

Abstract
Theoretical studies of proton-coupled electron transfer (PCET) reactions for model systems provide insight into fundamental concepts relevant to bioenergetics. A dynamical theoretical formulation for vibronically nonadiabatic PCET reactions has been developed. This theory enables the calculation of rates and kinetic isotope effects, as well as the pH and temperature dependences, of PCET reactions. Methods for calculating the vibronic couplings for PCET systems have also been developed and implemented. These theoretical approaches have been applied to a wide range of PCET reactions, including tyrosyl radical generation in a tyrosine-bound rhenium polypyridyl complex, phenoxyl/phenol and benzyl/toluene self-exchange reactions, and hydrogen abstraction catalyzed by the enzyme lipoxygenase. These applications have elucidated some of the key underlying physical principles of PCET reactions. The tools and concepts derived from these theoretical studies provide the foundation for future theoretical studies of PCET in more complex bioenergetic systems such as Photosystem II.
Keywords: proton-coupled electron transfer, proton transfer, electron transfer, hydrogen transfer
1. Introduction
Bioenergetic processes involving energy transformation play a vital role in all living organisms and structures. For example, Photosystem II (PSII) catalyzes the light-driven oxidation of water to molecular oxygen in plants.[1–3] In this process, absorption of light by the primary electron donor chlorophyll P680 leads to charge separation. A redox-active tyrosine residue YZ is thought to mediate the proton-coupled electron transfer (PCET) reaction between the oxygen evolving Mn4 cluster and P680.[4–10] The detailed mechanism of charge separation in PSII is still not well understood.
Given the large size and complexity of bioenergetic systems such as PSII, the fundamental aspects of the PCET mechanisms in these processes are more easily studied with model systems. Numerous experimental studies of model systems have been performed to elucidate the mechanism of tyrosine oxidation.[11–18] In some cases, photoexcitation of the metal in tyrosine-bound metal polypyridyl systems induces electron transfer from the tyrosine to the metal with concurrent proton transfer from the tyrosine to the solvent or the buffer.[14–18] Other experiments involve the chemical oxidation of phenols in solution.[11, 12] Often the data are interpreted in the context of Marcus theory for electron transfer.[19, 20]
A variety of theoretical approaches have been developed to study PCET reactions.[21–30] In the vibronically nonadiabatic formulation for PCET, [25–27] the active electrons and transferring proton are treated quantum mechanically, and the PCET reaction is described in terms of nonadiabatic transitions between pairs of reactant and product mixed electron-proton vibronic states. The dynamics of the proton donor-acceptor mode and the solvent/protein environment can be included in this formulation. Rate expressions based on linear response theory and the Golden Rule have been derived in various well-defined limits.[25,27] Methods for calculating the vibronic couplings in PCET systems have also been developed and implemented.[28,31] These theoretical approaches enable the calculation of rates and deuterium kinetic isotope effects (i.e., the ratio of the rate for hydrogen to the rate for deuterium), as well as their temperature and pH dependences.[32–35] Applications of these theoretical approaches to a wide range of systems have provided insight into fundamental concepts relevant to the PCET mechanisms in bioenergetic processes.
This review centers on PCET reactions that occur in a single chemical step (i.e., the electron and proton transfer simultaneously without an experimentally observable intermediate). In many cases, this type of PCET mechanism enables the system to avoid high-energy intermediates. Alternative sequential electron and proton transfer mechanisms[36] can be investigated with methods designed for studying single electron and single proton transfer reactions. The remainder of the review is organized as follows. Section II summarizes the vibronically nonadiabatic theoretical formulation for PCET reactions, providing the rate expressions in various limits and methods for calculating the vibronic couplings. Section III presents three applications of these theoretical approaches. The first application is the investigation of the detailed mechanism for tyrosyl radical generation in a tyrosine-bound rhenium polypyridyl complex that has recently been studied experimentally.[14,37] The second application is the calculation and analysis of the vibronic couplings for the phenoxyl/phenol and benzyl/toluene self-exchange reactions.[31] The third application is the study of the PCET reaction catalyzed by the enzyme soybean lipoxygenase, with an emphasis on the dynamical role of the enzyme and the magnitude and temperature dependence of the kinetic isotope effect.[34,35] Section IV presents a summary and discussion of potential future directions.
2. Theory
2.1 Rate calculations
This subsection summarizes the dynamical formulation for PCET reactions.[27] A PCET reaction is vibronically nonadiabatic when the vibronic coupling is significantly less than the thermal energy kBT. In this formulation, the active electrons and transferring proton are treated quantum mechanically, and the PCET reaction is described in terms of nonadiabatic transitions between pairs of reactant and product mixed electron-proton vibronic states. In the derivation of the rate expression, the nonadiabatic rate constant is expressed as the time integral of the probability flux correlation function, which in turn is expressed in terms of time correlation functions of the energy gap 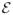 (i.e., the difference between the energies of the reactant and product states) and the R coordinate, where R is the distance between the proton donor and acceptor. The nonadiabatic coupling Vμν between the reactant and product vibronic states μ and ν is approximated to be of the form[38]
(i.e., the difference between the energies of the reactant and product states) and the R coordinate, where R is the distance between the proton donor and acceptor. The nonadiabatic coupling Vμν between the reactant and product vibronic states μ and ν is approximated to be of the form[38]
| (1) |
where R̄μ is the equilibrium value of the R coordinate on the reactant surface μ, is the vibronic coupling between states μ and ν at distance R̄μ, and αμν is a parameter that reflects the distance dependence of the vibronic coupling.
As will be discussed in the next subsection, the vibronic coupling can be approximated as Vμν (R) ≈ VelSμν (R) in the electronically nonadiabatic limit for proton transfer. Here Vel is an effective electronic coupling, and Sμν (R) is the overlap between the reactant and product proton vibrational wavefunctions at R. In the region about the equilibrium value of the R coordinate, this vibronic coupling can be approximated to be of the form in Eq. (1) with , where is the vibrational overlap at R̄μ.[31,38] The rate expressions given in this section are valid for any vibronic coupling with the form in Eq. (1).
Using linear response theory in conjunction with the Golden Rule and the form of the vibronic coupling given in Eq. (1), we derived a series of rate expressions based on well-defined approximations that have been tested for various systems. Two approximations that were found to be valid for the systems studied are: (1) only the initial value of the energy gap correlation function impacts the rate and (2) the R-coordinate correlation function can be represented by the analytical expression for an undamped classical mechanical harmonic oscillator. These two approximations lead to the rate expression[38,39]
| (2) |
where the summations are over the reactant and product vibronic states, Pμ is the Boltzmann probability for the reactant state, is the Boltzmann constant, is the driving force, Ω and M are the frequency and mass corresponding to the R mode, , and λμν is the reorganization energy. Related expressions have been derived for vibrationally nonadiabatic proton transfer reactions.[40–45]
Invoking the additional approximation that the R-coordinate time correlation function is approximately constant on the timescale of the probability flux correlation function decay leads to the simpler expression[35]
| (3) |
This rate expression is similar to the Marcus theory expression for electron transfer except for the factor of . It differs from Eq. (2) by the absence of the term λα in the two denominators. Thus, the approximation leading from Eq. (2) to Eq. (3) is equivalent to the assumption that λα ≪ λμν. If the R coordinate is assumed to be fixed during the PCET reaction, αμν= 0, and the first exponential factor in Eq. (3) becomes unity. In this limit, the expression in Eq. (3) becomes equivalent to the non-dynamical rate expressions derived previously with a multistate continuum theory for fixed R. [25] Further approximation that , in addition to αμν= 0, leads to the Marcus theory rate expression for nonadiabatic electron transfer modified by the inclusion of Franck-Condon overlap terms for the transferring hydrogen.[21,22]
An alternative derivation of Equation (3) provides additional connections to previous studies. Starting with the nonadiabatic PCET rate expression[25] derived for fixed R, the R-dependence of the vibronic coupling can be included according to the form in Eq. (1) and thermally averaged over a Boltzmann distribution for R. This procedure leads directly to Eq. (3) because
where
This approach is based on the assumptions that the reorganization energy and reaction free energy are independent of R, the R mode behaves as a classical harmonic oscillator, and the vibronic coupling depends exponentially on R. This relation provides a connection to the rate expression of Kuznetsov and Ulstrup[46] as implemented by Klinman and coworkers, [47,48] where the Marcus theory expression for nonadiabatic electron transfer is modified by the inclusion of R-dependent Franck-Condon overlap terms for the transferring hydrogen, thermally averaged over a Boltzmann distribution for R. Their rate expression leads to Eq. (3) if the overlap between the reactant and product proton vibrational wavefunctions is assumed to depend exponentially on R, as in Eq. (1) with . Thermal averaging over a Boltzmann distribution for R can also be performed numerically for a general form of the overlap between the reactant and product proton vibrational wavefunctions.
All of the quantities in the rate expressions above can be calculated from classical molecular dynamics simulations on the reactant surface and quantum mechanical calculations of the vibronic couplings. In linear response theory, the reorganization energy λμν can be expressed in terms of the energy gap variance as
| (4) |
and the driving force can be calculated as . The average R coordinate R̄ and the quantity kBT/MΩ2 = 〈δR2〉 can also be calculated directly from the molecular dynamics simulations. The mass M and frequency Ω can be determined independently by fitting the R-coordinate time correlation function from the classical molecular dynamics to the corresponding analytical expression for an undamped classical harmonic oscillator for the relevant time scale. The magnitude and distance dependence of the vibronic coupling (i.e., and αμν in Eq. (1)) can be determined from quantum mechanical methods described in the next section. For additional simplification, the reorganization energy can be assumed to be the same for all pairs of reactant and product states, so the reorganization energy and driving force can be calculated for the ground reactant and product states (i.e.,μ= ν=0). In this case, , where Δεμν is the difference between the product and reactant vibronic energy levels relative to the ground states. In addition, the exponential factor αμν can also be assumed to be the same for all pairs of reactant and product states.
The rate expression in Eq. (3) leads to a relatively simple expression for the KIE because only the first two factors depend on the isotope. If only the nonadiabatic transition between the two ground states is considered, the KIE can be approximated as[35]
| (5) |
where SH and SD are the overlaps of the hydrogen and deuterium wavefunctions, respectively, at R̄, and αH and αD represent the exponential dependence of this overlap on R for hydrogen and deuterium, respectively. Here we have assumed that , where the electronic coupling Vel is independent of isotope. The simplified expression for the KIE given in Eq. (5) provides insight into the magnitude and temperature dependence of the KIE for PCET systems. The effects of excited vibronic states can be included by summing over these states in Eq. (3).
The rate expressions above can be modified to include the quantum mechanical effects of the R coordinate motion. In this case, λα is replaced by in Eq. (2), and is replaced by in Eqs. (2) and (3). Analogous to the classical treatment of the R mode, the quantum mechanical version of Eq. (3) can also be obtained by starting with the nonadiabatic PCET rate expression[25] derived for fixed R and including the R-dependence of the vibronic coupling according to the form in Eq. (1), averaging over the quantum mechanical harmonic oscillator probability distribution function for R. In this case,
where
. Note that these modified expressions lead to the original expressions given above for the classical limit ĦΩ ≪ KBT.
2.2 Vibronic coupling calculations
According to the Golden Rule, the rate of a nonadiabatic PCET reaction is proportional to the square of the nonadiabatic vibronic coupling Vμν between the reactant and product diabatic vibronic states. This vibronic coupling is defined to be the Hamiltonian matrix element between the reactant and product mixed electronic-proton vibrational wavefunctions. The overall reaction is vibronically nonadiabatic with respect to the solvent or protein environment when this vibronic coupling is much less than kBT. The rate expressions given above for vibronically nonadiabatic PCET reactions depend on the magnitude and distance dependence of the vibronic coupling. Thus, the calculation of the vibronic coupling is essential for the determination of rates and KIEs for PCET reactions.
Even for vibronically nonadiabatic PCET reactions, the proton tunneling can be electronically nonadiabatic, electronically adiabatic, or in the intermediate regime. Here the electronically nonadiabatic and adiabatic limits for general PCET reactions refer to the relative timescales of the electrons and the transferring proton. The electrons respond instantaneously to the proton motion in the electronically adiabatic limit but not in the electronically nonadiabatic limit. In the electronically nonadiabatic limit, the vibronic coupling can be expressed as the product of the electronic coupling Vel and the Franck-Condon overlap Sμν of the reactant and product proton vibrational wavefunctions:
| (6) |
In the electronically adiabatic limit, the proton dynamics occur on the electronically adiabatic ground state potential energy surface, and the vibronic coupling can be calculated by standard semiclassical methods.[49,50] For a symmetric system, the vibronic coupling is half the splitting between the symmetric and antisymmetric proton vibrational states for the electronic ground state potential energy surface.
In general, PCET reactions can be in the intermediate regime between the electronically nonadiabatic and adiabatic limits. Georgievskii and Stuchebrukhov[28] derived a semiclassical expression for the general vibronic coupling :
| (7) |
where the factor κ is defined as
| (8) |
In Eq. (8), Γ(x) is the gamma-function and p is the proton adiabaticity parameter defined as
| (9) |
where vt is the tunneling velocity of the proton at the crossing point of the two proton potential energy curves and |ΔF| is the difference between the slopes of the proton potential energy curves at the crossing point. The tunneling velocity vt can be expressed in terms of the energy Vc at which the potential energy curves cross, the tunneling energy E, and the mass m of the proton:
| (10) |
In the electronically adiabatic limit, p≫1, κ = 1, and the vibronic coupling simplifies to . In the electronically nonadiabatic limit, p≪1, , and the vibronic coupling reduces to , as given in Eq. (6).
The adiabaticity of a general PCET reaction can be viewed in terms of the relative times of the proton tunneling and the electronic transition. Within the semiclassical framework, the time spent by the tunneling proton in the crossing region (i.e., the proton tunneling time) is
| (11) |
and the time required to change the electronic state (i.e., the electronic transition time) is
| (12) |
The adiabaticity parameter is simply the ratio of these two times:
| (13) |
When the proton tunneling time is much longer than the electronic transition time, the electronic states have enough time to mix completely and the proton transfer occurs on the electronically adiabatic ground state surface (i.e., the reaction is electronically adiabatic). When the proton tunneling time is much less than the electronic transition time, the reaction is electronically nonadiabatic because the electronic states no longer have enough time to mix completely during the proton tunneling process.
The vibronic couplings can be calculated with several different methods. As shown in Ref. [31], the input quantities for the semiclassical vibronic coupling expressions given above can be calculated with conventional electronic structure methods. Alternatively, the Hamiltonian matrix element between the reactant and product diabatic vibronic states can be calculated with the nuclear-electronic orbital (NEO) method.[51] In the NEO method, mixed nuclear-electronic wavefunctions are calculated with molecular orbital techniques. The diabatic vibronic states can be defined to be the two nonorthogonal localized nuclear-electronic wavefunctions obtained at the NEO-HF (Hartree-Fock) level. The Hamiltonian matrix element between these two diabatic vibronic states can be calculated with the NEO-NOCI (nonorthogonal configuration interaction) approach.[52] The NEO-NOCI method has been shown to agree well with the semiclassical methods for the phenoxyl/phenol system.[53]
3. Applications
3.1 PCET in tyrosine oxidation
Numerous experimental studies of model systems have been performed to study the fundamental mechanism of tyrosine oxidation.[11–18] For example, Sjodin et al. studied tyrosine-bound ruthenium-tris-bipyridine model systems with the flash-quench method.[15–18] In these experiments, the excited state of Ru is quenched by an external quencher, followed by electron transfer from the tyrosine to the photo-oxidized Ru. Sjödin et al. explained the pH dependence of the experimentally measured rate constant in terms of a PCET mechanism in which the proton transfers from tyrosine to bulk water.[15–18] Our theoretical calculations on this model system were consistent with this interpretation.[32] Alternative interpretations of this type of pH-dependence have also been proposed.[12,36] To avoid the use of external quenchers, Reece and Nocera designed and studied tyrosine-bound rhenium polypyridyl complexes.[14] In these experiments, tyrosine radicals are produced directly from the metal-to-ligand charge transfer (MLCT) excited state without an external quencher. The rate constant for emission quenching for this complex increases with pH in the range 4 < pH < 9 in the presence of phosphate buffer. In addition, the rate constant is independent of pH for 4 < pH < 8 in the absence of the phosphate buffer, and the dependence of the rate on phosphate buffer concentration is absent at low pH, where the dominant buffer species is H2PO4−.[54] These experimental observations are consistent with a PCET mechanism in which the proton is transferred from the tyrosine to the phosphate buffer species HPO42− for 4 < pH < 9.[54]
Recently, we used our multistate continuum theory[24–26] for PCET reactions to investigate the detailed mechanism for tyrosyl radical generation in the [Re(P-Y)(phen)(CO)3]PF6 system, where phen denotes 1,10-phenanthroline and P-Y denotes triphenylphosphine-tyrosine. This system is depicted in Figure 1. We evaluated two different models for tyrosyl radical generation in this system. In the phosphate-acceptor model, the phosphate buffer species HPO42− serves as the proton acceptor, and the pH-dependence of the overall rate arises from the titration between the HPO42− and H2PO4− forms of the phosphate buffer. In the water-acceptor model, bulk water serves as the proton acceptor. Our calculations indicate that the phosphate-acceptor model can successfully reproduce the experimentally observed pH-dependence of the overall rate and H/D KIE, whereas the water-acceptor model is not physically reasonable for this system. The phosphate buffer species HPO42− is favored over water as the proton acceptor in part because the proton donor-acceptor distance is ~0.2 Å smaller for the phosphate acceptor due to its negative charge. This smaller proton donor-acceptor distance leads to a larger vibronic coupling because of the greater overlap between the reactant and product proton vibrational wavefunctions. Other factors differentiating these two mechanisms include the slightly smaller outer-sphere reorganization energy for PCET in the phosphate-acceptor model and the differences in driving forces for PCET.
Figure 1.
Structure of the rhenium-tyrosine complex[14] hydrogen bonded to a phosphate HPO42− acceptor. The proton transfer and electron transfer reactions are indicated with arrows. Figure reproduced with permission from Ref. [37].
Here we review the results and analysis for the favored phosphate-acceptor model. In this model, the pH-dependence of the overall rate arises from the titration between the HPO42− and H2PO4− forms of the phosphate buffer, and the proton is assumed to transfer to HPO42− but not to H2PO4−. These assumptions are consistent with the experimental observations described above. Given these assumptions, the overall rate constant for tyrosine oxidation can be expressed as[54]
| (14) |
where [Phosphate]T is the total concentration of phosphate buffer and is the mole fraction of , which can be calculated as a function of pH using the relation
| (15) |
with pKa of 7.2 for HPO42−. In Eq. (14), is the bimolecular rate constant for the PCET reaction in which the proton is transferred to HPO42−, and kET is the rate constant for an ET reaction that is followed by rapid PT to the solvent.[54] The ET rate constant was determined to be kET = 1.0 ± 0.2 × 105 s−1 from experimental measurements in the absence of phosphate buffer. The bimolecular PCET rate constant was determined to be from measurements of kq as a function of phosphate buffer concentration at different pH values. The H/D KIE for the bimolecular PCET rate constant was determined to be ~3.0 from measurements of kq as a function of phosphate buffer concentration at high pH for the reaction in H2O and D2O.
We calculated the overall rate kq for hydrogen and deuterium transfer using the expression in Eq. (14) in conjunction with our PCET expression in Eq. (3) for αμν = 0 (i.e., for fixed proton donor-acceptor distance). The experimental and calculated data are depicted in Figure 2. We used the experimentally determined kET and fit the two coupling parameters in our model to reproduce the experimentally determined and H/D KIE for . The rate constant expressions given above apply to the unimolecular PCET reaction in a hydrogen-bonded complex. The bimolecular rate constant measured experimentally is related to the unimolecular rate constant according to , where Keq is the equilibrium association constant to form the hydrogen-bonded complex. For tyrosine and HPO42−, experiments[55] provide the estimate that Keq = 0.5 M−1, which is assumed to be the same in H2O and D2O for the KIE calculations.
Figure 2.

pH-dependence of the overall rate constant kq for the rhenium-tyrosine complex in H2O and D2O. The experimental data for kq measured with 10mM phosphate buffer in H2O (Figure 3 in Ref. [14]) are depicted with open circles (○). The rate constants for the phosphate-acceptor model with 10mM phosphate buffer calculated using Eq. (14) are depicted with thick and thin lines for the reaction in H2O and D2O, respectively. The rate constant for the reaction in H2O or D2O is plotted as a function of pH or pD, respectively, where the mole fraction or is calculated as a function of pH or pD using Eq. (15) with pKa = 7.2 or 7.8, respectively. Figure reproduced with permission from Ref. [37].
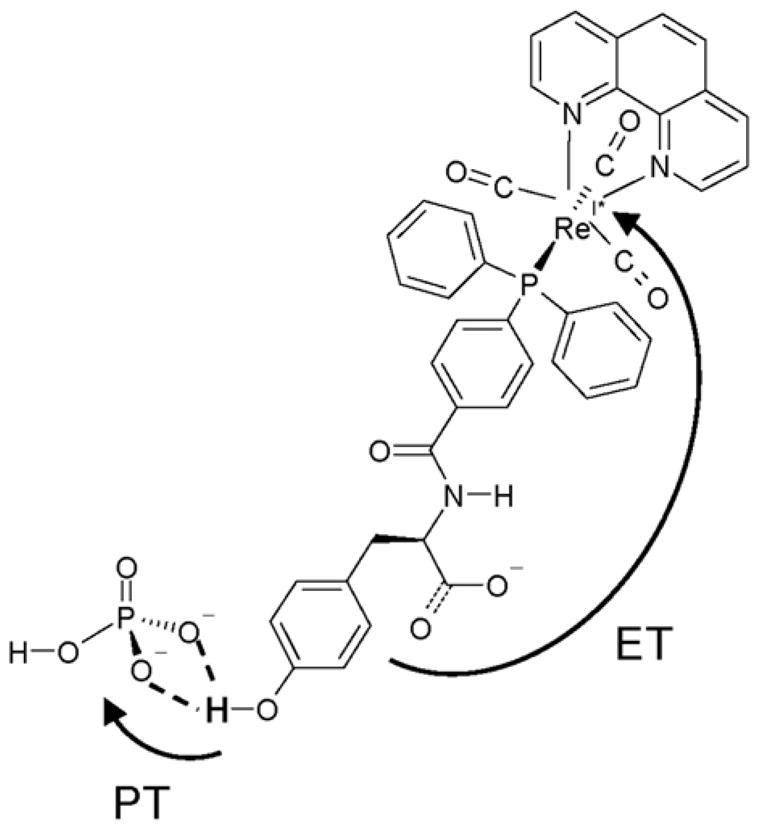
Analysis of the PCET calculations provides insight into the reorganization energies, reaction free energies, activation free energies, and vibronic couplings for the various pairs of reactant/product vibronic states for both hydrogen and deuterium transfer. Based on experimental measurements for a related system, [56] the inner-sphere reorganization energy is estimated to be 9.8 kcal/mol. The calculated outer-sphere reorganization energy for the overall PCET reaction was ~ 33 kcal/mol for all pairs of vibronic states. The dominant contribution to the rate arose from nonadiabatic transitions between the ground reactant state and the third product state for hydrogen transfer and the fourth product state for deuterium transfer. These contributions exceed the contributions from the nonadiabatic transition between the ground reactant and product states because the larger vibronic coupling overrides the slightly higher activation free energy barrier. The larger vibronic coupling is due mainly to the greater overlap between the reactant and product proton vibrational wavefunctions. This effect is more pronounced for deuterium than for hydrogen. The free energy surfaces and the corresponding proton vibrational wavefunctions are depicted in Figure 3.
Figure 3.
Analysis of the free energy surfaces for the PCET reaction in the phosphate-acceptor model for the rhenium-tyrosine complex. In the center frame are slices of the two-dimensional ET diabatic free energy surfaces as functions of the solvent coordinates. The slices were obtained along the line connecting the minima of the lowest energy reactant (I) and product (II) two-dimensional free energy surfaces. In the left frame is the reactant (I) proton potential energy curve and the corresponding proton vibrational wavefunction as a function of the proton coordinate evaluated at the minimum of the ground state reactant free energy surface. In the right frame is the product (II) proton potential energy curve and the corresponding proton vibrational wavefunction as a function of the proton coordinate evaluated at the minimum of the ground state product free energy surface. Figure reproduced with permission from Ref. [37].
These calculations provide insight into the fundamental mechanism of tyrosyl radical generation. Such insights have implications for PCET in PS II. For example, the conclusion that the phosphate serves as a proton acceptor is consistent with experimental observations in D1-His190 mutants of PSII, where the rates of P680+• reduction by Yz increased dramatically in the presence of imidazole and other small organic bases.[57,58]
3.2 Vibronic couplings for phenoxyl/phenol and benzyl/toluene self-exchange reactions
We have calculated the vibronic couplings for the phenoxyl/phenol and the benzyl/toluene self-exchange reactions. As discussed above, the vibronic couplings significantly impact the rates and kinetic isotope effects, as well as their temperature dependences, for general PCET reactions. Although the splittings between the ground and excited electronic states are significantly larger than the thermal energy kBT at room temperature for these systems, the vibronic couplings for both systems were found to be smaller than kBT, indicating that the reactions are vibronically nonadiabatic with respect to a solvent environment. The transition state geometries of the phenoxyl/phenol and the benzyl/toluene systems are qualitatively different. The phenoxyl/phenol transition state has C2 symmetry, and the O···H···O bond is approximately planar with the phenol rings and represents a strong hydrogen bond. The benzyl/toluene transition state has C2h symmetry, and the C···H···C bond is orthogonal to the planes of the benzene rings and does not form a strong hydrogen bond.
As shown in Figure 4, the electronic wavefunctions for the two systems are qualitatively different at the transition state. In the phenoxyl/phenol system, the two highest-energy occupied molecular orbitals are dominated by 2p orbitals on the donor and acceptor oxygen atoms that are perpendicular to the hydrogen donor-acceptor axis. In the ground state, the doubly occupied molecular orbital corresponds to π-bonding, and the singly occupied molecular orbital corresponds to π-antibonding. In the benzyl/toluene system, the two highest-energy occupied molecular orbitals are dominated by σ orbitals on the donor and acceptor carbon atoms and are oriented along the hydrogen donor-acceptor axis. In the ground state, the highest doubly occupied molecular orbital corresponds to σ-bonding, and the singly occupied molecular orbital corresponds to σ-antibonding. Previously Mayer, Borden, and coworkers[59] used these differences in the singly occupied molecular orbitals of the transition state wavefunctions to designate the phenoxyl/phenol and benzyl/tolune systems as PCET and hydrogen atom transfer (HAT), respectively.
Figure 4.
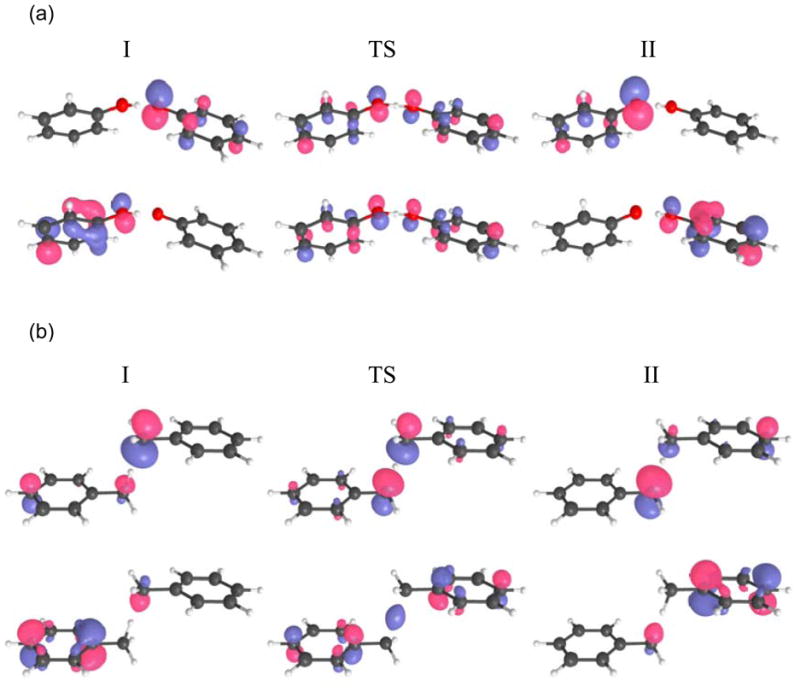
The two highest-energy occupied electronic molecular orbitals for (a) the phenoxyl/phenol and (b) the benzyl/toluene system. The electronic wavefunctions for diabatic states I and II are calculated at the minima of the ground state electronically adiabatic potential energy curves shown in Figure 5, and the electronic wavefunctions for the transition states (TS) are calculated at the maxima of these potential energy curves. For both systems, the ground state electronic wavefunction is predominantly single configurational, and the lower molecular orbital is doubly occupied, while the upper molecular orbital is singly occupied. Figure reproduced with permission from Ref. [31].
Figure 5 depicts the potential energy curves along the transferring hydrogen coordinate for the phenoxyl/phenol and the benzyl/toluene systems. The CASSCF electronically adiabatic ground and excited state curves, as well as the electronically diabatic curves corresponding to the reactant and product electron transfer states, are depicted. Note that the splitting between the electronically adiabatic ground and excited states is more than an order of magnitude larger for the benzyl/toluene system than for the phenoxyl/phenol system. As a result, the diabatic curves are very similar to the adiabatic curves for the phenoxyl/phenol system but are significantly different from the adiabatic curves for the benzyl/toluene system.
Figure 5.
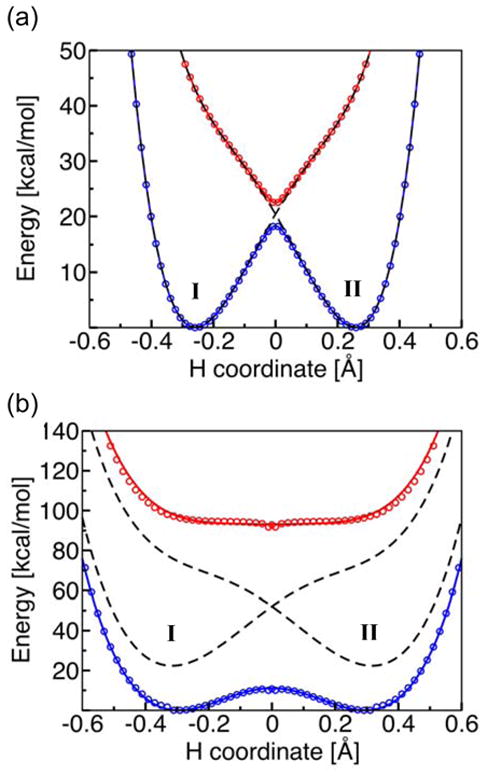
State-averaged CASSCF ground and excited state electronically adiabatic potential energy curves along the transferring hydrogen coordinate for (a) the phenoxyl/phenol and (b) the benzyl/toluene system. The coordinates of all nuclei except the transferring hydrogen correspond to the transition state geometry. The proton donor-acceptor distances are 2.40 Å and 2.72 Å, respectively, for the phenoxyl/phenol and the benzyl/toluene system. The CASSCF results are depicted as open circles that are blue for the ground state and red for the excited state. The black dashed lines represent the diabatic potential energy curves corresponding to the two localized diabatic electron transfer states I and II. The mixing of these two diabatic states with the electronic coupling Vel leads to the CASSCF ground and excited state electronically adiabatic curves depicted with solid colored lines following the colored open circles. For the phenoxyl/phenol system, the solid colored lines and the black dashed lines are nearly indistinguishable because the adiabatic and diabatic potential energy curves are virtually identical except in the transition state region. Figure reproduced with permission from Ref. [31].
The fundamental nature of the proton tunneling is different for the two systems. For the phenoxyl/phenol system, the adiabaticity parameter p is very small, , and τe ≈ 80τp. In this case, the electronic transition time is significantly greater than the proton tunneling time. As a result, the electrons are not able to rearrange fast enough for the proton to move on the electronically adiabatic ground state surface, and the proton transfer reaction is electronically nonadiabatic. For the benzyl/toluene system, the adiabaticity parameter p is larger, κ ≈ 1, and τp ≈ 4τe. In this case, the electronic transition time is less than the proton tunneling time. Thus, the electrons can respond instantaneously to the proton motion, and the proton moves on the electronically adiabatic ground state surface. This analysis indicates that the proton tunneling is electronically nonadiabatic for the phenoxyl/phenol system but electronically adiabatic for the benzyl/toluene system.
Figure 6 illustrates the physical principles underlying the electronically nonadiabatic and adiabatic limits. For the electronically nonadiabatic phenoxyl/phenol reaction, the vibronic coupling is the product of the electronic coupling between the diabatic states and the overlap of the reactant and product proton vibrational wavefunctions corresponding to these diabatic states. For the electronically adiabatic benzyl/toluene reaction, the vibronic coupling is half the energy splitting between the states corresponding to the symmetric and antisymmetric proton vibrational wavefunctions for the electronically adiabatic ground state.
Figure 6.
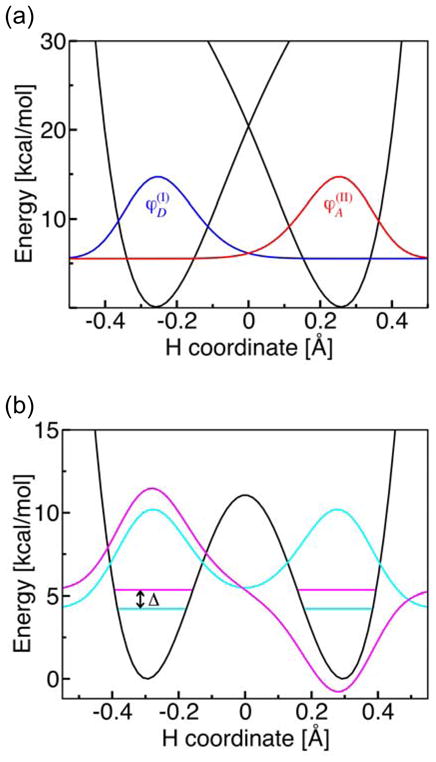
(a) Diabatic potential energy curves corresponding to the two localized diabatic electron transfer states I and II and the corresponding proton vibrational wavefunctions (blue) and (red) for the phenoxyl/phenol system. Since this reaction is electronically nonadiabatic, the vibronic coupling is the product of the electronic coupling Vel and the overlap of the reactant and product proton vibrational wavefunctions . (b) Electronically adiabatic ground state potential energy curve and the corresponding proton vibrational wavefunctions for the benzyl/toluene system. Since this reaction is electronically adiabatic, the vibronic coupling is equal to half of the energy splitting Δ between the symmetric (cyan) and antisymmetric (magenta) proton vibrational states for the electronic ground state potential energy surface. For illustrative purposes, the excited vibrational state is shifted up in energy by 0.8 kcal/mol. Figure reproduced with permission from Ref. [31].
We also examined the dependence of the vibronic coupling on the proton donor-acceptor distance and the dependence of the KIE on both the magnitude and the distance dependence of the vibronic coupling. The vibronic coupling decreases exponentially with the proton donor-acceptor distance for both electronically adiabatic and electronically nonadiabatic reactions in the range of chemically relevant distances. For a given proton donor-acceptor distance, the vibronic couplings are significantly smaller for deuterium than for hydrogen because of the smaller overlap between the reactant and product proton vibrational wavefunctions for deuterium. Moreover, the value of the exponential decay parameter α is larger for deuterium than for hydrogen because the overlap between the reactant and product deuterium wavefunctions falls off faster with distance than the corresponding overlap for the hydrogen wavefunctions. These trends are directly relevant to the study of general PCET reactions.
This type of analysis provides a new perspective on the distinction between PCET and HAT reactions. A conventional method for distinguishing PCET from HAT is that the electron and proton are transferred between different donors and acceptors (or different sets of orbitals) for PCET. Within this framework, our analysis suggests that PCET reactions are electronically nonadiabatic, whereas HAT reactions are electronically adiabatic. These two mechanisms can be differentiated by calculating the adiabaticity parameter, which depends on the electronic coupling and other quantities that can be determined with quantum chemistry methods.
Furthermore, the distinction between electronic adiabaticity and nonadiabaticity has important experimental consequences because the vibronic couplings can be substantially different in the electronically adiabatic and nonadiabatic limits. As discussed above, the magnitude and distance dependence of the vibronic coupling can significantly impact the magnitudes and temperature dependences of the rates and the KIEs. Thus, the calculation of the vibronic coupling in the correct limit, or in the intermediate regime, is critical for the interpretation of experimental data and the generation of experimentally testable predictions.
3.3 PCET catalyzed by lipoxygenase
We have examined the dynamical behavior and the temperature dependence of the kinetic isotope effects for the PCET reaction catalyzed by the enzyme soybean lipoxygenase (SLO).[35] Lipoxygenases catalyze the oxidation of unsaturated fatty acids. Kinetic studies have shown that the hydrogen abstraction step in SLO is rate-limiting above 32 °C.[60] In this step, the pro-S hydrogen atom from carbon atom C11 of the linoleic acid substrate is transferred to the Fe(III)–OH cofactor, forming a radical intermediate substrate and Fe(II)–OH2.[61] Quantum mechanical calculations[62] and analysis of the thermodynamic properties based on electrochemical data[34,48] indicate that the hydrogen abstraction step occurs by a PCET mechanism, in which the electron transfers from the π-system of the substrate to the iron of the cofactor, while the proton transfers from the C11 carbon of the substrate to the hydroxyl ligand of the cofactor. This PCET reaction is depicted in Figure 7. The SLO reaction has been studied with a variety of theoretical approaches.[34,35,47,48,61–70]
Figure 7.

The hydrogen abstraction step of the reaction catalyzed by soybean lipoxygenase with its natural substrate linoleic acid. In this step, a hydrogen is abstracted from the linoleic acid to the iron cofactor. Figure reproduced with permission from Ref.[35].
Our calculations of this reaction were based on the vibronically nonadiabatic formulation for PCET reactions that includes the quantum mechanical effects of the active electrons and the transferring proton, as well as the motions of all atoms in the complete solvated enzyme system.[27,39] As described above, the rate is represented by the time integral of a probability flux correlation function that depends on the vibronic coupling, the average of the energy gap and R coordinate, and the time correlation functions of the energy gap and R coordinate. The vibronic couplings were estimated to within a constant factor by calculating the overlaps between reactant and product hydrogen vibrational wavefunctions for model systems, and the other quantities were calculated from classical molecular dynamics simulations of the entire system.
We tested the underlying assumptions of this vibronically nonadiabatic formulation for the SLO enzyme reaction and investigated the dynamical behavior of the protein, substrate, and cofactor. Our estimates of the contributions to the vibronic couplings for this PCET reaction indicated that this reaction is vibronically nonadiabatic. Moreover, our molecular dynamics simulations of the entire solvated enzyme system provided an estimate of 39 kcal/mol for the total reorganization energy. We found that the dynamical behavior (i.e., the time dependence) of the probability flux correlation function is dominated by the equilibrium protein and solvent motions and is not significantly influenced by the proton donor-acceptor motion. The magnitude of the overall rate, however, is strongly influenced by the frequency of the proton donor-acceptor motion, the magnitude and distance dependence of the vibronic coupling, and the protein/solvent reorganization energy.
These calculations reproduced the experimentally observed[48,61] magnitude and temperature dependence of the KIE for the SLO enzyme reaction without fitting any parameters directly to the experimental kinetic data. A comparison of the experimental and calculated data is depicted in Figure 8. The temperature dependence of the KIE is determined mainly by the effective proton donor-acceptor frequency, which can be calculated from the R coordinate variance in the molecular dynamics simulations, and the distance dependence of the vibronic couplings for hydrogen and deuterium, which can be calculated from the vibrational wavefunctions for model systems. The ratio of the overlaps of the hydrogen and deuterium vibrational wavefunctions strongly impacts the magnitude of the KIE but does not significantly influence the temperature dependence. These trends are summarized in the relatively simple, approximate expression for the KIE given in Eq. (5). For the SLO reaction, the large magnitude of the KIE arises mainly from the dominance of tunneling between the ground vibronic states and the relatively large ratio of the overlaps between the corresponding hydrogen and deuterium vibrational wavefunctions. The weak temperature dependence of the KIE is due in part to the dominance of the local component of the proton donor-acceptor motion.
Figure 8.

Temperature dependence of the KIE for soybean lipoxygenase obtained with the rate expression in Eq. (2) including the excited vibronic states. The experimental data[48] are depicted with circles. Figure reproduced with permission from Ref. [35].
4. Summary and conclusions
In this review, we described the vibronically nonadiabatic theoretical formulation for PCET reactions and presented rate expressions in various limits, as well as methods for calculating the vibronic couplings. We illustrated this methodology through three different applications. In the first application, we investigated tyrosyl radical generation in a tyrosine-bound rhenium polypyridyl complex. The experimental data and theoretical calculations indicate that the electron transfers from the tyrosine to the photoexcited rhenium concurrently with the proton transferring to the phosphate buffer. In the second application, we calculated the vibronic couplings for the phenoxyl/phenol and benzyl/toluene self-exchange reactions and found that the proton transfer reaction is electronically nonadiabatic for phenoxyl/phenol, which was designated as PCET, but electronically adiabatic for benzyl/toluene, which was designated as HAT. The analysis of these couplings provided a new diagnostic for differentiating between the conventionally defined PCET and HAT reactions. In the third application, we simulated the PCET reaction catalyzed by the enzyme lipoxygenase. The simulations elucidated the dynamical role of the enzyme and the physical basis for the experimental observations of an unusually large KIE with relatively weak temperature dependence.
These theoretical studies provide insight into fundamental concepts relevant to PCET mechanisms in bioenergetic processes. The feedback between experiment and theory plays an essential role in these types of studies. In principle, these theoretical approaches could be applied to model systems that are more directly related to specific biological systems such as PSII. The calculation of reorganization energies, vibronic couplings, rates, and KIEs for PCET reactions in complex bioenergetic systems would provide new insights into the overall mechanisms. Such calculations represent an exciting and challenging direction for future research.
Acknowledgments
We are grateful for support of this work by NSF grant CHE-05-01260 and NIH grant GM56207.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Debus RJ. Biochimica et Biophysica Acta. 1992;1102:269. doi: 10.1016/0005-2728(92)90133-m. [DOI] [PubMed] [Google Scholar]
- 2.Barber J, Andersson B. Nature. 1994;370:31. [Google Scholar]
- 3.Diner BA, Babcock GT. In: Oxygenic Photosynthesis: The Light Reactions. Ort DR, Yocum CF, editors. Kluwer; Dordrecht, The Netherlands: 1996. pp. 213–47. [Google Scholar]
- 4.Tommos C, Tang X-S, Warncke K, Hoganson CW, Styring S, McCracken J, Diner BA, Babcock GT. Journal of the American Chemical Society. 1995;117:10325–35. [Google Scholar]
- 5.Hoganson CW, Lydakis-Simantiris N, Tang X-S, Tommos C, Warncke K, Babcock GT, Diner BA, McCracken J, Styring S. Photosynthesis Research. 1995;47:177–84. doi: 10.1007/BF00020428. [DOI] [PubMed] [Google Scholar]
- 6.Hoganson CW, Babcock GT. Science. 1997;277:1953–56. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 7.Blomberg MRA, Siegbahn PEM, Styring S, Babcock GT, Akermark B, Korall P. Journal of the American Chemical Society. 1997;119:8285–92. [Google Scholar]
- 8.Kuhne H, Brudvig GW. Journal of Physical Chemistry B. 2002;106:8189–96. [Google Scholar]
- 9.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Nature. 2005;438:1040–44. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 10.Groot ML, Pawlowicz NP, van Wilderen LJGW, Breton J, van Stokkum IHM, van Grondelle R. Proceedings of the National Academy USA. 2005;102:13087–92. doi: 10.1073/pnas.0503483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhile IJ, Mayer JM. Journal of the American Chemical Society. 2004;126:12718–19. doi: 10.1021/ja031583q. [DOI] [PubMed] [Google Scholar]
- 12.Fecenko CJ, Meyer TJ, Thorp HH. Journal of the American Chemical Society. 2006;128:11020–21. doi: 10.1021/ja061931z. [DOI] [PubMed] [Google Scholar]
- 13.Costentin C, Robert M, Saveant J-M. Journal of the American Chemical Society. 2006;128:4552–53. doi: 10.1021/ja060527x. [DOI] [PubMed] [Google Scholar]
- 14.Reece SY, Nocera DG. Journal of the American Chemical Society. 2005;127:9448–58. doi: 10.1021/ja0510360. [DOI] [PubMed] [Google Scholar]
- 15.Sjodin M, Styring S, Akermark B, Sun L, Hammarstrom L. Journal of the American Chemical Society. 2000;122:3932–36. [Google Scholar]
- 16.Sjodin M, Styring S, Akermark B, Sun L, Hammarstrom L. Phil Trans R Soc Lond B. 2002;357:1471–79. doi: 10.1098/rstb.2002.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjodin M, Ghanem R, Polivka T, Pan J, Styring S, Sun L, Sundstrom V, Hammarstrom L. Phys Chem Chem Phys. 2004;6:4851–58. [Google Scholar]
- 18.Sjodin M, Styring S, Wolpher H, Xu Y, Sun L, Hammarstrom L. Journal of the American Chemical Society. 2005;127:3855–63. doi: 10.1021/ja044395o. [DOI] [PubMed] [Google Scholar]
- 19.Marcus RA. Annual Reviews of Physical Chemistry. 1964;15:155–96. [Google Scholar]
- 20.Marcus RA, Sutin N. Biochimica et Biophysica Acta. 1985;811:265–322. [Google Scholar]
- 21.Cukier RI. Journal of Physical Chemistry. 1996;100:15428–43. [Google Scholar]
- 22.Cukier RI, Nocera DG. Annual Reviews of Physical Chemistry. 1998;49:337–69. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- 23.Cukier RI. Biochimica et Biophysica Acta-Bioenergetics. 2004;1655:37–44. doi: 10.1016/j.bbabio.2003.06.011. [DOI] [PubMed] [Google Scholar]
- 24.Hammes-Schiffer S. Accounts of Chemical Research. 2001;34:273–81. doi: 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]
- 25.Soudackov A, Hammes-Schiffer S. Journal of Chemical Physics. 2000;113:2385–96. [Google Scholar]
- 26.Soudackov A, Hammes-Schiffer S. Journal of Chemical Physics. 1999;111:4672–87. [Google Scholar]
- 27.Soudackov A, Hatcher E, Hammes-Schiffer S. Journal of Chemical Physics. 2005;122:014505. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]
- 28.Georgievskii Y, Stuchebrukhov AA. Journal of Chemical Physics. 2000;113:10438–50. doi: 10.1063/1.1796751. [DOI] [PubMed] [Google Scholar]
- 29.Mayer JM. Annual Review of Physical Chemistry. 2004;55:363–90. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- 30.Costentin C, Robert M, Saveant J-M. Journal of Electroanalytical Chemistry. 2006;588:197–206. [Google Scholar]
- 31.Skone JH, Soudackov AV, Hammes-Schiffer S. Journal of the American Chemical Society. 2006;128:16655. doi: 10.1021/ja0656548. [DOI] [PubMed] [Google Scholar]
- 32.Carra C, Iordanova N, Hammes-Schiffer S. Journal of the American Chemical Society. 2003;125:10429–36. doi: 10.1021/ja035588z. [DOI] [PubMed] [Google Scholar]
- 33.Hammes-Schiffer S, Iordanova N. Biochimica et Biophysica Acta-Bioenergetics. 2004;1655:29–36. doi: 10.1016/j.bbabio.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Hatcher E, Soudackov AV, Hammes-Schiffer S. Journal of the American Chemical Society. 2004;126:5763–75. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- 35.Hatcher E, Soudackov AV, Hammes-Schiffer S. Journal of the American Chemical Society. 2007;129:187–96. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- 36.Costentin C, Robert M, Saveant J-M. Journal of the American Chemical Society. 2007;129:5870–79. doi: 10.1021/ja067950q. [DOI] [PubMed] [Google Scholar]
- 37.Ishikita H, Soudackov AV, Hammes-Schiffer S. Journal of the American Chemical Society. 2007 doi: 10.1021/ja072708k. (in press) [DOI] [PubMed] [Google Scholar]
- 38.Hatcher E, Soudackov A, Hammes-Schiffer S. Chemical Physics. 2005;319:93–100. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]
- 39.Hatcher E, Soudackov A, Hammes-Schiffer S. Journal of Physical Chemistry B. 2005;109:18565–74. doi: 10.1021/jp052909f. [DOI] [PubMed] [Google Scholar]
- 40.Borgis D, Lee S, Hynes JT. Chemical Physics Letters. 1989;162:19–26. [Google Scholar]
- 41.Borgis D, Hynes JT. Journal of Chemical Physics. 1991;94:3619–28. [Google Scholar]
- 42.Suarez A, Silbey R. Journal of Chemical Physics. 1991;94:4809–16. [Google Scholar]
- 43.Trakhtenberg LI, Klochikhin VL, Pshezhetsky SY. Chemical Physics. 1982;69:121–34. [Google Scholar]
- 44.Kiefer PM, Hynes JT. Solid State Ionics. 2004;168:219–24. [Google Scholar]
- 45.Kiefer PM, Hynes JT. Journal of Physical Chemistry A. 2004;108:11793–808. [Google Scholar]
- 46.Kuznetsov AM, Ulstrup J. Canadian Journal of Chemistry. 1999;77:1085–96. [Google Scholar]
- 47.Knapp MJ, Klinman JP. European Journal of Biochemistry. 2002;269:3113–21. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 48.Knapp MJ, Rickert KW, Klinman JP. Journal of the American Chemical Society. 2002;124:3865–74. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 49.Vorotyntsev MA, Dogonadze RR, Kuznetsov AM. Dokl Akad Nauk SSSR. 1973;209:1135. [Google Scholar]
- 50.Fain B. Theory of Rate Processes in Condensed Media. Springer; New York: 1980. [Google Scholar]
- 51.Webb SP, Iordanov T, Hammes-Schiffer S. Journal of Chemical Physics. 2002;117:4106–18. [Google Scholar]
- 52.Skone JH, Pak MV, Hammes-Schiffer S. Journal of Chemical Physics. 2005;123:134108. doi: 10.1063/1.2039727. [DOI] [PubMed] [Google Scholar]
- 53.Ludlow MK, Skone JH, Hammes-Schiffer S. Journal of Physical Chemistry B. 2007 doi: 10.1021/jp0753474. (submitted) [DOI] [PubMed] [Google Scholar]
- 54.Reece SY, Nocera DG. personal communication.
- 55.Alev-Behmoaras T, Toulme JJ, Helene C. Photochemistry and Photobiology. 1979;30:533–39. [Google Scholar]
- 56.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. Journal of the American Chemical Society. 2006;128:13654–55. doi: 10.1021/ja0636688. [DOI] [PubMed] [Google Scholar]
- 57.Hays A-MA, Vassiliev IR, Golbeck JH, Debus RJ. Biochemistry. 1998;37:11352–65. doi: 10.1021/bi980510u. [DOI] [PubMed] [Google Scholar]
- 58.Hays A-MA, Vassiliev IR, Golbeck JH, Debus RJ. Biochemistry. 1999;38:11851–65. doi: 10.1021/bi990716a. [DOI] [PubMed] [Google Scholar]
- 59.Mayer JM, Hrovat DA, Thomas JL, Borden WT. Journal of the American Chemical Society. 2002;124:11142–47. doi: 10.1021/ja012732c. [DOI] [PubMed] [Google Scholar]
- 60.Glickman MH, Klinman JP. Biochemistry. 1995;34:14077–92. doi: 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]
- 61.Rickert KW, Klinman JP. Biochemistry. 1999;38:12218–28. doi: 10.1021/bi990834y. [DOI] [PubMed] [Google Scholar]
- 62.Lehnert N, Solomon EI. J Biol Inorg Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- 63.Kuznetsov AM, Ulstrup J. Canadian Journal of Chemistry. 1999;77:1085–96. [Google Scholar]
- 64.Tresadern G, McNamara JP, Mohr M, Wang H, Burton NA, Hillier IH. Chemical Physics Letters. 2002;358:489–94. [Google Scholar]
- 65.Olsson MHM, Siegbahn PEM, Warshel A. Journal of the American Chemical Society. 2004;126:2820–28. doi: 10.1021/ja037233l. [DOI] [PubMed] [Google Scholar]
- 66.Olsson MHM, Siegbahn PEM, Warshel A. Journal of Biological Inorganic Chemistry. 2004;9:96–99. doi: 10.1007/s00775-003-0503-2. [DOI] [PubMed] [Google Scholar]
- 67.Siebrand W, Smedarchina Z. Journal of Physical Chemistry B. 2004;108:4185–95. [Google Scholar]
- 68.Borowski T, Krol M, Chruszcz M, Broclawik E. Journal of Physical Chemistry B. 2001;105:12212–20. [Google Scholar]
- 69.Borowski T, Broclawik E. Journal of Physical Chemistry B. 2003;107:4639–46. [Google Scholar]
- 70.Tejero I, Eriksson LA, Gonzalez-Lafont A, Marquet J, Lluch JM. Journal of Physical Chemistry B. 2004;108:13831–38. [Google Scholar]