

Abstract
We have demonstrated that polyamide nucleic acids complementary to the transactivation response (TAR) element of HIV-1 LTR inhibit HIV-1 production when transfected in HIV-1 infected cells. We have further shown that anti-TAR PNA (PNATAR) conjugated with cell-penetrating peptide (CPP) is rapidly taken up by cells and exhibits strong antiviral and anti-HIV-1 virucidal activities. Here, we pharmacokinetically analyzed 125I-labeled PNATAR conjugated with two CPPs: a 16-mer penetratin derived from antennapedia and a 13-mer Tat peptide derived from HIV-1 Tat. We administered the 125I-labeled PNATAR–CPP conjugates to male Balb/C mice through intraperitoneal or gavage routes. The naked 125I-labeled PNATAR was used as a control. Following a single administration of the labeled compounds, their distribution and retention in various organs were monitored at various time points. Regardless of the administration route, a significant accumulation of each PNATAR-CPP conjugate was found in different mouse organs and tissues. The clearance profile of the accumulated radioactivity from different organs displayed a biphasic exponential pathway whereby part of the radioactivity cleared rapidly, but a significant portion of it was slowly released over a prolonged period. The kinetics of clearance of individual PNATAR-CPP conjugates slightly varied in different organs, while the overall biphasic clearance pattern remained unaltered regardless of the administration route. Surprisingly, unconjugated naked PNATAR displayed a similar distribution and clearance profile in most organs studied although extent of its uptake was lower than the PNATAR-CPP conjugates.
Introduction
Peptide nucleic acids (PNAs) are a new class of antisense DNA analogs that have no sugar phosphate backbone and display high affinity for complementary RNA and DNA sequences (Nielsen et al., 1991; UHLMANN, 1998; Koppelhus and Nielsen, 2003). We have shown that PNAs targeted to the primer binding site and A-loop sequences in the U5-PBS region of the HIV-1 genome block the initiation of reverse transcription (Lee et al., 1998), strongly destabilize the priming of natural 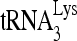 primer with the viral genome, and inhibit HIV-1 replication (Kaushik and Pandey, 2002). We have further demonstrated that anti-HIV-1 PNA targeted to the transactivation response (TAR) element of HIV-1 LTR when transfected into HIV-1-infected cells prevents Tat–TAR interaction and blocks not only Tat-mediated transactivation of HIV-1 LTR transcription (Mayhood et al., 2000), but also HIV-1 production (Kaushik et al., 2002a, 2002b). These studies demonstrate the therapeutic potential of PNA for intervention in HIV-1 replication and production by targeting multiple critical regions of the viral genome. However, poor cellular uptake of PNA has been a major impediment in the development of this class of compounds as therapeutic agents. Various approaches have been adopted for the efficient biodelivery of PNA into cells, including conjugation to lipophilic moieties (Ljungstrom et al., 1999; Muratovska et al., 2001), cell-specific receptor ligands (Basu and Wickstrom, 1997; Boffa et al., 2000; Zhang et al., 2001), carrier peptides (Cutrona et al., 2000), neamine (Riguet et al., 2004), and guanidine-based PNA (Zhou et al., 2003) electrostatically bound with the polymeric core shell microsphere (Chiarantini et al., 2005) or loaded into the autologous red blood cells (Chiarantini et al., 1995; Turrini et al., 1991).
primer with the viral genome, and inhibit HIV-1 replication (Kaushik and Pandey, 2002). We have further demonstrated that anti-HIV-1 PNA targeted to the transactivation response (TAR) element of HIV-1 LTR when transfected into HIV-1-infected cells prevents Tat–TAR interaction and blocks not only Tat-mediated transactivation of HIV-1 LTR transcription (Mayhood et al., 2000), but also HIV-1 production (Kaushik et al., 2002a, 2002b). These studies demonstrate the therapeutic potential of PNA for intervention in HIV-1 replication and production by targeting multiple critical regions of the viral genome. However, poor cellular uptake of PNA has been a major impediment in the development of this class of compounds as therapeutic agents. Various approaches have been adopted for the efficient biodelivery of PNA into cells, including conjugation to lipophilic moieties (Ljungstrom et al., 1999; Muratovska et al., 2001), cell-specific receptor ligands (Basu and Wickstrom, 1997; Boffa et al., 2000; Zhang et al., 2001), carrier peptides (Cutrona et al., 2000), neamine (Riguet et al., 2004), and guanidine-based PNA (Zhou et al., 2003) electrostatically bound with the polymeric core shell microsphere (Chiarantini et al., 2005) or loaded into the autologous red blood cells (Chiarantini et al., 1995; Turrini et al., 1991).
We have used cell-penetrating peptides (CPPs) as the vector to deliver PNA cargo into cells. Anti-HIV-1 PNAs conjugated with CPP are efficiently taken up by cells; they also effectively block HIV-1 replication and viral production by HIV-1 infected cells (Kaushik et al., 2002a; Chaubey et al., 2005; Tripathi et al., 2005). We have further shown that anti-HIV-1 PNATAR-CPP conjugates are potent virucidal agents that are able to penetrate virion particles and render them noninfectious (Chaubey et al., 2005; Tripathi et al., 2007). Unexpectedly, PNATAR-CPP conjugates are nontoxic and well tolerated at higher doses when administered in mice (Chaubey et al., 2008). With the improved solubility and cellular uptake of PNATAR-CPP conjugates, further studies pertaining to their pharmacokinetic behavior, in vivo tissue distribution, and immune response, are necessary to establish PNA as potential therapeutic agents. In the present study, we have evaluated the tissue distribution and pharmacokinetic behavior of 125I-labeled PNATAR-CPP conjugates and naked unconjugated 125I-labeled PNATAR when administered to male Balb/C mice via intraperitoneal (IP) or gavage routes.
Methods and Materials
Synthesis of PNATAR-CPP conjugates
Cys-PNATAR monomers and NPYS-Cys-derivative of Penetratin and Tat peptide (Fig. 1) were synthesized by Bio-Synthesis (Lewisville, TX, USA). We conjugated the NPYS-Cys-peptides with Cys-PNATAR using standard disulfide chemistry as described by Kaushik et al. (2002a), Koppelhus et al. (2002), and Tripathi et al. (2005). The C-terminal of PNATAR had a terminal tyrosine residue for iodination with 125I (Fig. 1). In PNATAR-O-penetratin purchased from Bio-Synthesis, penetratin peptides (43–58) were synthesized in tandem after the synthesis of PNATAR carrying ethylene glycol linker-Tyr at the N-terminus (Fig. 1).
FIG. 1.

Sequence of anti-transactivation response PNA (PNATAR) and its cell-penetrating peptide (CPP) conjugates. (1) Unconjugated 16-mer naked PNATAR; (2) PNATAR conjugated with 13-mer Tat peptide via S-S linkage; (3) PNATAR conjugated with penetratin via S-S linkage; (4) PNATAR covalently linked with penetratin via ethylene glycol (egl) linker (O-linker).
Iodination of PNATAR-CPP conjugates
We radiolabeled the PNATAR-CPP conjugates and PNATAR with 125I, using Na125I and a chloramine-T-labeling kit from ICN (Costa Mesa, California). For radiolabeling, 0.5 nmol of the conjugate and 0.28 nmol of 125I (0.5 mCi) were reacted in the presence of 2.8 nmol of chloramine-T for 1 minutes; labeling was quenched by the addition of 62 nmol of sodium metabisulphite according to manufacturer's protocol. We purified the labeled conjugate by NAP-10 gel filtration (GE Healthcare Bio-Sciences, Piscataway, NJ, USA), then by C18 reverse phase columns. The labeled conjugate was quantified by absorption at 260 nm. The specific radioactivity was adjusted to the desired concentration by adding unlabeled conjugate.
Animals
Male Balb/c mice 6–8 weeks old were purchased from Taconic Farms (Germantown, NY, USA) and quarantined for 2 weeks before the start of the experiment. The weight variation of individual mice was within ±10% of the mean body weight. The mice were fed lab-diet certified rodent diet. Food and water were available ad libitum. The mice were housed in a room with a temperature of 70 ± 3°F, relative humidity of 31–62%, and a 12-hours light/dark cycle. All treatment of mice was done according to the Guide for the Care And Use of Laboratory Animals, as per a protocol approved by the Institutional Animal Care and Use Committee of the UMDNJ. Every precaution was taken to ensure the welfare of animals at all times during the entire course of study.
Pharmacokinetic protocol
Mice were randomly assigned to eight time groups of five mice per group; each group was housed in a separate cage for pharmacokinetic studies. Each mouse was given an IP injection of 200 μL of a solution containing 36 nmol of labeled PNATAR or its conjugate derivative (1.5 × 106 CPM) in phosphate-buffered saline (PBS). Similar dose was given for oral gavage. The mice were euthanized at various times. We then collected different organs and tissues (kidney, intestine, stomach, liver, spleen, lungs, brain, heart, and skeletal muscle), rinsed them in PBS, wiped them with filter paper, and weighed them, after which the accumulated radioactivity in each sample was counted. Blood was also collected from each animal, weighed, and counted for radioactivity. The radioactivity was counted with a Packard Cobra model 5003 automatic gamma counter equipped with a 3-inch sodium iodide well crystal (125I T1/2 ∼ 60 d; energy ∼ 35 keV; overall counting efficiency ∼ 0.88).
To determine the radioactivity excreted in urine, groups of three mice were placed in a polycarbonate cage lined with filter paper (Schleicher & Schuell Bioscience, NH, USA). Animals were moved to fresh cages at 4-hours intervals. Filter papers from the emptied cage were cut into pieces, after which their radioactivity was counted. Feces were also collected and counted separately without any further treatment.
Pharmacological data analysis
The radioactivity accumulated in a mouse organ tissue and the total organ weight were used to calculate the percent of administered radioactivity per gram of tissue (%AA/g) at each time point where
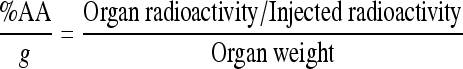 |
(1) |
The resulting data for each radiolabeled compound were least squares fitted to a two-component exponential function expressed as
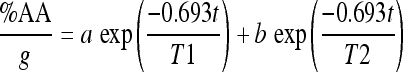 |
(2) |
where T1 and T2 are the biological half-times of the fast- and slow-clearing components, respectively. The two-component fitting procedure was performed using SigmaPlot software from SPSS (Chicago, IL, USA). The pharmacokinetic coefficients a and b, along with their associated half-time parameters T1 and T2, are given in the tables for different organs (Tables 1–4). The uncertainties associated with each parameter (Δ) in the fitting procedure are listed in the same tables. R2 is the coefficient of determination; the most common measure of how well a regression model describes the data. R2 values near 1 indicate that the equation is a good description of the relationship between the dependent and independent variables.
Table 1.
Pharmacokinetic Coefficients of Unconjugated and Conjugated PNA in Mouse Organs/Tissues after IP Administration: Accumulation of Rapid and Slow Clearance Components
| |
Accumulation of rapid clearance component a (%AA/g) |
Accumulation of slow clearance component b (%AA/g) |
||||
|---|---|---|---|---|---|---|
| Organ/Tissues | PNATAR | PNATAR-Tat | PNATAR-Penetratin | PNATAR | PNATAR-Tat | PNATAR-Penetratin |
| Kidney | 8.70 (±3.6) | 5.77 (±5.0) | 3.90 (±0.7) | 13.37 (±0.59) | 14.73 (±1.72) | 2.7 (±0.33) |
| Intestine | 18.45 (±1.6) | 4.73 (±1.2) | 12.00 (±5.0) | 0.13 (±0.01) | 0.26 (±0.19) | 0.18 (±0.07) |
| Liver | 1.16 (±0.3) | 1.99 (±0.8) | 2.40 (±0.45) | 0.24 (±0.04) | 0.46 (±0.22) | 0.32 (±0.08) |
| Blood | 2.47 (±1.2) | 3.94 (±2.4) | 4.10 (±0.95) | 0.53 (±0.30) | 0.12 (±0.01) | 0.08 (±0.10) |
| Spleen | 4.27 (±0.6) | 1.57 (±0.6) | 0.88 (±0.23) | 0.28 (±0.07) | 0.39 (±0.09) | 0.26 (±0.07) |
| Heart | 0.91 (±0.2) | 1.30 (±0.2) | 0.79 (±0.18) | 0.05 (±0.0X) | 0.16 (±0.05) | 0.07 (±0.01) |
| Brain | 0.09 (±0.03) | 0.14 (±0.05) | 0.14 (±0.10) | 0.01 (±0.0X) | 0.03 (±0.004) | 0.014 (±0.008) |
| Lungs | 3.76 (±0.7) | 3.83 (±0.30) | 0.93 (±0.28) | 0.07 (±0.0X) | 0.26 (±0.08) | 0.07 (±0.01) |
| Skeletal muscle | 0.36 (±0.1) | 0.88 (±0.30) | 2.50 (±0.30) | 0.13 (±0.01) | 0.16 (±0.04) | 0.07 (±0.02) |
A single dose of 125I-labeled PNA was IP administered to Balb/C mice as described in Materials and Methods. Mice were euthanized at different time points and the radioactive PNA compounds in different organs and tissues were monitored. Accumulation of radioactivity in individual organs or tissues was calculated as a percent of injected activity per gram tissue (%IA/g). Maximum accumulation is expressed as a percent of injected activity per gram (%IA/g). a = pharmacokinetic coefficient for maximum accumulation by the rapid-release pathway; b = pharmacokinetic coefficient for maximum accumulation by the slow-release pathway.
Table 4.
Pharmacokinetic Coefficients of Unconjugated and Conjugated PNA in Mouse Organs/Tissues after Oral Administration: Half-Times of Biphasic Clearance Pattern
| |
Half-time of rapid clearance T1 (hours) |
Half-time of slow clearance T2 (hours) |
||||
|---|---|---|---|---|---|---|
| Organ/Tissues | PNATAR | PNATAR-Penetratin (S-S) | PNATAR-Penetratin (-O-) | PNATAR | PNATAR-Penetratin (S-S) | PNATAR-Penetratin (-O-) |
| Kidney | 0.5 (±0.1) | 0.8 (±0.31) | 0.7 (±0.1) | 51.0 (±116) | 495.0 (±2581) | 330.0 (±2389) |
| Intestine | 0.5 (±0.1) | 1.5 (±0.46) | 0.7 (±0.1) | 187.0 (±334) | 169.0 (±58) | 277.0 (±255) |
| Stomach | 0.8 (±0.2) | 2.9 (±0.73) | 0.5 (±0.1) | 65.0 (±12) | 63.0 (±20) | 136.0 (±109) |
| Liver | 0.5 (±0.1) | 1.3 (±0.24) | 0.7 (±0.1) | 30.0 (±22) | 99.0 (±100) | 866.0 (±5739) |
| Spleen | 0.6 (±0.1) | 1.7 (±0.15) | 1.0 (±0.1) | X | X | 2.0 (x) |
| Blood | 0.5 (±0.1) | 1.5 (±0.19) | 0.8 (±0.1) | X | 72.0 (±73) | 27.0 (±9.0) |
| Lungs | 0.5 (±0.1) | 0.9 (±0.07) | 0.8 (±0.1) | 38.0 (±40) | 40.0 (±22) | 102.0 (±184) |
| Brain | 0.5 (±0.1) | 1.3 (±0.50) | 0.4 (±0.2) | X | 26.0 (±23) | 28.0 (x) |
| Heart | 0.6 (±0.1) | 1.5 (±0.15) | 1.1 (±0.3) | X | 23.0 (±12) | 8.1 (±3.6) |
| Sk Muscle | 0.9 (±0.1) | 2.0 (±0.20) | 1.1 (±0.1) | X | X | 24.0 (x) |
The half-time of the biphasic clearance pattern, comprised of a fast-release (T1) and slow-release component (T2). For each time point, four mice were injected. The values in italics in parenthesis are the results of uncertainties in the fitting procedure by a two-component function, as noted in the Results. The goodness of fit (R2) values ranged from 0.85 to 1. Each “X” indicates that, in the specified organs or tissues, no significant data could be obtained to determine b and T2 values for PNATAR and its CPP conjugates.
Tissue stability of PNATAR-peptide conjugate
Balb-C mice IP injected with PNATAR-S-S-penetratin in saline solution were euthanized at different time points. Kidney and liver tissues were collected, washed with saline solution and homogenized in a buffer (1.5 mL/gm tissue) consisting of 20 mM Tris, pH 7.5, 0.1M EDTA, and 0.15% NP-40. Samples were centrifuged at 14,000 rpm for 10 minutes. The supernatant was acidified to pH 4.0 with 5% TFA and centrifuged again at 14,000 rpm for 10 minutes. The clear supernatant was applied on disposable Sep-Pak reverse phase C18 cartridges (Waters, Milford, MA, USA) preequilibrated with 5% acetonitrile in 0.1% TFA. The column was washed extensively with 5% acetonitrile/0.1% TFA. The PNA bound to the column was eluted with 60% acetonitrile in 0.1% TFA at 50°C. An aliquot of each sample was analyzed by mass spectrometry using Applied Biosystems 4700 Proteomics Analyzer 7000. The remaining samples were lyophilized, dissolved in PBS, and subjected to gel-retardation assay as described in Chaubey et al. (2007).
Results
Distribution of unconjugated naked PNATAR and its CPP conjugates in different organs after administration by IP injection or gavage
We administered 125I-labeled naked PNATAR or PNATAR-CPP conjugated with either penetratin or Tat peptide to male Balb/C mice by IP injection. For gavage, we used 125I-labeled naked PNATAR and two derivatives of PNATAR-penetratin conjugates. One of these derivatives had disulfide linkage between PNATAR and the peptide; the other had covalent linkage via an o-linker (Fig. 1). The main reason for not including PNATAR-Tat conjugate in the gavage is that Lys and Arg rich Tat peptide may be susceptible to trypsin in the intestine.
We then sacrificed the mice at different time intervals and counted the radioactivity accumulated in each organ or tissue (Fig. 2). We observed that significant amounts of IP-administered PNATAR-CPP conjugates, as well as naked PNATAR, had been rapidly taken up and distributed in all mouse organs and tissues except the brain and skeletal muscle (Fig. 2A). We recorded maximum accumulation of these compounds within one hour after their administration; after that, the accumulated radioactivity declined with time. The accumulations of both naked PNATAR and PNATAR–Tat conjugate were higher in the kidneys than in other organs, while the maximum accumulation of PNATAR-penetratin conjugate occurred in the intestine, followed, in order, by the liver, blood, and spleen (Fig. 2A). Accumulations of these compounds were lowest in the brain and skeletal muscle.
FIG. 2.

Accumulation and biodistribution of naked PNATAR and its cell-penetrating peptide conjugates in mice organs and tissues: 125I-labeled unconjugated or conjugated PNAs were dissolved in phosphate-buffered saline and given, in a single dose of 200 μl, to Balb/C mice by intraperitoneal injection (A) or oral gavage (B). At different points after this administration, the mice were euthanized and their organs and tissues individually collected and their accumulated radioactivity counted with a gamma counter. After that there was a steady decline in the activity in each organ and tissue.
We noted a similar pattern when these compounds were administered orally (Fig. 2B). Surprisingly, the biodistribution of the uptake of unconjugated naked PNATAR was similar to that of PNATAR-CPP conjugates. Although cellular uptake of unconjugated naked PNATAR was negligible in a cell-culture system, its uptake, biodistribution, and retention in vivo was significant, suggesting the possible involvement of some unknown mechanism that may facilitate its uptake and biodistribution.
Clearance profile of PNATAR and its CPP conjugates after IP administration or gavage
We monitored the clearance profiles of IP and orally administered PNATAR and its CPP conjugates from the different organs and tissues as a function of time. We observed biphasic clearance of both PNATAR and its CPP conjugates in all of the organs and tissues examined. Figure 3 shows the representative clearance pattern of PNATAR-penetratin from blood. As shown in Fig. 3A, a significant portion of the IP-administered PNATAR-penetratin accumulated in the blood was cleared rapidly with a half-time (T1) of 0.64 h. The rate constant of clearance (0.693/T1) for the rapidly cleared component was 1.083 hour−1. This component is designated as “a,” which is an extrapolated value obtained from a y-intercept representing the percent of administered radioactivity (%AA) subjected to rapid clearance. The second component, designated as “b,” represents %AA of the component slowly cleared from the blood, with a half-time (T2) of 55.9 hours and a clearance rate constant (0.693/T2) of 0.0124 hour−1. We noted similar biphasic clearance profile for orally administered PNATAR-penetratin from the blood (Fig. 3B).
FIG. 3.

Metabolic clearance of PNATAR-penetratin from mouse blood. 125I-labeled PNATAR-penetratin was administered either by (A) intraperitoneal injection or (B) oral gavage. Blood samples collected from the tail vein at the indicated times were counted for accumulated radioactivity and normalized to the percent-injected activity. The values fitted well in a two-component system in the sigma plot shown. The symbols represent the average injected activity per gram (%IA/g) from 4 animals at each time point. The uncertainties in the measurements are shown by vertical lines (otherwise within the size of the symbol).
The profile of the clearance of each test compound from different mouse organs was also biphasic, albeit with variable T1 and T2 values and clearance rate constants. Results obtained with each test compound administered by the IP route or gavage are depicted, respectively, in Tables 1–4. Table 1 shows the percent of IP-administered radioactivity (%AA) of each compound subject to rapid or slow clearance. Tables 3 and 4 depict half-times of both components (T1 and T2) from the respective organs and tissues. As shown in Table 1, the percent of IP-administered naked PNATAR that accumulated in organs as rapid-clearance component “a” varied from 0.09 to 18.45, the lowest being in the brain and the highest in the intestine. PNATAR-Tat and PNATAR-penetratin displayed an approximately similar pattern, with varying degrees of accumulation as a rapid-clearance component in different organs and tissues. Slow-clearance components of all three compounds were highest in the kidney, ranging from 2.7% for PNATAR-penetratin to 13.3–14.7% for naked PNATAR and PNATAR-Tat conjugate (Table 1). In other organs and tissues, the extent of accumulation of the slow-release components was lower and significantly varied for each of the test compounds. Half-time values (T1) for PNATAR and its peptide conjugates varied marginally in different organs and tissues except the kidney, where PNATAR-Tat had the highest T1 value, 10.4 hours, albeit with very large standard deviation. PNATAR-penetratin and naked PNATAR had T1 values of 6.4 and 1.33 hours, respectively (Table 1). The half-time T2 values of the slow-clearance components of all three test compounds were significantly large (Table 2). The T2 value for PNATAR-penetratin in blood was significantly higher than those for the other two test compounds. In the spleen, the %AA and the T1 and T2 values for naked PNATAR were greater than the values for the other two test compounds. It would be interesting to determine whether any of the test compounds elicit an immunological response by examining stimulation of spleenocytes in in vitro cell culture upon in vivo exposure to these compounds.
Table 3.
Pharmacokinetic Coefficients of Unconjugated and Conjugated PNA in Mouse Organs/Tissues after Oral Administration: Accumulation of Radio and Slow Clearance Components
| |
Accumulation of rapid clearance component a (%AA/g) |
Accumulation of slow clearance component b (%AA/g) |
||||
|---|---|---|---|---|---|---|
| Organ/Tissues | PNATAR | PNATAR-Penetratin (S-S) | PNATAR-Penetratin (-O-) | PNATAR | PNATAR-Penetratin (S-S) | PNATAR-Penetratin (-O-) |
| Kidney | 8.9 (±2.2) | 2.1 (±0.77) | 5.9 (±0.4) | 0.12 (±0.05) | 0.26 (0.08) | 0.19 (±0.1) |
| Intestine | 18.4 (±1.4) | 26.0 (±6.0) | 98.0 (±3.7) | 0.03 (±0.01) | 0.05 (0.001) | 0.03 (±0.001) |
| Stomach | 58.0 (±12) | 16.0 (±2.1) | 112.0 (±7.8) | 0.07 (±0.01) | 0.12 (0.02) | 0.05 (±0.004) |
| Liver | 6.0 (±1.1) | 0.98 (±0.15) | 3.4 (±0.1) | 0.07 (±0.01) | 0.08 (0.01) | 0.03 (±0.003) |
| Spleen | 2.8 (±0.3) | 0.69 (±0.04) | 2.0 (±0.1) | x | X | 0.11 (—) |
| Blood | 11.0 (±1.1) | 1.8 (±0.18) | 7.6 (±0.4) | x | 0.04 (0.004) | 0.05 (±0.01) |
| Lungs | 8.2 (±0.8) | 48.0 (±3.7) | 3.7 (±0.2) | 0.04 (±0.02) | 0.07 (0.02) | 0.01 (±0.002) |
| Brain | 0.4 (±0.1) | 0.08 (±0.02) | 0.34 (±0.2) | x | 0.01 (0.003) | 0.05 (—) |
| Heart | 3.6 (±0.3) | 0.74 (±0.05) | 2.4 (±0.4) | x | 0.05 (0.01) | 0.04 (±0.01) |
| Skeletal Muscle | 0.8 (±0.1) | 0.37 (±0.03) | 1.1 (—) | x | X | 0.01 (—) |
A single dose of 125I labeled PNA was administered to Balb/C mice by gavage as described in the Materials and Methods. Mice were euthanized at different time points and the radioactive PNA compounds in different organs and tissues were monitored. Accumulation of radioactivity in individual organs or tissues was calculated as percent of injected activity per gram of tissue (%IA/g). Maximum accumulation is expressed as percent of injected activity per gram (%IA/g). a = pharmacokinetic coefficient for maximum accumulation by the rapid-release pathway; b = pharmacokinetic coefficient for maximum accumulation by the slow-release pathway.
Table 2.
Pharmacokinetic Coefficients of Unconjugated and Conjugated PNA in Mouse Organs/Tissues after IP Administration: Half-Time of Biphasic Clearance Pattern
| |
Half-time of rapid clearance T1 (hours) |
Half-time of slow clearance T2 (hours) |
||||
|---|---|---|---|---|---|---|
| Organ/Tissues | PNATAR | PNATAR-Tat | PNATAR-Penetratin | PNATAR | PNATAR-Tat | PNATAR-Penetratin |
| Kidney | 1.33 (±0.6) | 10.40 (±6.4) | 6.40 (±2.9) | 25.67 (±2.4) | 26.66 (±7.0) | 45 (±5.6) |
| Intestine | 0.97 (±0.1) | 0.95 (±0.2) | 0.67 (±0.2) | 93.68 (±34.2) | 77.02 (±156.6) | 44 (±31) |
| Liver | 1.06 (±0.3) | 0.97 (±0.4) | 0.98 (±0.2) | 56.82 (±22.8) | 57.29 (±38.4) | 39 (±12) |
| Blood | 0.78 (±0.3) | 0.58 (±0.3) | 0.64 (±0.1) | 24.41 (±7.4) | 35.73 (±5.7) | 56 (±19) |
| Spleen | 0.63 (±0.1) | 0.84 (±0.3) | 1.30 (±0.4) | 119.52 (±42.2) | 42.53 (±12.5) | 23 (±5.0) |
| Heart | 1.33 (±0.3) | 1.29 (±0.3) | 2.00 (±0.7) | 45.91 (±19.8) | 24.49 (±10.4) | 43 (±11) |
| Brain | 1.16 (±0.4) | 0.94 (±0.3) | 4.80 (±9.1) | 10.46 (±4.8) | 16.43 (±4.2) | 22 (±41) |
| Lungs | 0.70 (±0.1) | 0.89 (±0.1) | 3.00 (±1.3) | 59.25 (±15.7) | 31.8 (±13.7) | 110 (±84) |
| Skeletal muscle | 1.37 (±0.5) | 1.24 (±0.5) | 1.90 (±0.3) | 30.01 (±5.9) | 44.15 (±31.2) | 45 (±29) |
The half-time of the biphasic clearance pattern is comprised of a fast-release (T1) and a slow-release component (T2). For each time point, five mice were injected. The values in italics placed in parentheses are the results of uncertainties in the fitting procedure by a two-component function, as mentioned in the Results. The goodness of fit (R2) values ranged from 0.86 to 1.
The biphasic clearance data and the respective half-time values for orally administered compounds are shown, respectively, in Tables 3 and 4. The distribution of the test compounds administered via gavage displayed a very different pattern from that of IP-administered compounds (Table 3). The extrapolated value of “a” obtained from the y-intercept of the sigma plot was large in the stomach and intestine. This suggests that a significant portion of the administered compound either remained unabsorbed in the stomach or intestine and excreted via the fecal route or was absorbed and rapidly cleared. However, the first possibility must be ruled out since there was no significant difference in the fecal excretion pattern of all PNA compounds administered by either route. In this context, the slow-release component “b” is of interest. It is the component that is expected to have therapeutic value with regard to these compounds.
Stability of IP-administered PNATAR-penetratin conjugate in mouse organs
After IP injection, PNATAR conjugate was isolated from kidneys and livers collected from euthanized mice at different times. After their binding to a 32P-labeled target sequence (82 mer TAR DNA), the isolated PNATAR conjugates were subjected to mass spectroscopy and a gel-retardation assay. Mass spectroscopy of the conjugate isolated from the liver and kidney indicated several mass peaks of the conjugate due to truncation of its peptide components (Fig. 4A). Gel retardation of the 32P-labeled target sequence suggested that the PNA component of the conjugate had remained intact (Fig. 4B). PNA alone has been shown to be highly stable in tissue and cellular extracts since it is completely resistant to proteases and nucleases (Demidov et al., 1994; Jansen and Richardson 2000).
FIG. 4.

Mass spectroscopy (A) and gel retardation analysis (B) of PNATAR-S-S-penetratin isolated from mouse organs. Following intraperitoneal (IP) injection of the conjugate, mice were euthanized at different times, and the conjugate accumulated in kidney and liver tissue was isolated. Mass spectrometry analysis of the isolated compound from kidney following 6 h of IP administration (top panel A) was done on an Applied Biosystems 4700 Proteomics Analyzer 7000. For gel retardation (bottom panel B), 5′-32P-labeled 82-mer TAR-DNA (5 nM) was incubated with the isolated PNATAR conjugate in the binding buffer (50 mM Tris.HCl, pH 7.8, and 5 mM MgCl2) for 30 min at room temperature. Gel loading dye containing 0.25% bromophenol blue and 30% glycerol was added to it and loaded on a native 6% polyacrylamide gel. The gel was run for 3 h at 150V in 1X Tris-borate-EDTA buffer, dried, and subjected to Phosphor-Imager analysis (Molecular Dynamics).
Excretion of the conjugates
As described in Methods, we also measured the excretion of 125I-labeled test compounds in urine and feces. The rates of urinary excretion of both naked PNATAR and PNATAR-Tat were lower than the rate of excretion of PNATAR-penetratin conjugate (Table 5). During the first 4 h after IP administration, about 45% of PNATAR-penetratin was excreted in the urine, while only 9% of PNATAR-Tat and 10% of naked PNA were excreted; 8.6% of orally administered PNATAR-penetratin was excreted. In contrast, there was no significant difference in the fecal excretion pattern of any of the PNA compounds administered by either route (Table 6).
Table 5.
Excretion Profile of PNA Conjugates Administered by Intraperitoneal Injection
| |
Naked PNATAR |
PNATAR-Tat |
PNATAR-Penetratin (S-S) |
|||
|---|---|---|---|---|---|---|
| |
% AA excreted in |
% AA excreted in |
% AA excreted in |
|||
| åTime (hours) | Urine | Feces | Urine | Feces | Urine | Feces |
| 4 | 9.5 | 1.24 | 4.8 | 0.8 | 28.8 | 0.74 |
| 8 | 0.72 | 0.52 | 1.9 | 1.9 | 7.37 | 1.16 |
| 12 | 0.21 | 0.11 | 1.17 | 1.17 | 7.28 | 1.59 |
| 16 | 0.1 | 0.03 | 0.21 | 0.10 | 0.99 | 0.36 |
| 20 | 0.02 | 0.02 | 0.16 | 0.01 | 0.21 | 0.04 |
| 24 | 0.04 | 0.01 | 0.03 | 0.02 | 0.28 | 0.03 |
| 48 | 0.01 | 0.06 | 0.05 | 0.01 | 0.81 | 0.24 |
| 72 | 0.20 | 0.10 | 0.40 | 0.17 | 0.43 | 0.18 |
Groups of three male Balb/C mice were given 125I-labeled naked PNA or PNA conjugates and placed in separate polycarbonate cages lined with thick Whatman filter papers. Animals were transferred to fresh cages at an interval of 4 hours. The filter papers soaked in the urine were collected, cut into pieces, and counted for radioactivity. Feces were counted without further processing. Values are expressed as the percent of administered activity (%AA) excreted in urine or feces.
Table 6.
Excretion Profile of PNA Conjugates Administered by Gavage
| |
Naked PNATAR |
PNATAR-Penetratin (S-S) |
PNATAR-Penetratin (-O-) |
|||
|---|---|---|---|---|---|---|
| |
% AA excreted in |
% AA excreted in |
% AA excreted in |
|||
| Time (hours) | Urine | Feces | Urine | Feces | Urine | Feces |
| 1 | 8 | 0 | 5.7 | 0.04 | 5.03 | 0.16 |
| 2 | 0.5 | 0.1 | 2.0 | 0.39 | 4.32 | 2.84 |
| 4 | 11.8 | 19.3 | 0.1 | 2.77 | 8.92 | 2.2 |
| 8 | 0.5 | 3.5 | 13.3 | 9.55 | 10.73 | 21.8 |
| 16 | 6.4 | 1.43 | 12.0 | 10.30 | 0.5 | 1.0 |
| 32 | 4.9 | 3.9 | 12.6 | 8.30 | 2.1 | 6.3 |
| 64 | 1 | 11.1 | 5.0 | 12.30 | 5.00 | 1.1 |
Groups of three male Balb/C mice were given 125I-labeled naked PNA or PNA conjugates and placed in separate polycarbonate cages lined with thick Whatman filter papers. Animals were transferred to fresh cages at an interval of 4 hours. The filter papers soaked in the urine were collected, cut into pieces, and counted for radioactivity. Feces were counted without further processing. Values are expressed as the percent of administered activity (%AA) excreted in urine or feces.
Discussion
The potential of PNAs as antisense therapeutic agents has been recognized for quite some time. However, their solubility and cellular uptake has been a consistent impediment to their development for this purpose (Wittung et al., 1995). Several efforts to improve their in vitro cellular uptake by conjugating PNAs with various cell-permeating peptides, lipids, or antibiotics have had mixed results (Koppelhus and Nielsen, 2003). Although it has been reported that naked PNA can be taken up in vivo (McMahon et al., 2002, Pierce et al., 2005, Boffa et al., 2007), its cellular uptake in in vitro cell-culture systems has been negligible (Kaushik et al., 2002a; Chaubey et al., 2005; Tripathi et al., 2005).
Earlier, we demonstrated that PNATAR targeted to the TAR element of HIV-1 LTR efficiently prevents Tat–TAR interaction by sequestering the TAR sequence and blocks Tat-mediated transactivation of HIV-1 LTR transcription (Mayhood et al., 2000). We further demonstrated that PNATAR conjugated with CPP efficiently enters cells and blocks Tat-mediated transactivation of HIV-1 transcription and replication (Kaushik et al., 2002b, Chaubey et al., 2005, Tripathi et al., 2005). Besides antiviral activity, PNATAR-CPP conjugates also have potent anti-HIV-1 virucidal activity that, on brief exposure, renders HIV-1 virions noninfectious. The cellular uptake of naked PNAs in cell culture is negligible. However, after conjugation with CPP, they are rapidly internalized into cells and interact with their target sequences.
To evaluate in vivo uptake and distribution of PNATAR and its CPP conjugates, we introduced 125I radiolabel into the PNA moiety via Tyr residue that had been added in tandem at the N-terminal of PNA during synthesis. The penetratin or Tat peptide to which naked PNATAR was conjugated is devoid of Tyr residue, so that radio-iodination is selective to the Tyr-PNATAR moiety of the conjugate. The radiolabeled naked Tyr-PNATAR is stable when incubated with mouse blood serum or liver tissue extract for 24 hours at room temperature (unpublished results). We also evaluated the stability of the peptide moiety of the labeled conjugate, which, on cellular uptake, was either shortened or cleaved by cellular proteases (Fig. 4).
IP administration of 125I-labeled PNATAR and its CPP conjugates demonstrated that both unconjugated and conjugated PNATAR are taken up from the peritoneal cavity to different organs and tissues. However, overall accumulation and the clearance profiles of these compounds varied in different organs, probably due to differences in membrane transport systems and their cellular constitution (McMahon et al., 2002; Trehin et al., 2004). The maximum accumulation of PNA compounds occurred at all sites within the first 2–3 h after administration, irrespective of whether it was done by IP injection or oral gavage (Fig. 2). The clearance pattern of each accumulated PNATAR derivative displayed a biphasic profile consisting of rapid-clearance and slow-clearance components (Fig. 3). The biological half-lives of PNA compounds in the two phases are represented by T1 and T2 (Fig. 3). The slow-component pathway (T2) of any potential drug candidate is the desired mode of clearance because it ensures longer biological residence of the drug in the body, extending the time of its pharmacological effectiveness.
Surprisingly, the distribution, and clearance profile of naked PNA in mice after IP administration did not significantly differ from those of PNA-CPP conjugates. In an earlier study, we showed that in a cell-culture system, the cellular uptake of naked PNA is negligible (Chaubey et al., 2007). However, after the conjugation of naked PNA with CPP, cellular uptake of the PNA-CPP conjugate was rapid and homogenous (Chaubey et al., 2005, Tripathi et al., 2005, 2007). The higher T2 values observed for PNATAR-penetratin in kidney and blood suggest that after initial rapid internalization of this compound, its peptide moiety may have been cleaved, resulting in slow clearance from the cells. Similarly, higher or somewhat similar T2 values for PNATAR-Tat in liver, intestine, and kidney (Table 2)could be possible following degradation of the Tat peptide moiety of the conjugate. Tat peptide, being rich in Arg and Lys residues (Fig. 1), could be susceptible to trypsin-like proteases in mouse organs (Walsh, 1970; Zamecnik et al., 1986).
We used PNATAR-penetratin (S-S) and PNATAR- penetratin (-O-), as well as naked PNATAR, in our pharmacokinetic studies of compounds administered by the oral route. Our finding that all three compounds were significantly taken up and distributed throughout the major internal organs of mice after gavage indicates the existence of an uptake mechanism for these conjugates in the epithelial membrane of the intestine. This initial study of PNATAR conjugated to penetratin by -S-S- linker was encouraging in that all organs and tissue except the spleen and skeletal muscle displayed a slow-release pathway. However, when a stable ethylene glycol linker (-O-) was used in place of the cleavable -S-S-linker, no significant change in clearance profile was noted in most of the organs and tissues studied (Table 4).
The peptide transporter system in the epithelial or brush border membranes of the intestine that are implicated in the transport of di-, tri- or tetra- peptides may have an important function in the uptake of PNATAR-CPP conjugates administered via the oral route (Fei et al., 1994; Liang et al., 1995). This system facilitates the uptake of dietary protein by the small intestine, as well as the absorption of orally active β-lactam antibiotics and other peptide-like drugs (Liang et al., 1995). In the kidney, peptide transporters help to reabsorb filtered peptides, peptide-derived antibiotics, and peptidomimetics drugs (Liang et al., 1995; Bordin et al., 2002). It is possible that the CPP moiety of the PNA-penetratin conjugate undergoes partial proteolysis at the brush-border membrane, resulting in a PNATAR-CPP conjugate with a reduced length of CPP that may be an ideal substrate for peptide transporters. Although unconjugated naked PNATAR is taken up and distributed in different organs irrespective of the route of administration, the extent of the uptake and distribution of its CPP conjugates is higher than that of the naked PNATAR. The possible mechanism of uptake of naked PNA could be via unknown Toll-like receptors induced or present on the surface of intestinal epithelial cells (Furuka et al., 2006). It should be possible to deliver anti-HIV-1 PNAs specifically in the lymph nodes/macrophages by conjugating them with ligands that are exclusively recognized by macrophage receptors such as formyl peptide receptors, mannose receptors and Fc receptors. A natural ligand, fMLF (formyl methionyl-leucyl-phenylalanine) conjugated with PEG nanocarrier has been shown to specifically accumulate in the IP macrophages (Wan et al., 2007). This could be a potential therapeutic strategy to deliver anti-HIV-1 PNAs specifically to macrophages to achieve their effective concentrations in the cells and block HIV-1 replication and production of virion particles. We have recently shown that PNATAR-CPP conjugates are not toxic up to a single 300-mg dose/kg of body weight of mice. Repeated doses of 100 mg/kg of body weight are also well tolerated (Chaubey et al. 2008). The reduced in vivo toxicity and stability of PNATAR and its CPP conjugates, as well as their slow release and clearance from different organs, suggest the possible therapeutic potential of these compounds.
Acknowledgment
This research was supported by a grant from the NIAID/NIH (AI42520).
References
- BASU S. WICKSTROM E. Synthesis and characterization of a peptide nucleic acid conjugated to a D-peptide analog of insulin-like growth factor 1 for increased cellular uptake. Bioconjug. Chem. 1997;8:481–488. doi: 10.1021/bc9700650. [DOI] [PubMed] [Google Scholar]
- BOFFA L.C. CUTRONA G. CILLI M. MATIS S. DAMONTE G. MARIANI M.R. MILLO E. MORONI M. RONCELLA S. FEDELI F. FERRARINI M. Inhibition of Burkitt's lymphoma cells growth in SCID mice by a PNA specific for a regulatory sequence of the translocated c-myc. Cancer Gene Ther. 2007;14:220–226. doi: 10.1038/sj.cgt.7701002. [DOI] [PubMed] [Google Scholar]
- BOFFA L.C. SCARFI S. MARIANI M.R. DAMONTE G. ALLFREY V.G. BENATTI U. MORRIS P.L. Dihydrotestosterone as a selective cellular/nuclear localization vector for anti-gene peptide nucleic acid in prostatic carcinoma cells. Cancer Res. 2000;60:2258–2262. [PubMed] [Google Scholar]
- BORDIN L. BRUNATI A.M. DONELLA-DEANA A. BAGGIO B. TONINELLO A. CLARI G. Band 3 is an anchor protein and a target for SHP-2 tyrosine phosphatase in human erythrocytes. Blood. 2002;100:276–282. doi: 10.1182/blood.v100.1.276. [DOI] [PubMed] [Google Scholar]
- CHAUBEY B. TRIPATHI S. DÉSIRÉ J. BAUSSANNE I. DDÉCOUT J. PANDEY V.N. Mechanism of RNA cleavage catalyzed by sequence specific polyamide nucleic acidneamine conjugate. Oligonucleotides. 2007;17:302–313. doi: 10.1089/oli.2007.0085. [DOI] [PubMed] [Google Scholar]
- CHAUBEY B. TRIPATHI S. GANGULY S. HARRIS D. CASALE R.A. PANDEY V. N. A PNA-transportan conjugate targeted to the TAR region of the HIV-1 genome exhibits both antiviral and virucidal properties. Virology. 2005;331:418–428. doi: 10.1016/j.virol.2004.10.032. [DOI] [PubMed] [Google Scholar]
- CHAUBEY B. TRIPATHI S. PANDEY V.N. Single acute-dose and repeat-doses toxicity of anti HIV-1 PNATAR–Penetratin Conjugate after Intraperitoneal Administration to Mice. Oligonucleotides. 2008;18:9–20. doi: 10.1089/oli.2007.0088. [DOI] [PubMed] [Google Scholar]
- CHIARANTINI L. CERASI A. FRATERNALE A. MILLO E. BENATTI U. SPARNACCI K. LAUS M. BALLESTRI M. TONDELLI L. Comparison of novel delivery systems for antisense peptide nucleic acids. J. Control. Release. 2005;109:24–36. doi: 10.1016/j.jconrel.2005.09.013. [DOI] [PubMed] [Google Scholar]
- CHIARANTINI L. ROSSI L. FRATERNALE A. MAGNANI M. Modulated red blood cell survival by membrane protein clustering. Mol. Cell Biochem. 1995;144:53–59. doi: 10.1007/BF00926740. [DOI] [PubMed] [Google Scholar]
- CUTRONA G. CARPANETO E.M. ULIVI M. RONCELLA S. LANDT O. FERRARINI M. BOFFA L.C. Effects in live cells of a c-myc anti-gene PNA linked to a nuclear localization signal. Nat. Biotechnol. 2000;18:300–303. doi: 10.1038/73745. [DOI] [PubMed] [Google Scholar]
- DEMIDOV V.V. POTAMAN V.N. FRANK-KAMENETSKIL M.D. EGHLOM M. BUCHARD O. SONNICHSEN S.H. NIELSEN P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994;48:1310–1313. doi: 10.1016/0006-2952(94)90171-6. [DOI] [PubMed] [Google Scholar]
- FEI Y.J. KANAI Y. NUSSBERGER S. GANAPATHY V. LEIBACH F.H. ROMERO M.F. SINGH S.K. BORON W.F. HEDIGER M.A. Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature. 1994;368:563–566. doi: 10.1038/368563a0. [DOI] [PubMed] [Google Scholar]
- FURUTA T. KIKUCHI T. AKIRA S. WATANABE N. YOSHIKAWA Y. Roles of the small intestine for induction of toll-like receptor 4-mediated innate resistance in naturally acquired murine toxoplasmosis. Int. Immunol. 2006;18:1655–1662. doi: 10.1093/intimm/dxl099. [DOI] [PubMed] [Google Scholar]
- JANSEN K. RICHELSON E. Detection of peptide nucleic acids in tissue extracts of treated animal by gel mobility shift assay. J. Biochem. Bioph. Meth. 2000;42:31–34. doi: 10.1016/s0165-022x(99)00046-9. [DOI] [PubMed] [Google Scholar]
- KAUSHIK N. BASU A. PALUMBO P. MYERS R.L. PANDEY V.N. Anti-TAR polyamide nucleotide analog conjugated with a membrane-permeating peptide inhibits human immunodeficiency virus type 1 production. J. Virol. 2002a;76:3881–3891. doi: 10.1128/JVI.76.8.3881-3891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAUSHIK N. BASU A. PANDEY V.N. Inhibition of HIV-1 replication by anti-trans-activation responsive polyamide nucleotide analog. Antiviral Res. 2002b;56:13–27. doi: 10.1016/s0166-3542(02)00024-4. [DOI] [PubMed] [Google Scholar]
- KAUSHIK N. PANDEY V.N. PNA targeting the PBS and A-loop sequences of HIV-1 genome destabilizes packaged tRNA3 (Lys) in the virions and inhibits HIV-1 replication. Virology. 2002;303:297–308. doi: 10.1006/viro.2002.1630. [DOI] [PubMed] [Google Scholar]
- KOPPELHUS U. AWASTHI S.K. ZACHAR V. HOLST H.U. EBBESEN P. NIELSEN P.E. Cell-dependent differential cellular uptake of PNA, peptides, and PNA-peptide conjugates. Antisense Nucleic Acid Drug Dev. 2002;12:51–63. doi: 10.1089/108729002760070795. [DOI] [PubMed] [Google Scholar]
- KOPPELHUS U. NIELSEN P.E. Cellular delivery of peptide nucleic acid (PNA) Advanced Drug Deliver. Rev. 2003;55:267–280. doi: 10.1016/s0169-409x(02)00182-5. [DOI] [PubMed] [Google Scholar]
- LEE R. KAUSHIK N. MODAK M.J. VINAYAK R. PANDEY V.N. Polyamide nucleic acid targeted to the primer binding site of the HIV-1 RNA genome blocks in vitro HIV-1 reverse transcription. Biochemistry. 1998;37:900–910. doi: 10.1021/bi972197m. [DOI] [PubMed] [Google Scholar]
- LIANG R. FEI Y.J. PRASAD P.D. RAMAMOORTHY S. HAN H. YANG-FENG T.L. HEDIGER M.A. GANAPATHY V. LEIBACH F.H. Human intestinal H+/peptide cotransporter. Cloning, functional expression, and chromosomal localization. J. Biol. Chem. 1995;270:6456–6463. doi: 10.1074/jbc.270.12.6456. [DOI] [PubMed] [Google Scholar]
- LJUNGSTROM T. KNUDSEN H. NIELSEN P.E. Cellular uptake of adamantyl conjugated peptide nucleic acids. Bioconjug. Chem. 1999;10:965–972. doi: 10.1021/bc990053+. [DOI] [PubMed] [Google Scholar]
- MAYHOOD T. KAUSHIK N. PANDEY P.K. KASHANCHI F. DENG L. PANDEY V.N. Inhibition of Tat-mediated transactivation of HIV-1 LTR transcription by polyamide nucleic acid targeted to TAR hairpin element. Biochemistry. 2000;39:11532–11539. doi: 10.1021/bi000708q. [DOI] [PubMed] [Google Scholar]
- MCMAHON B.M. MAYS D. LIPSKY J. STEWART J.A. FAUQ A. RICHELSON E. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev. 2002;12:65–70. doi: 10.1089/108729002760070803. [DOI] [PubMed] [Google Scholar]
- MURATOVSKA A. LIGHTOWLERS R.N. TAYLOR R.W. TURNBULL D.M. SMITH R.A. WILCE J.A. MARTIN S.W. MURPHY M.P. Targeting peptide nucleic acid (PNA) oligomers to mitochondria within cells by conjugation to lipophilic cations: implications for mitochondrial DNA replication, expression and disease. Nucleic Acids Res. 2001;29:1852–1863. doi: 10.1093/nar/29.9.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIELSEN P.E. EGHOLM M. BERG R.H. BUCHARDT O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- PIERCE T.L. WHITE A.R. TREGEAR G.W. SEXTON P.M. Peptide-oligonucleotide hybrids in antisense therapy. Mini. Rev. Med. Chem. 2005;5:41–55. doi: 10.2174/1389557053402846. [DOI] [PubMed] [Google Scholar]
- RIGUET E. TRIPATHI S. CHAUBEY B. DESIRE J. PANDEY V.N. DECOUT J.L. A peptide nucleic acid-neamine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J. Med. Chem. 2004;47:4806–4809. doi: 10.1021/jm049642d. [DOI] [PubMed] [Google Scholar]
- TREHIN R. NIELSEN H.M. JAHNKE H.G. KRAUSS U. BECKSICKINGER A.G. MERKLE H.P. Metabolic cleavage of cell-penetrating peptides in contact with epithelial models: human calcitonin (hCT)-derived peptides, Tat (47–57) and penetratin (43–58) Biochem J. 2004;382(Pt 3):945–956. doi: 10.1042/BJ20040238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIPATHI S. CHAUBEY B. BARTON B.E. PANDEY V.N. Anti HIV-1 virucidal activity of polyamide nucleic acid-membrane transducing peptide conjugates targeted to primer binding site of HIV-1 genome. Virology. 2007;363:91–103. doi: 10.1016/j.virol.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIPATHI S. CHAUBEY B. GANGULY S. HARRIS D. CASALE R.A. PANDEY V.N. Anti-HIV-1 activity of anti-TAR polyamide nucleic acid conjugated with various membrane transducing peptides. Nucleic Acids Res. 2005;33:4345–4356. doi: 10.1093/nar/gki743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURRINI F. ARESE P. YUAN J. LOW P.S. Clustering of integral membrane proteins of the human erythrocyte membrane stimulates autologous IgG binding, complement deposition, and phagocytosis. J. Biol. Chem. 1991;266:23611–23617. [PubMed] [Google Scholar]
- UHLMANN E. Peptide nucleic acids (PNA) and PNA-DNA chimeras: from high binding affinity towards biological function. Biol. Chem. 1998;379:1045–1052. [PubMed] [Google Scholar]
- WALSH K. Trypsinogens and trypsins of various species. In: Perlman G.A.L., editor. Methods in Enzymology. Academic Press; New York: 1970. 19 pp. [Google Scholar]
- WAN L. POOYAN S. HU P.J. LEIBOWITZ M.J. STEIN S. SINKO P.J. Peritoneal macrophage uptake, pharmacokinetics and biodistribution of macrophage-targeted PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarriers for improving HIV drug delivery. Pharmaceut. Res. 2007;24:2110–2119. doi: 10.1007/s11095-007-9402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WITTUNG P. KAJANUS J. EDWARDS K. NIELSEN P. NORDEN B. MALMSTORM B.G. Phospholipid membrane permeability of peptide nucleic acid. FEBS Lett. 1995;365:27–29. doi: 10.1016/0014-5793(95)00409-3. [DOI] [PubMed] [Google Scholar]
- ZAMECNIK P.C. GOODCHILD J. TAGUCHI Y. SARIN P.S. Inhibition of replication and expression of human T-cell lymphotropic virus type III in cultured cells by exogenous synthetic oligonucleotides complementary to viral RNA. Proc. Natl. Acad. Sci. USA. 1986;83:4143–4146. doi: 10.1073/pnas.83.12.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X. SIMMONS C.G. COREY D.R. Liver cell specific targeting of peptide nucleic acid oligomers. Bioorg. Med. Chem. Lett. 2001;11:1269–1272. doi: 10.1016/s0960-894x(01)00198-6. [DOI] [PubMed] [Google Scholar]
- ZHOU P. WANG M. DU L. FISHER G.W. WAGGONER A. LY D.H. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA) J. Am. Chem. Soc. 2003;125:6878–6879. doi: 10.1021/ja029665m. [DOI] [PubMed] [Google Scholar]