell cycle arrest (permitting DNA repair) or apoptosis (in case of irreparable DNA damage). Thus, cell cycle arrest can result from Chk1/2-dependent activating phosphorylation of p53 tumor suppressor gene or inactivating phosphorylation of Cdc25 family phosphatases which lead, respectively, to an upregulation of cyclin-dependent kinase inhibitor p21Waf1, or inhibition of CDK2 activity by rendering it in an inactive (phosphorylated) state. Alternatively, Chk1/2-dependent activation of p53 can induce apoptosis via upregulation and activation of a subset of pro-apoptotic proteins. A similar apoptotic effect may result from ATM-dependent phosphorylation of the nonreceptor tyrosine kinase c-Abl,24 which phosphorylates and activates the p53-related protein p73,25,26 leading to induction of pro-apoptotic genes in a manner that overlaps with the effect of p53 (reviewed in ref. 27). Interestingly, besides promoting cell cycle arrest, DDR-triggered inhibition of CDK2 may also contribute to the induction of pro-apoptotic genes by rendering the transcription factor FOXO-1 in its active (unphosphorylated) state.21 In accordance with the data obtained by Oka et al.1 colon tumorigenesis is associated with a gradual activation of the upstream DDR elements (ATM, Chk2 and H2AX) but, paradoxically, this does not result in an increased apoptosis induction, suggesting that malignant transformation may result in impairments of the DDR pathways downstream from Chk1/2 and the ATM-cAbl pathway (indicated by red crosses).
In the early stages of tumorigenesis, one molecular lesion appears to be sufficient to alter the growth properties of the transformed cell. For instance, colorectal carcinogenesis may start as an inherited, or more commonly environmentally acquired, mutation of the APC (Adenomatous Polyposis Coli) gene, which propels excessive, but not invasive, growth of the cells in colonic mucosa.11 But this single molecular lesion predisposes a cell to further sequential accumulation of other mutations, including those of p53, and the cells acquire invasive properties (Fig. 2). This is known as genomic instability, and is generally due to the faulty repair of DNA lesions, which may be incidental to the replicative process itself or due to mutagens in the environment.12 While normal cells are not subject to this hazard, as they have functioning cell cycle checkpoints that prevent cells with DNA damage from entering S phase, as recognized back in 1973;13 transformed cells are at risk for further damage, since these check points are usually not functional.14-16
Figure 2.
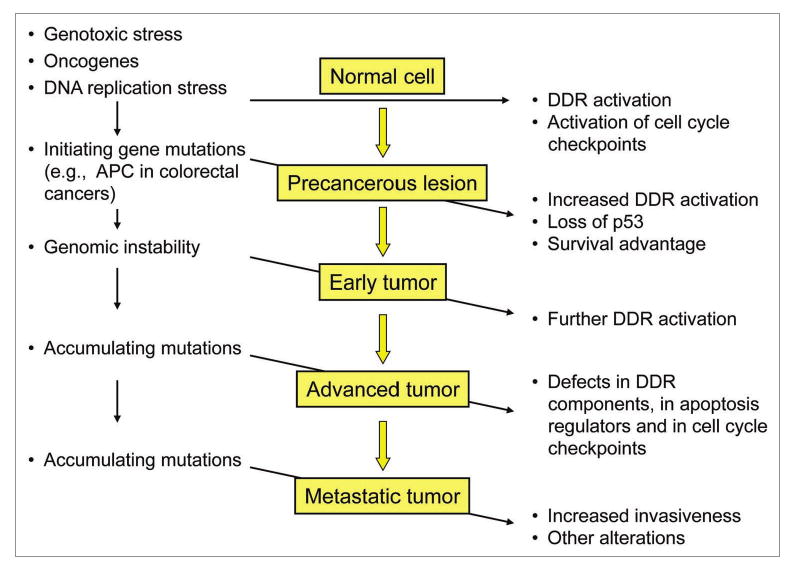
Possible role of DDR impairment in tumorigenesis. DDR protects normal cells from consequences of DNA damage through the activation of cell cycle checkpoints which prevent entry into the S phase of cells with damaged DNA. In the early stages of tumorigenesis, initiating mutations and associated DNA replication stress cause a marked activation of DDR. This frequently provides a sufficient barrier to the progression of precancerous lesions into malignant tumors by inducing cell cycle arrest and eliminating malignant cells by apoptosis induction. However, genomic instability and accumulating mutations can still lead to a more aggressive cancer, in spite of further activation of the upstream DDR components, as shown by Oka et al. in this issue of CB&T.1 This loss of the protecting function of the DDR may be explained by the accumulating defects in the downstream apoptosis-inducing and cell cycle-inhibitory pathways that are normally activated by DNA damage.
Yet in spite of this grim scenario, not everyone develops aggressive tumors. This is known to be due, in part, to elimination of transformed cells by the immune system, which recognizes new cell surface components. It has, however, also been proposed by groups led by Bartek17 and by Halazonetis18 that additional defenses against tumor progression are provided by DDR. These groups have provided evidence, based partly on studies of excised human bladder tumors at various stages of neoplastic progression,19 and precancerous lesions of lung and skin,20 that DDR may provide a barrier to neoplastic progression in early tumorigenesis. This seems reasonable, because sensors such as ATM/ATR can detect DNA damage and alert the DNA repair machinery, as well as Chk1/Chk2 and p53 or the related protein p73, to induce cell cycle arrest or apoptosis, and thus provide selective pressure against early neoplastic progression. However, the study by Oka et al. reported in this issue of Cancer Biology & Therapy1 raises the possibility that DDR may, under some circumstances, actually enhance the likelihood of malignant transformation.
Oka et al. observed the presence of activated markers of DDR in tumors from 55 consecutive cases of colorectal carcinoma. The markers, p-ATM, γH2AX and p-Chk2 protein levels, were found to be present in minimal amounts in normal tissue adjacent to the tumors, but their level increased in parallel with the progression from adenoma to advanced carcinoma. Intriguingly, no apoptosis, indicative of major DNA damage, was detected. The authors suggest that this is due to a malfunction of the apoptotic cascade or to an impairment of the downstream components of DDR. These were not identified, but the authors speculate that the impairment of DDR was due to the inability of the transcription factor FOXO-1 to induce apoptosis of cells with severe DNA damage. Normally, Chk2-dependent Cdk2 inactivation during DDR renders FOXO-1 in an unphosphorylated “pro-apoptotic” state;21 however, this as well as other downstream DDR pathways might be disabled in advanced cancers (Figs. 1 and 2). Thus, DDR may prevent, but under different conditions promote, tumor progression by allowing cells with increasingly severe DNA damage to survive under some circumstances.
Of course, this concept requires further documentation and an exploration of the possible ways of targeting DDR for treatment and/or chemoprevention of cancer. Therapeutically relevant DDR inhibition approaches have been described by Ljungman in a recent review.22 On the other hand, facilitation of DDR, as a cellular defense system, may have a role in cancer prevention. Indeed, recent findings suggest that frequently consumed plant antioxidants may cause DNA damage23 and therefore their regular ingestion may reduce cancer incidence, acting, at least in part, by DDR activation. Thus, studies by Oka et al. may have clinical and preventive importance by expanding our knowledge of the DDR.
References
- 1.Oka K, Tanaka T, Enoki T, Yoshimura K, Ohshima M, Kubo M, et al. DNA damage signaling is activated during cancer progression in human colorectal carcinoma. Cancer Biol Ther. 2010 doi: 10.4161/cbt.9.3.10751. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 3.Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nature Genetics. 2002;30:290–4. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 4.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–5. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 7.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 8.el-Deiry WS, Harper JW, O'Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–74. [PubMed] [Google Scholar]
- 9.Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, et al. Immediate early upregulation of bax expression by p53 but not TGFbeta1: a paradigm for distinct apoptotic pathways. Oncogene. 1994;9:1791–8. [PubMed] [Google Scholar]
- 10.Burns TF, El-Deiry WS. Microarray analysis of p53 target gene expression patterns in the spleen and thymus in response to ionizing radiation. Cancer Biol Ther. 2003;2:431–43. doi: 10.4161/cbt.2.4.478. [DOI] [PubMed] [Google Scholar]
- 11.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 12.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Studzinski GP, Gierthy JF. Selective inhibition of the cell cycle of cultured human diploid fibroblasts by aminonucleoside of puromycin. Journal of Cellular Physiology. 1973;81:71–83. doi: 10.1002/jcp.1040810109. [DOI] [PubMed] [Google Scholar]
- 14.Gierthy JF, Studzinski GP. Absence of aminonucleoside-sensitive steps in the cell cycle of SV40-transformed human fibroblasts. Cancer Res. 1973;33:2673–6. [PubMed] [Google Scholar]
- 15.Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z. Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes. Cancer Res. 1992;52:470–3. [PubMed] [Google Scholar]
- 16.Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: protection of normal cells and unshielding of cancer cells. Cell Cycle. 2002;1:375–82. doi: 10.4161/cc.1.6.259. [DOI] [PubMed] [Google Scholar]
- 17.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–9. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 18.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 19.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 20.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 21.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–7. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 22.Ljungman M. Targeting the DNA damage response in cancer. Chem Rev. 2009;109:2929–50. doi: 10.1021/cr900047g. [DOI] [PubMed] [Google Scholar]
- 23.Pesakhov S, Khanin M, Studzinski GP, Danilenko M. Distinct combinatorial effects of the plant polyphenols curcumin, carnosic acid and silibinin on proliferation and apoptosis in acute myeloid leukemia cells. Nutr Cancer. doi: 10.1080/01635581003693082. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387:516–9. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 25.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–9. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 26.Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814–7. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- 27.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–21. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]