


Abstract
Linkage and association analyses were performed to identify loci affecting disease susceptibility by scoring previously characterized sequence variations such as microsatellites and single nucleotide polymorphisms. Lack of markers in regions of interest, as well as difficulty in adapting various methods to high-throughput settings, often limits the effectiveness of the analyses. We have adapted the Escherichia coli mismatch detection system, employing the factors MutS, MutL and MutH, for use in PCR-based, automated, high-throughput genotyping and mutation detection of genomic DNA. Optimal sensitivity and signal-to-noise ratios were obtained in a straightforward fashion because the detection reaction proved to be principally dependent upon monovalent cation concentration and MutL concentration. Quantitative relationships of the optimal values of these parameters with length of the DNA test fragment were demonstrated, in support of the translocation model for the mechanism of action of these enzymes, rather than the molecular switch model. Thus, rapid, sequence-independent optimization was possible for each new genomic target region. Other factors potentially limiting the flexibility of mismatch scanning, such as positioning of dam recognition sites within the target fragment, have also been investigated. We developed several strategies, which can be easily adapted to automation, for limiting the analysis to intersample heteroduplexes. Thus, the principal barriers to the use of this methodology, which we have designated PCR candidate region mismatch scanning, in cost-effective, high-throughput settings have been removed.
INTRODUCTION
DNA sequence variation is both a source of human disease and a means by which disease mechanisms may be elucidated. Linkage analysis (which compares variation among affected relatives) and association tests (which compare variation among affected individuals and controls) are the two major approaches to identifying genes and chromosomal regions affecting human disease susceptibility. Each of these approaches primarily relies on scoring DNA sequence variation in the form of short tandem repeat polymorphisms (primarily microsatellites) or single nucleotide polymorphisms (SNPs). As human genetic research progresses towards a more comprehensive analysis of complex genetic disorders, the number (density) of such markers and the effectiveness with which they are scored in individuals must increase dramatically.
Linkage analysis of adult genetic disorders by genotyping microsatellites often suffers from incomplete information, requiring identity-by-state rather than identity-by-descent (ibd) analysis. While this may be largely overcome by using more markers in the regions of interest, this reduces the efficiency, especially if the analysis mandates examination of particular candidate gene regions for which marker occurrence is infrequent and/or uninformative. The need for manual interpretation and human error checking of genotyping data is also time consuming, affecting the throughput considerably. A more recent technical approach has been the typing of multiple SNPs. However, the strategies now employed using these markers require an exact knowledge of SNP sequence attributes and location. Since the number of SNPs required for proposed susceptibility studies may be quite large (1,2), the typing of these markers for a sufficiently robust analysis by currently available methods is expensive and often beyond the reach of the typical academic laboratory. Currently available commercial software packages and substantial literature on the subject provide only partial solutions to overcoming the problems inherent in conventional genotyping methodologies.
Within the past few years various techniques have been developed to detect or score sequence variation, particularly in PCR products. These methods can be divided into two categories: (i) those detecting unknown sequence variants, including chemical mismatch cleavage (CMC; 3–5), denaturing gradient gel electrophoresis (DGGE; 6), single-stranded conformation polymorphism (SSCP; 7), detection of virtually all mutations–SSCP (DOVAM-S; 8) and others; (ii) those scoring known sequence variants, such as TaqMan (9), molecular beacon hybridization (10), Invader (11), allele-specific PCR (12), degenerate oligonucleotide primed PCR (DOC–PCR) (13), mutation detection by electrocatalysis at DNA-modified electrodes (14), arrayed primer extension (APEX) (15) and chip-based genotyping by mass spectrometry (16) (for reviews see 17–20). None of these strategies performs equally well for scoring genotypes and for mutation detection. To perform both tasks a technique should be sensitive enough to detect virtually all mutation types and quantitative enough that data may be translated into allele-sharing status. Moreover, for high-throughput purposes the method should be easy to optimize for automated typing of many loci.
Genomic mismatch scanning (GMS) is a hybridization-based technique designed to enrich ibd regions between two individuals without the need for genotyping or sequencing (21). In other words, genetic variation may be exploited without the effort and expense of characterizing it carefully. Regions of ibd, once selected by GMS, can then be used for mapping by hybridization to a microarray containing ordered clones of genomic DNA (21–26). GMS employs the Escherichia coli mismatch repair enzymes mutH, mutL and mutS (27) to identify DNA regions that contain mismatches in DNA fragments from different sources (cases, relatives, controls, etc.). MutS has increased binding affinity for single base mismatches and 1–4 nt insertions or deletions (28). Only C-C mismatches are weakly recognized. Following MutS binding to heteroduplex DNA, MutL is recruited and activated. In the presence of ATP the complex then binds and activates MutH, a latent endonuclease that cleaves DNA 5′ to a nearby d(GATC) site (29). The mismatch and cleavage sites may be separated by as much as 1 kb (30,31).
We are currently conducting a study of genetic susceptibility to cancer in affected sibling pairs and have decided to investigate the possibility of replacing microsatellite genotyping with a method based on quantitative mismatch detection using the MutHLS enzymes. Two previous studies have shown that mismatch scanning using bacterial Mut enzymes could be used for mutation detection on PCR products (32,33), opening the way to performing genotyping on PCR-amplified candidate regions. However, strategies and experimental conditions that are critical to performing quantitative genotyping have not yet been elaborated.
Using an adaptation of the Taguchi method (34) we have conducted a comprehensive biochemical optimization of the technique. We have quantified the effects of such factors as choice of polymerase, monovalent cation concentration, ADP/ATP ratio and position of the MutH recognition signal for different target DNAs ranging from 260 to 1250 bp. We found that the optimal experimental conditions for a given target region are independent of sequence environment and depend mostly on the PCR product size. Direct correlations of the KCl and MutL concentrations with the length of the target DNA were observed. Therefore, near optimal assay conditions for a new target region could be predicted solely on the basis of the PCR product size. Our modifications, which we now collectively designate PCR candidate region mismatch scanning (PCR–CRMS), have simplified the mismatch scanning assay, rendered it quantitative and demonstrated its potential for cost-effective, high-throughput genotyping.
MATERIALS AND METHODS
Materials
His-Bind Quick Columns were purchased from Novagen (Madison, WI). The Centriplus concentrators were from Amicon (Beverly, MA). The NAP-25 columns were from Pharmacia. The QIAquick PCR Purification Kit was from Qiagen (Valencia, CA). Dynabeads M-280 Streptavidin was purchased from Dynal AS (Oslo, Norway). AmpliTaq Gold was purchased from Roche Pharmaceuticals. The Expand High Fidelity PCR System, ATP (lithium salt), ADP and IPTG were obtained from Boehringer Mannheim. GeneScan-500 (TAMRA) size standards were purchased from PE Applied Biosystems. The CDKN1A PAC clone 431A14 was obtained from the Roswell Park Cancer Institute (Buffalo, NY).
Expression of the His6-MutHLS proteins
We used a modification of the procedure provided by Feng and Winkler (35). Briefly, a fresh colony of each strain (TX3149, TX3150 or TX3151) was used to inoculate 40 ml of LB medium containing 50 µg/ml carbenicillin. The culture was incubated at 37°C and shaken at 250 r.p.m. for 6–8 h to mid-exponential phase (OD660 nm of the 0.5–0.6). The cells were then harvested by centrifugation at 5000 g for 10 min and the pellets were resuspended in 4 ml of LB medium for overnight storage at 4°C. The next morning 2 × 2.5 l flasks, containing 500 ml of LB medium with 10 µg/ml carbenicillin, were inoculated using 2 ml of the cell preparation from the previous evening. The cell suspension was incubated at 37°C with rotary shaking (250 r.p.m.) until the OD660 nm reached 0.5–0.6 (4–6 h). Then 120 mg IPTG (1 mM final) was added and the cells were allowed to overexpress the recombinant proteins for 3 h. The culture was then chilled on ice for 5 min and centrifuged at 5000 g for 20 min at 4°C. The pellets were pooled and washed twice with 50 ml of ice-cold water following a final centrifugation at 3000 g for 10 min at 4°C. The pellet was then stored at –70°C until the protein purification step.
Large-scale one-step purification of the His6-MutHLS proteins
All steps were carried out at 4°C or on ice. The frozen cell extract was thawed on ice using 60 ml of 1× binding buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl, pH 7.9) supplemented with the protease inhibitors PMSF (1 mM), antipain (50 µg/ml), benzamidine (1 mM), leupeptin (2.5 µg/ml) and pepstatin A (2.5 µg/ml). Cells were disrupted using an ultrasound sonicator (4 × 20 s) at 40% power level and 50% pulse (Sonifier 450; Branson). The cell lysate was then centrifuged at 35 000 g for 20 min at 4°C. The cell extract supernatants were filtered using a 60 ml syringe and a 0.45 µm filter. The filtrates for MutS and MutL (60 ml) were then loaded onto two pre-equilibrated His-Bind Quick Columns (30 ml filtrate/column) following the manufacturer’s instructions. For MutH, four columns were loaded using 15 ml filtrate/column. The columns were then equilibrated using 30 ml of 1× binding buffer and washed once with 50 ml of wash buffer (60 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl, pH 7.9) mixed with binding buffer (1:1) and further washed using 13 ml of a solution of wash buffer with binding buffer (3:1). The His-tagged proteins were then eluted twice with 7 ml of elution buffer (300 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl, pH 7.9). The eluted fractions were concentrated for 30–60 min using Centriplus YM-50 concentrators. The buffer was then changed, using a NAP-25 column (Pharmacia) with buffer A (20 mM Tris–HCl, pH 8, 1 mM EDTA, 1 mM DTT, 200 mM KCl and 20% glycerol). To the eluted protein solutions (3.75 ml) was added 2.5 ml of buffer B containing 94% glycerol instead of 20%. The final buffer composition of the protein samples was 50% glycerol, 20 mM Tris–HCl, pH 8, 1 mM EDTA, 1 mM DTT and 200 mM KCl. The enzyme preparations were then aliquoted and stored at –70°C.
Amplification of target DNA
Target DNAs as well as the reference DNA were PCR amplified, the latter using a FAM-labeled forward primer and a biotin-labeled reverse primer. The locus chosen as the target to optimize the method included part of intron 2, exon 3 and the proximal 3′-UTR of the human CDKN1A gene (Fig. 1). This region was selected for the presence of known RFLPs (36; G.Larson, L.Geller, S.D.Flanagan, G.Zhang, J.Longmate and T.G.Krontiris, manuscript in preparation) and the availability of several previously genotyped human genomic DNA samples. The forward primer sequence used was 5′-TCCTCAGTTGGGCAGCTCCG-3′. The reverse primer sequence was: for the 260 bp target, 5′-GCCAGGGTATGTACATGAGGAG-3′; for the 516 bp target, 5′-CGCCTGTGACAGCGATGG-3′; for the 969 bp target, 5′-GCTGAGAGGGTACTGAAGGGA-3′. For amplification of the target with a GATC site 64 bp from the 5′-end of the fragment the forward primer sequence was 5′-TCTTCTTGGCCTGGCTGAC-3′. The 260 bp PCR amplification was performed in a total volume of 20 µl, using 200 µM dNTPs, 250 nM each primer, 1.5 mM MgCl2 and 25 ng DNA. Either AmpliTaq Gold (PE Applied Biosystems) or the Expand High Fidelity enzyme preparation (Roche Pharmaceuticals) was used in the buffers provided by the vendor. PCR reactions were carried out with a first cycle of 96°C for 2 min, 60°C for 45 s and 72°C for 45 s and 26–29 further cycles of 94°C for 30 s, 60°C for 45 s and 72°C for 45 s, with a 3 min final extension. For the 516 and 969 bp amplicons a final MgCl2 concentration of 2.1 mM was used along with the Expand High Fidelity PCR System. The 516 bp product was amplified with a first cycle of 96°C for 2 min and 68°C for 1 min and 26–29 further cycles at 94°C for 30 s and 68°C for 1 min. The 969 bp amplicon was obtained using the previous PCR conditions with an annealing/extension time of 80 s.
Figure 1.
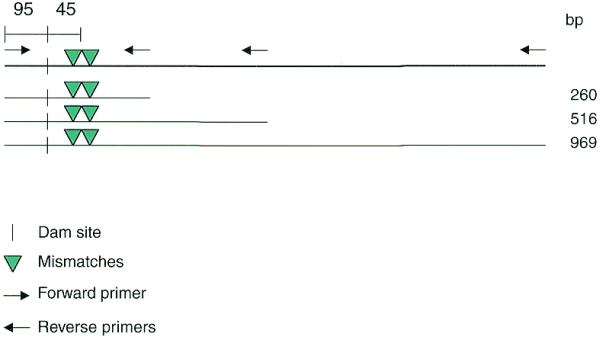
Schematic representation of target PCR products used to optimize PCR–CRMS. PCR primers were designed to amplify DNA fragments from exon 3 of the human CDKN1A gene. Known polymorphisms are designated by green arrowheads. All the targets were PCR amplified using the same forward primer (→). Three different lengths of target DNA, 260, 516 and 969 bp, were amplified using specific reverse primers (←). Therefore, all targets carry the same mutations, along with the same dam reporter site (GATC, vertical bars). The dam sites were 95 bp from the end of each amplicon and 45 bp away from the first mismatch. The two polymorphic sites were 13 bp apart.
The promoter–5′-UTR region of CDKN1A was amplified using the forward primer sequence 5′-CTGCTCCACCGCACTCTGG-3′ and the reverse primer 5′-TCCGCTCCCATCTACCTCAC-3′. Amplification was performed using the Expand High Fidelity enzyme preparation along with the buffer supplied by the manufacturer. The cycling conditions were one cycle at 96°C for 2 min and 68°C for 100 s followed by 29 further cycles at 94°C for 30 s and 68°C for 100 s, with a 3 min final extension at 68°C.
Three different regions of the androgen receptor locus, varying in length and nucleotide sequence, were amplified as PCR–CRMS targets from a single male DNA sample: exon 5 (285 bp), the 5′-UTR (537 bp) and exon 1 (956 bp). The forward primers were, respectively, 5′-CAACCCGTCAGTACCCAGACTGACC-3′, 5′-AAGGCAGTCAGGTCTTCAGTAGC-3′ and 5′-CACTTGCATCTGCCACCTTTAC-3′; the reverse primers were 5′-AGCTTCACTGTCACCCCATCACCATC-3′, 5′-CACTTCGCGCACGCTCTG-3′ and 5′-GGAGGTGGAGAGCAAATGCA-3′. Amplification was performed using the Expand High Fidelity enzyme preparation along with the buffer supplied by the manufacturer, supplemented with 500 ng/µl BSA. The cycling conditions were as follows: an initial cycle of 96°C for 2 min followed by 45 s at the annealing temperature of 62 (285 and 537 bp targets) or 60°C (956 bp targets) and 72°C for 40 (285 bp), 45 (537 bp) or 90 s (956 bp). Thirty-one cycles followed at 94°C for 30 s, at the annealing temperature above for 45 (285 and 537 bp) or 60 s (956 bp) and at 72°C for 40 (285 bp), 45 (537 bp) or 90 s (956 bp). All the reactions were ended with a 3 min final extension at 72°C.
PCR primers for investigating the effects of GATC position on PCR–CRMS efficiency
The reverse primer used was the same as for the 260 bp target DNA amplification. For amplification of targets with a GATC site 64, 45, 30 and 15 bp from the end of the fragment the forward primer sequences were, 5′-TCTTCTTGGCCTGGCTGAC-3′, 5′-TTCTGCTGTCTCTCCT CAGATTTC-3′, 5′-TCAGATTTCTACCACTCCAAACG-3′ and 5′-TCCAAACGCCGGCT GACT-3′, respectively.
Single-strand fluorescent DNA (ssf) probe preparation
For the purposes of heteroduplex selection and detection of homozygote variations we purified a ssf probe. This was achieved using high affinity binding of the unwanted biotin-containing DNA strand to streptavidin beads. An aliquot of 40 µl of the PCR product harboring a FAM molecule at the 5′-end of one strand and a biotin molecule at the 5′-end of the complementary strand was first purified from salts and residual primers using a QIAquick PCR purification kit using the manufacturer’s protocol. DNA was eluted in 35 µl of elution buffer (EB) (10 mM Tris–HCl, pH 8.5). The ssf probe was purified using Dynabeads M-280 Streptavidin. Briefly, the paramagnetic beads (17.5 µl) were equilibrated in 17.5 µl of 2× binding buffer (BB) (10 mM Tris–HCl, pH 7.8, 1 mM EDTA, 2 M NaCl). The beads, resuspended in 35 µl of BB, were gently mixed with an equal volume of DNA at room temperature for 15 min. Following paramagnetic separation, the beads were washed using 35 µl of BB and resuspended in 15 µl of freshly prepared 0.1 M NaOH for 10 min. The solution was then magnetically separated from the beads and transferred into a second tube containing 7.5 µl of 0.2 M HCl. Then 1.88 µl of 1 M Tris–HCl, pH 8, was quickly added to the ssf probe solution. Finally, the ssf probe was further purified using a QIAquick PCR purification kit and eluted with 25 µl of EB.
PCR–CRMS assay
All assays were carried out in 0.2 ml thin-walled test tubes. Solutions (10 µl) containing 250 fmol DNA corresponding to 3 µl of PCR product were first heat denatured and reannealed, in PCR–CRMS buffer, then preincubated at 37°C until enzyme addition. The 10× PCR–CRMS buffer contained 200 mM Tris–HCl, pH 8, 100 µM EDTA, 7 mM DTT, 60 mM MgCl2, 1 mg/ml BSA, 100 µM ADP and 50–500 mM KCl. When the 260 bp product was used as target, 5% DMSO was added to the solution. The samples were heat denatured and reannealed, using a Robocycler (Stratagene), at 99°C for 10 min, immediately followed by a 15 min incubation at 60°C. The tubes were preincubated at 37°C for 5–10 min. An aliquot of 1–1.5 µl of the purified His6-MutS enzyme (260–390 ng, 275–410 nM) was first added for higher specificity (without ATP) for 20 min at 37°C. The endonuclease reaction was initiated by adding a cocktail of 1 µl of His6-MutH (25 ng, 85 nM), 1–1.5 µl of His6-MutL (120–180 ng, 180–270 nM) and 0.15 µl of 100 mM ATP (final concentration 1.5 mM). The incubation was continued for 20 min at 37°C. The final KCl concentration varied from 60 to 110 mM.
The self-reannealed reactions (mutation detection mode) were terminated using 10 µl of deionized formamide containing 25 mM EDTA and 0.05% bromophenol blue, then kept on ice until loaded on an 8 M urea–polyacrylamide gel. Following electrophoresis the gel was stained with Vistra Green (Amersham Life Science) and scanned using a Fluorimager SI scanner (Vistra Fluorescence). Fractions cleaved were quantified using ImageQuaNT software.
The fluorescence-based typing reactions (genotyping mode) were treated as above except that 10–25 fmol ssf probe was added to the solution prior to the denaturation/reannealing step. Reactions were stopped using 0.5 µl of 0.5 M EDTA, followed by a 30 min evaporation under low atmosphere. The resulting 2 µl solution was electrophoresed on a glycerol-tolerant 6% polyacrylamide–8 M urea gel containing 100 mM Tris–HCl, 28.75 mM taurine and 500 µM EDTA at 45°C, 2500 V for 5 h on the ABI Prism 377 DNA Sequencer. The fraction digested was then quantitated from GeneScan electropherograms. For analysis of the 5′-UTR and part of the promoter region of the CDKN1A gene a native gel system was used for better resolution of large DNA fragments (1257 bp). The gel solution was the same as above, but lacking urea. Electrophoresis was for 12 h at 1000 V and 30°C.
Optimization of PCR–CRMS using the Taguchi method
A modified Taguchi method (34; P.Lundberg and M.Beaulieu, manuscript in preparation) was used to determine the optimal experimental conditions necessary to perform PCR–CRMS on different target DNAs. We used arrays of four parameter by three parameter values to measure the effects and interactions of specific reaction components simultaneously. The end point for PCR–CRMS optimization purposes was the highest ratio between the fraction of endonuclease activity obtained from heterozygote and homozygote DNA samples. The optimal level for each component, by Taguchi analysis, was finally assayed, along with two or three slightly different conditions, to confirm the values as the most appropriate experimental conditions.
RESULTS
PCR–CRMS using self-reannealed PCR products (mutation detection mode)
To test the feasibility of employing mismatch scanning for high-throughput genotyping we first chose the terminal exon of CDKN1A for the initial analysis (Fig. 1) because of the occurrence of suitable polymorphisms and dam sites. Following PCR amplification of test DNAs we denatured and reannealed aliquots of the products individually and then added the MutH, MutL and MutS enzymes. The fraction of reannealed PCR product cleaved by MutH was obtained from a quantitative fluorescence scan of the polyacrylamide gel. Figure 2 shows a typical PCR–CRMS assay (mutation detection mode) performed on 260 bp CDKN1A amplicons. Using a modified Taguchi protocol (see Materials and Methods) our goal was at least a 3:1 ratio of heterozygote to homozygote (background) cleavage and a heterozygote cleavage as close to the theoretical 50% as possible. In the example shown only 5% of the homozygous sample was cleaved, while the heterozygous sample was cleaved at the 50% level expected. Thus, the signal-to-noise ratio was suitably high and heterozygote recognition/cleavage excellent. The conditions required for this performance are listed in Table 1. Optimal specificity required preincubation of MutS and target DNA with ADP as well as addition of DMSO to the reaction.
Figure 2.
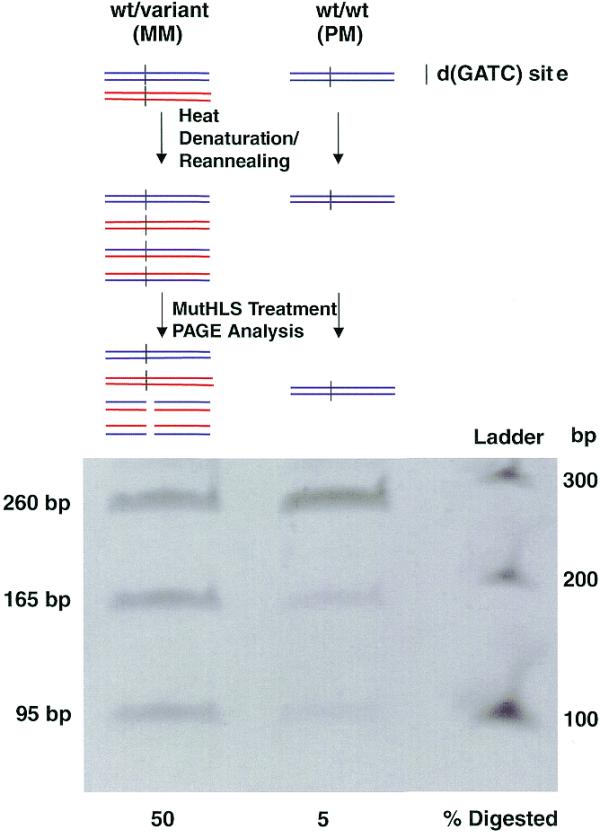
PCR–CRMS assay with self-annealed PCR products (mutation detection mode). A PCR product, either heterozygous (left, top) or homozygous (right, top), is heat denatured and reannealed to itself. The heterozygous sample is expected to generate equal amounts of homoduplex (PM) and heteroduplex (MM). However, the homozygous sample generates only one homoduplex (PM) molecule. MM duplexes are specific targets for activated MutH. Following MutHLS treatment 50% of the heterozygous sample was digested; only a 5% background level of cleavage was observed with the homozygous sample.
Table 1. Optimized parameters for different target DNA sizes.
| Target DNA (bp) | 260 | 516 | 969 | 1257, 5′ UTR | ||||
| Analysis mode |
Mutation detection |
Genotyping |
Mutation detection |
Genotyping |
Mutation detection |
Genotyping |
Mutation detection |
Genotyping |
| KCl (mM) | 85 | 60 | 95 | 75 | 110 | 95 | 122 | 102 |
| DMSO (%) | 5 | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| ADP (µM) | 100 | 10 | 0 | 0 | 0 | 0 | 0 | 0 |
| MutL (nM) | 180 | 180 | 225 | 225 | 270 | 270 | 288 | 288 |
Fixed concentration of target DNA (25 nM), MutH (85 nM), MutS (275 nM), BSA (100 µg/ml), MgCl2 (6 mM), DTT (1 mM) Tris buffer (20 mM), EDTA (0.4 mM), ATP (1.5 mM) and Expand buffer (0.3×) were used. Mutation detection, mutation detection mode. Genotyping, genotyping mode.
When PCR–CRMS was first attempted with 516 and 969 bp PCR products at the same locus (Fig. 1) the experimental conditions optimal for the 260 bp target appeared to be inappropriate for the longer targets; it became necessary to re-optimize the assay for each target. The Taguchi method was again successfully applied to estimate the effects of individual components (Table 1 and Fig. 3). Interestingly, the larger targets did not require DMSO or preincubation with ADP. Moreover, it was apparent that the salt concentration was a key determinant in obtaining specificity. For example, the heterozygote:homozygote cleavage ratio obtained using the 969 bp target increased nearly 3-fold on simply increasing the KCl concentration by 20 mM (Fig. 3B).
Figure 3.
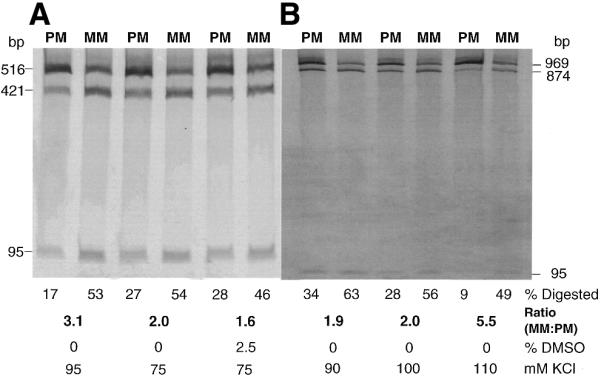
Optimization of PCR–CRMS with longer targets amplified from the CDKN1A locus. The assay conditions were further optimized to accommodate the 516 bp (A) and 969 bp (B) PCR products. Optimal conditions were predicted using the Taguchi method. Homozygous (PM) and heterozygous samples (MM) were used as negative and positive controls, respectively. Product digestion is quantitated as the percentage of the fragment cleaved relative to input. MM:PM, a measure of signal to noise, represents the ratio of the heterozygous fraction digested to the homozygous fraction digested. Also shown below are the final concentrations of DMSO (%) and KCl (mM) added to the reaction.
Factors affecting optimization of PCR–CRMS
We investigated separately the effects of several factors that, in our initial work, appeared to be of critical importance in increasing the cleavage of true mismatches while maintaining low background cleavage rates. Because errors during PCR amplification could potentially increase the background MutH cleavage during mismatch scanning (32), we first tested a wide variety of thermostable DNA polymerases to establish which was best suited for target amplification and mutation detection. A comparison of the two best enzymes, AmpliTaq Gold and the Expand High Fidelity enzyme cocktail, is shown in Figure 4. Homozygous DNA amplified with AmpliTaq Gold was cleaved at a 2- to 3-fold higher rate than that amplified with Expand High Fidelity. Therefore, we confirmed that the error rate of polymerases contributed to a high background, with the Expand High Fidelity enzyme cocktail providing the degree of proofreading required for successful application of PCR–CRMS.
Figure 4.
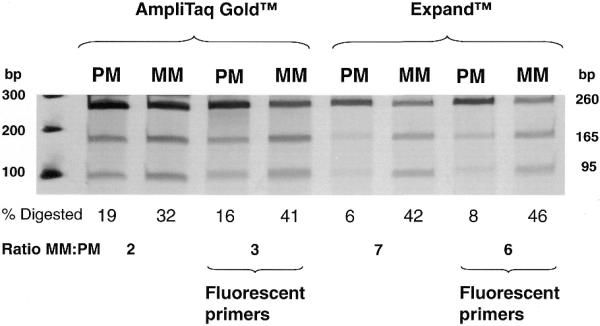
Effects of polymerase type and fluorescence-tagged primers on PCR–CRMS signal-to-noise ratio. Either AmpliTaq Gold or Expand enzyme was used to PCR amplify target DNAs. For each enzyme either an unlabeled or a FAM-labeled forward primer was used for PCR amplification. As in Figure 2, percent cleavage and MM:PM ratio are given below each reaction set.
A series of experiments were performed to titrate the optimal KCl concentration. The results of one such trial are depicted in Figure 5A. At 55 mM salt concentration both the heteroduplex DNA target (mismatch, MM) as well as the homoduplex target (perfect match, PM) were entirely cleaved. The addition of only 5 mM KCl (60 mM total) reduced non-specific cleavage of the PM DNA to 27%. At 85 mM both MM cleavage (53%) and the specific:non-specific ratio (53%:7%) were optimal. Another experiment using four different target amplicons within a completely distinct locus (human androgen receptor) was performed using a KCl concentration ranging from 70 to 105 mM (representing the linear range observed in Fig. 5A). The results (Fig. 5B) show that a linear relationship exists between the measured background activity and the length of target fragment over a range from 285 to 1257 bp. Using the data pooled from Figure 5B and Table 1 we plotted the KCl concentration yielding optimal cleavage rate ratios against DNA fragment length. Figure 5C shows a linear correlation (r2 = 0.91), from which equation 1 was derived.
Figure 5.

Effect of KCl concentration on PCR–CRMS. (A) KCl titration performed on the 260 bp PCR product as target. Forward arrowhead, homozygous samples (PM); backward arrowhead, heterozygous sample (MM). (B) Correlation of KCl with length of target DNA. PCR–CRMS was performed on homozygous amplicons of the indicated size from different loci. Non-specific cleavage at various KCl concentrations is given on the vertical axis. (C) Near linear correlation of KCl for optimal cleavage rate ratios with the length of eight different target DNAs. The correlation coefficient (r2) and linear regression equation are shown.
[KCl]10% max background level = 0.043 × fragment length (bp) + 72.6 mM 1
To assess the validity of the relation in 1 we optimized a PCR–CRMS assay for a new locus (386 bp long from human chromosome 22) using KCl concentration increments of 5 mM. The optimal salt concentration was experimentally determined to be 90 mM (data not shown); that predicted from 1 was 89.2 mM.
Another parameter, the optimal amount of MutL, also demonstrated a direct dependence on target length (Table 1 and Fig. 6). We observed a direct correlation of the MutL optimum with the logarithm of the fragment size in base pairs (r2 = 1). Equation 2 was derived from these data.
Figure 6.

MutL concentration correlation with the length of the target DNA. The optimal MutL enzyme concentration was determined for different target DNAs varying in size using a modified Taguchi optimization procedure (see Materials and Methods; 34). A linear correlation of the apparent concentration of MutL enzyme with the logarithm of PCR product length was observed. The correlation coefficient and derived equation are shown.
[MutL]app. (nM) = 158 × log10[fragment length (bp)] – 203 2
Since the assays were performed using enzyme fractions with partial purity (one-step purification; see Materials and Methods), equation 2 was expressed as ‘apparent’ concentration of MutL and must, therefore, be readjusted with new batches of enzyme. However, the knowledge of this correlation provides an important tool for easy optimization of new target regions.
Although we were able to show that optimization of PCR–CRMS with PCR products led to results quantitative enough for genotyping and allele-sharing work, maximal flexibility of this method for genotyping required one further condition: the efficiency of cleavage had to remain high even if the dam (GATC) recognition site was very close to the end of the target DNA fragment (see Discussion). Another group showed that MutH cleavage efficiency dropped dramatically as the dam site moved closer than 200 bp to the fragment end; under the reaction conditions employed in that study only 20% efficiency was possible at 200 bp (32). As shown in Figures 2 and 3, with our optimization, particularly of KCl concentration, we were able to obtain quantitative cleavage with the dam site 95 bp from the end. By moving the upstream primer closer to the dam site we also tested the efficiency of cleavage when the site was 64, 45, 30 and 15 bp away from the end. A 50% relative cleavage efficiency was still possible at 64 bp from the target fragment end (Table 2). At 45 bp relative cleavage dropped to 13%; no cleavage was detected with dam sites 30 and 15 bp from the end. Therefore, PCR–CRMS may still be useful in mutation detection applications when the dam site is as little as 45 bp away from the end of the target fragment, but quantitative genotyping will probably require distances >64 bp.
Table 2. Effect of GATC site position on PCR–CRMS efficiency.
| Distance (bp)a |
95 |
64 |
45 |
30 |
15 |
| Relative activity | 1 | 0.5 | 0.13 | BDL | BDL |
aDistance between the GATC site and the end of the PCR product.
BDL, below detection limit.
Fluorescence-based PCR–CRMS (genotyping mode)
In preparing for a fluorescence-based assay we first asked whether incorporation of a primer with a fluorescent tag into a PCR-amplified target would interfere with the MutHLS enzymes. As shown in Figure 4, labeled and unlabeled PCR products exhibited the same performance with PCR–CRMS. Therefore, the presence of a fluorescent label at one end of the PCR-amplified target DNA did not increase the non-specific cleavage rate, making possible the use of PCR–CRMS on an automated detection platform such as the ABI 377.
A schematic representation of our heteroduplex selection strategy using a labeled reference probe is shown in Figure 7A. The test DNA, from a locus of interest, is PCR amplified from genomic DNA extracted from peripheral blood leukocytes. The reference probe is produced from DNA with a known genotype using the same primer sequences, however, one is tagged with a fluorescent label and the other with a biotin molecule. The ssf probe is purified using paramagnetic streptavidin beads and NaOH treatment. An aliquot of the probe is then mixed with the test sample PCR product in a ratio of 1:5–1:10. The solution is heat denatured and reannealed in the assay buffer. Reannealing yields six different species: four unlabeled homoduplexes and two fluorescence-labeled heteroduplexes. Following MutHLS treatment the reaction products are loaded on an ABI 377 sequencing gel. Only labeled heteroduplexes are detected; quantitation of percent cleavage is obtained directly from the GeneScan electropherogram.
Figure 7.
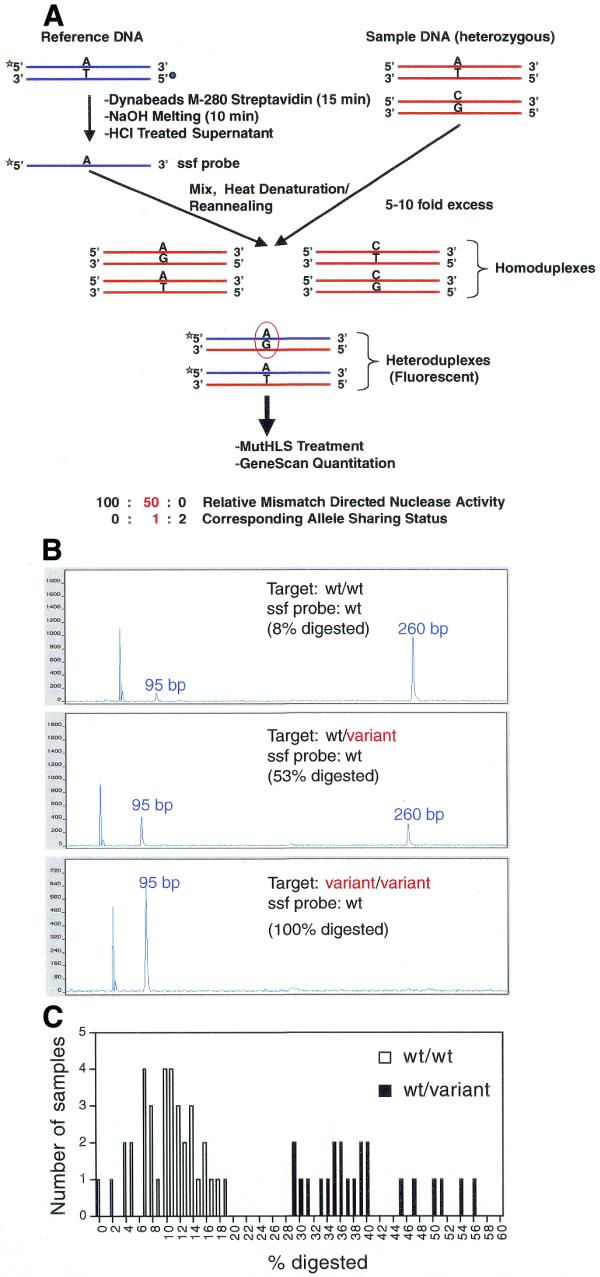
PCR–CRMS genotyping mode. (A) Schematic representation of the PCR–CRMS strategy employing ssf probe. A target locus of interest is PCR amplified with standard primers (red strands). A reference probe is also amplified using a fluorescence-labeled forward primer and a biotin-tagged reverse primer. Following purification of single-strand, labeled reference probe by streptavidin binding, the ssf probe (dark blue strands) is mixed with the test sample PCR products in a ratio of 1:5 to 1:10. The solution is heat denatured and reannealed in assay buffer. The fluorescent heteroduplexes thus formed are the targets of PCR–CRMS and are the only duplexes detected using the ABI 377 automatic sequencer, forced to hybridize with the minus strand of the unknown sample forming heteroduplexes. Quantitative analysis of the electropherogram (GeneScan) provides the extent of mismatch-directed cleavage; in genotyping mode this corresponds to allele-sharing status. The green and blue circles represent the fluorescent label and biotin tag, respectively. (B) Three typical electropherograms of PCR–CRMS products representing the ratios of PCR–CRMS products obtained with a wild-type ssf probe and three types of genotypes following electrophoresis on the ABI 377 automatic sequencer. Upper, homozygous wild-type DNA (wt/wt); middle, heterozygous wild-type/variant DNA (wt/variant); lower, homozygous variant DNA (variant/variant). Percent cleavage for each reaction is provided in each panel. (C) Frequency distribution of the percent digestion data obtained from a blind PCR–CRMS assay performed on 58 previously genotyped DNAs. The ssf probe was wild-type. The open and solid bars represent samples previously genotyped as homozygotes and heterozygotes, respectively.
Figure 7B depicts electropherograms from three target DNAs sampled with a ssf probe prepared from the 260 bp amplicon of CDKN1A exon 3. One DNA was homozygous for the wild-type allele (top), one heterozygous for the wild-type and variant alleles (middle) and one homozygous for the variant allele (bottom). As predicted, only 8% of the probe was cleaved when hybridized with homozygous wild-type DNA (wt/wt) and 53% with heterozygous DNA (wt/v). The entire probe was cleaved in the presence of a homozygous variant DNA sample (v/v). This result indicated that the ssf probe assay could be quite quantitative, raising the possibility of using a mismatch detection strategy for genotyping
To test the feasibility of using PCR–CRMS for genotyping of a larger number of samples we assayed blind 58 human genomic DNA samples previously genotyped at CDKN1A exon 3 by DNA sequencing. Figure 7C depicts the frequency distribution of cleavage fractions obtained from each of these samples. All samples previously known to be homozygous for the wild-type allele in the region interrogated were grouped together in the range 1–19% cleavage (open bars). The heterozygous samples for that same region were also grouped together by the assay. However, in this instance all the heterozygous DNA samples were cleaved at a rate of 30–56% (solid bars). On average, in the setting of a multi-sample assay, homozygous DNAs were cleaved at a rate of 10%, compared to 40% for the heterozygous DNAs.
The transition to a fluorescence-based assay required further optimization of the reaction conditions. While longer targets could be assayed by our heteroduplex selection strategy, using the corresponding ssf probe and the ABI 377 automated sequencer, the KCl concentration for a given target fragment size was always reduced by 10–20 mM when compared to self-annealing assays with unlabeled probes. Table 1 summarizes this and other parameters.
Direct fluorescence-based genotyping with patient (sib–sib) samples
Throughput and flexibility of genotyping might be increased by labeling one sample DNA and testing it directly against another sample DNA (e.g. a family member) without the use of a reference probe. In this fashion DNAs from family members, such as affected siblings, could be compared to one another for the accumulation of allele-sharing data. To evaluate the performance of PCR–CRMS under these conditions we performed an assay in which an unpurified, double-stranded fluorescent (dsf) probe was prepared by amplifying genomic DNA from a previously known homozygote. This DNA was reannealed with crude PCR products from genomic DNA of a homozygote (Fig. 8A) or heterozygote (Fig. 8B) in the ratio 1:30 and PCR–CRMS was performed. A simple dilution of unpurified dsf probe was sufficient to maintain the quantitative aspect of the assay. The cleavage of labeled probe in the presence of a homozygous sample was only 11%; the heterozygous sample cleavage was 40%, for a MM:PM ratio of nearly 4. This result demonstrated the feasibility of comparing relatives using PCR–CRMS.
Figure 8.
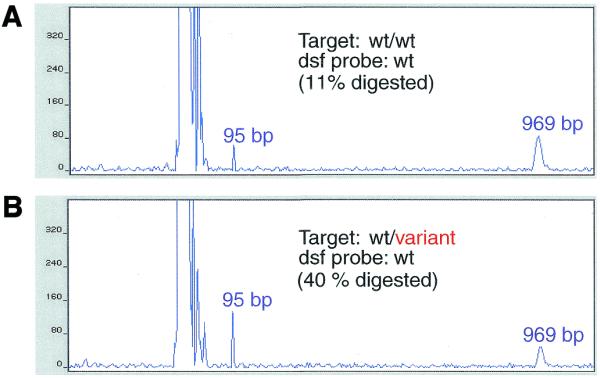
Direct comparison of two sample DNAs (dsf probe). A 969 bp target region from CDKN1A (see Fig. 1) was PCR amplified from DNA of several individuals with known genotypes. Amplification in one case employed a fluorescence-labeled primer; the other DNAs were amplified with standard primers. The dsf probe for a wild-type homozygote, without further purification, was denatured and reannealed in the presence of a 30-fold excess of unlabeled PCR product from a wild-type homozygote (A) or a wild-type/variant heterozygote (B). The electropherograms depict the input sample (969 bp), cleavage fragment (95 bp) and a large excess of unincorporated, labeled primer to the left of the 95 bp peak. Percent cleavage for each reaction is also provided in each panel.
Determination of allele-sharing status in complex haplotypes
To assess the reliability of the assay in quantitating differences among complex haplotypes we moved the target sequence upstream to the promoter region of CDKN1A. A 1257 bp fragment at that location contained three GATC sites, as well as eight SNPs. These SNPs defined seven haplotypes (L.Geller, G.P.Larson and T.G.Krontiris, in preparation). We used either self-annealing of homo- and heterozygote DNA samples or a heteroduplex selection assay that employed haplotype A1 as the ssf probe; both methods gave the expected results (Fig. 9).
Figure 9.

Genotyping of complex haplotypes in a self-annealing and genotyping assay. PCR–CRMS was performed on eight previously genotyped samples with complex SNP haplotypes within the promoter region and 5′-UTR of CDKN1A. The haplotypes (A1, A1a, A2a, A3, C1, D1 and D1a; L.Geller. G.P.Larson and T.G.Krontiris, manuscript in preparation) are indicated on the left of the figure. Relative locations of the SNPs (vertical bars, homozygous variants; half-vertical bars, heterozygous variants) and the reporter sites (diamonds) are schematically represented in the middle portion of the figure. At the fragment termini and over the SNP and reporter sites are numbers corresponding to a PAC reference clone DNA sequence (NCBI accession no. Z85996). At the right of the figure are columns listing the percent digestion observed in the self-annealing and genotyping assays, as well as the corresponding percent cleavage expected for each type of assay (theoretical maximum).
Eight different genomic DNA samples previously haplotyped at the CDKN1A promoter region were PCR amplified. As expected, the two homozygous DNA samples, A1/A1 and C1/C1, were cleaved at the lowest rates of 11 and 12%. Five of six heterozygous samples (A1/A2a, A1/A3, A1a/A3, A1/C1 and D1a/C1) were cleaved at an average rate of 51% (self-annealing) (Fig. 9). Only one heterozygous genotype, carrying a SNP almost 900 bp away from the nearest reporter site, was cleaved at lower rates of ∼35%. We believe that this rate could probably be attributed to the relatively long distance separating the mismatch from the nearest available GATC site. Moreover, detailed examination of the cleavage products revealed that all three reporter sites were used as target sites for the activated MutH (not shown). We concluded that PCR–CRMS was effective even when multiple SNPs and reporter sites were present in the target sequence under analysis.
DISCUSSION
We have demonstrated that the E.coli mismatch detection enzymes MutS, MutL and MutH may be employed for quantitative genotyping of patient DNA samples. The method is easily adapted to automated sequencers for high-throughput usage; massed-tagged primers can, in principle, enable adaptation to genotyping by mass spectroscopy as well. Thus, PCR–CRMS may supplement or even replace microsatellite genotyping in family-based genetic analyses such as genome scans of affected sibling pairs. Our successful adaptation of mismatch scanning coincides with the appearance of the comprehensive human genome sequence, allowing the choice of any marker region coupled to a reporter dam site at any desired position in the PCR fragment. Although the commercial availability of the required mismatch enzymes has been unreliable in the past, we purified all three proteins successfully to near homogeneity using a simple, one-step, Ni2+-chelation affinity batch protocol. Therefore, PCR–CRMS is accessible to individual research laboratories, as well as to academic or commercial consortia.
We have examined several blocks to the efficient use of mismatch scanning for genotyping: (i) the effect of polymerase errors during PCR amplification of genomic target regions on background levels of mismatch detection; (ii) the degree of difficulty in optimizing the mismatch detection reaction for each new genome segment scanned; (iii) the placement of dam recognition sites in regions convenient for analysis without loss of MutH cleavage efficiency; (iv) the isolation of intersample heteroduplexes for analysis without employing complex selection strategies. Each of these difficulties may be overcome with straightforward solutions that are amenable to automation.
For example, previous reports on the use of MutS (33) or MutHLS (32) enzymes for mutation detection of PCR products revealed conflicting results on the use of different DNA polymerases for target amplification. One report suggested that Pfu polymerase does not perform better than Taq polymerase in terms of the signal-to-noise ratio. This was quite an unexpected result, since Taq DNA polymerase lacks 3′→5′ proofreading exonuclease activity and, therefore, exhibits a higher misincorporation rate (37,38). A second report examined the effect of using Pfu, Vent or Taq polymerases and found an almost direct correlation of MutHLS non-specific cleavage of homozygous DNA samples with the previously established frequency of polymerase errors during PCR amplification. To determine the best conditions for PCR–CRMS we measured the cleavage activity on PCR products amplified with the AmpliTaq Gold and the Expand High Fidelity enzyme systems. The latter system is composed of an enzyme mix containing thermostable Taq and Pwo DNA polymerases and is designed to give PCR products from genomic DNA with high yield and fidelity (39). Our results confirmed that the Expand system, with an error rate of 8.5 × 10–6, was superior to AmpliTaq Gold, with an error rate of 2.6 × 10–5. In fact, using these error rate values a 260 bp amplicon would contain ∼6.5% (Expand) and 20% (Taq) of its fragments with at least one misincorporation. Interestingly, these numbers corresponded to the background levels obtained in Figure 4. This result sets a minimally acceptable level of replication error for satisfactory application of PCR–CRMS.
One of our most important conclusions in these studies was that optimization of the mismatch detection reaction was dependent upon salt and MutL concentration in a predictable relationship to target fragment length. A previous report, using human repair enzymes, demonstrated a clear maximum of mismatch repair at 130 mM KCl, with the efficiency of the reaction dropping precipitously at both lower and higher salt concentrations (40). Using our experimental conditions with bacterial enzymes and a 260 bp PCR product it appeared that a higher salt concentration (150 mM) resulted in a low efficiency of cleavage. The use of a lesser amount of KCl (60 mM) was too permissive and specificity for the mismatch was lost. The optimal salt concentration for the 260 bp fragment was 85 mM. This result was in agreement with a previous study postulating weaker hMutSα–DNA interaction with increasing salt concentration (40). The same group demonstrated that at low KCl concentration (≤50 mM) both homo- and heteroduplex DNA activated hMutSα ATPase activity to the same degree. By analogy to the human enzyme it seems that regulation of bacterial MutH cleavage discrimination by KCl was effective at the MutS–DNA binding step.
The optimal experimental conditions for longer PCR products amplified from the CDKN1A locus differed mainly by a proportionally higher salt concentration. This was an unexpected result, since the mismatches and reporter site sequence environment used were identical. In addition, when different regions of the human androgen receptor gene locus were tested the same correlation was observed. These results were in agreement with data reported by Blackwell et al. (40), who found a significant and reproducible DNA chain length effect on kcat, ATP of hMutSα. The authors suggested that a proportional increase in ATPase activity with DNA chain length was the expected result for the translocation mechanism model (40–43), rather than the molecular switch model (44). Under our experimental conditions the interrogation of longer PCR products resulted in higher background cleavage activity, which seemed consistent with the hypothesis that MutS locates the target mismatch by moving along the DNA helix following a translocation mechanism. On the other hand, this result was inconsistent with the switch model, which does not invoke a functional role for sequences external to the mismatch.
The results obtained following optimization using the Taguchi method, done on targets differing in their size and sequence environment, demonstrated the necessity of determining the range of MutL concentrations to be used for optimal cleavage of mismatch-containing DNA while keeping the background activity as low as possible. We found a direct logarithmic correlation (r2 = 1) between length of the target and optimal MutL concentration. As observed with the salt concentration effect, this phenomenon was independent of the sequence environment with G+C content varying from 45 to 55%. We believe that this direct relation with substrate length further supports the idea that a translocation mechanism is involved in mismatch scanning.
Two previous studies (45,46) revealed that MutL alone could stimulate some MutH endonuclease activity via direct physical interaction in the presence of ATP. These studies also demonstrated that addition of MutS to the reaction further stimulated MutH activity, specifically for mismatch-containing DNA. Therefore, the basal activity measured on perfectly matched DNA samples was explained by the spontaneous capacity of MutL to activate MutH without MutS, emphasizing the importance of determining the appropriate concentration of MutL to obtain quantitative genotype and allele-sharing information. The Taguchi method is particularly suited to these determinations.
The relative placement of the GATC recognition sequence can be an important determinant of PCR–CRMS flexibility. Keeping cleavage close to the end of the target fragment will be important if the technique is to be adapted to mass spectrometry, where current technology is optimal for fragments smaller than 100–120 nt in length. Our studies have demonstrated that cleavage efficiency can be maintained when the dam site is within 85–100 nt of the end of the target fragment. Much shorter distances may be employed if mutation detection, rather than genotyping, is the goal.
One final concern about the adaptability of mismatch scanning for genotyping is whether intersample heteroduplex selection can be accomplished in a manner simple enough for automation. As we have shown, two different schemes that involve diluting out a labeled reference strand with PCR products from other test DNAs may be adopted. One reference probe may be used for many samples or one sample of each relative pair may serve as the reference. Either approach, or both in combination, appears promising for high-throughput genotyping.
As discussed earlier, one type of mismatch that cannot be effectively detected is C-C mispairing. Two recent publications have demonstrated that the point mutation C→G is one of the least frequent occurring in the human genome, at 4.7–5.0% of events (47,48). For mutation detection purposes this means that, on average, only 1 of 20 mutations present in the human genome would not be detected using a single probe. However, the possibility of interrogating the other strand, where a C→G mutation would be seen as a G-G mismatch, should alleviate this problem. The use of a probe labeled on both ends with different dyes should be feasible, so that both strands can be interrogated at once. This strategy should further improve the sensitivity of the method by detecting virtually all types of mutations. In any event, for genotyping purposes lack of C-C mismatch detection is not an issue, since other variants would suffice.
Finally, a major attraction of mismatch scanning in the characterization of human genetic variation is the ability to exploit such variation with minimal effort, especially when contrasted with microsatellite genotyping. This ease of use, together with the expectation of a very high density of useful SNPs in the human genomic sequence (1,2), will provide great flexibility in the choice of target regions for PCR–CRMS genotyping.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Malcolm E. Winkler for providing the bacterial strains expressing E.coli MutH, MutL and MutS. We also thank Dr Wei Yang for advice on protein purification. M.B. received a fellowship from the Medical Research Council of Canada. This work was supported by funds from the Beckman Research Institute of the City of Hope, as well as from PHS grants CA45052 and AG15720.
References
- 1.Halushka M.K., Fan,J.B., Bentley,K., Hsie,L., Shen,N., Weder,A., Cooper,R., Lipshutz,R. and Chakravarti,A. (1999) Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nature Genet., 22, 239–247. [DOI] [PubMed] [Google Scholar]
- 2.Cargill M., Altshuler,D., Ireland,J., Sklar,P., Ardlie,K., Patil,N., Lane,C.R., Lim,E.P., Kalayanaraman,N., Nemesh,J., Ziaugra,L., Friedland,L., Rolfe,A., Warrington,J., Lipshutz,R., Daley,G.Q. and Lander,E.S. (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nature Genet., 22, 231–238. [DOI] [PubMed] [Google Scholar]
- 3.Cotton R.G., Rodrigues,N.R. and Campbell,R.D. (1988) Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations. Proc. Natl Acad. Sci. USA, 85, 4397–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellis T.P., Humphrey,K.E., Smith,M.J. and Cotton,R.G. (1998) Chemical cleavage of mismatch: a new look at an established method. Hum. Mutat., 11, 345–353. [DOI] [PubMed] [Google Scholar]
- 5.Lambrinakos A., Humphrey,K.E., Babon,J.J., Ellis,T.P. and Cotton,R.G. (1999) Reactivity of potassium permanganate and tetraethylammonium chloride with mismatched bases and a simple mutation detection protocol. Nucleic Acids Res., 27, 1866–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers R.M., Maniatis,T. and Lerman,L.S. (1987) Detection and localization of single base changes by denaturing gradient gel electrophoresis. Methods Enzymol., 155, 501–527. [DOI] [PubMed] [Google Scholar]
- 7.Orita M., Iwahana,H., Kanazawa,H., Hayashi,K. and Sekiya,T. (1989) Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc. Natl Acad. Sci. USA, 86, 2766–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Q., Feng,J., Buzin,C., Wen,C., Nozari,G., Mengos,A., Nguyen,V., Liu,J., Crawford,L., Fujimura,F.K. and Sommer,S.S. (1999) Detection of virtually all mutations-SSCP (DOVAM-S): a rapid method for mutation scanning with virtually 100% sensitivity. Biotechniques, 26, 932, 936,–938, 940–942. [DOI] [PubMed] [Google Scholar]
- 9.Heid C.A., Stevens,J., Livak,K.J. and Williams,P.M. (1996) Real time quantitative PCR. Genome Res., 6, 986–994. [DOI] [PubMed] [Google Scholar]
- 10.Tyagi S., Bratu,D.P. and Kramer,F.R. (1998) Multicolor molecular beacons for allele discrimination. Nature Biotechnol., 16, 49–53. [DOI] [PubMed] [Google Scholar]
- 11.Kwiatkowski R.W., Lyamichev,V., de Arruda,M. and Neri,B. (1999) Clinical, genetic and pharmacogenetic applications of the Invader assay. Mol. Diagn ., 4, 353–364. [DOI] [PubMed] [Google Scholar]
- 12.Ugozzoli L. and Wallace,R.B. (1992) Application of an allele-specific polymerase chain reaction to the direct determination of ABO blood group genotypes. Genomics, 12, 670–674. [DOI] [PubMed] [Google Scholar]
- 13.Cheng J., Waters,L.C., Fortina,P., Hvichia,G., Jacobson,S.C., Ramsey,J.M., Kricka,L.J. and Wilding,P. (1998) Degenerate oligonucleotide primed-polymerase chain reaction and capillary electrophoretic analysis of human DNA on microchip-based devices. Anal. Biochem., 257, 101–106. [DOI] [PubMed] [Google Scholar]
- 14.Boon E.M., Ceres,D.M., Drummond,T.G., Hill,M.G. and Barton,J.K. (2000) Mutation detection by electrocatalysis at DNA-modified electrodes. Nature Biotechnol., 18, 1096–1100. [DOI] [PubMed] [Google Scholar]
- 15.Kurg A., Tonisson,N., Georgiou,I., Shumaker,J., Tollett,J. and Metspalu,A. (2000) Arrayed primer extension: solid-phase four-color DNA resequencing and mutation detection technology. Genet. Test., 4, 1–7. [DOI] [PubMed] [Google Scholar]
- 16.Tang K., Fu,D.J., Julien,D., Braun,A., Cantor,C.R. and Koster,H. (1999) Chip-based genotyping by mass spectrometry. Proc. Natl Acad. Sci. USA, 96, 10016–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glavac D. and Dean,M. (1995) Applications of heteroduplex analysis for mutation detection in disease genes. Hum. Mutat., 6, 281–287. [DOI] [PubMed] [Google Scholar]
- 18.Dianzani I., Landegren,U., Camaschella,C., Ponzone,A., Piazza,A. and Cotton,R.G. (1999) Fifth International Mutation Detection Workshop, May 13–16, 1999, Vicoforte, Italy. Hum. Mutat., 14, 451–453. [DOI] [PubMed] [Google Scholar]
- 19.Cotton R.G. (1999) Mutation detection by chemical cleavage. Genet. Anal., 14, 165–168. [DOI] [PubMed] [Google Scholar]
- 20.Taylor G.R. and Deeble,J. (1999) Enzymatic methods for mutation scanning. Genet. Anal., 14, 181–186. [DOI] [PubMed] [Google Scholar]
- 21.Nelson S.F., McCusker,J.H., Sander,M.A., Kee,Y., Modrich,P. and Brown,P.O. (1993) Genomic mismatch scanning: a new approach to genetic linkage mapping. Nature Genet., 4, 11–18. [DOI] [PubMed] [Google Scholar]
- 22.McAllister L., Penland,L. and Brown,P.O. (1998) Enrichment for loci identical-by-descent between pairs of mouse or human genomes by genomic mismatch scanning. Genomics, 47, 7-11. [DOI] [PubMed] [Google Scholar]
- 23.Cheung V.G. and Nelson,S.F. (1998) Genomic mismatch scanning identifies human genomic DNA shared identical by descent. Genomics, 47, 1–6. [DOI] [PubMed] [Google Scholar]
- 24.Cheung V.G., Gregg,J.P., Gogolin-Ewens,K.J., Bandong,J., Stanley,C.A., Baker,L., Higgins,M.J., Nowak,N.J., Shows,T.B., Ewens,W.J., Nelson,S.F. and Spielman,R.S. (1998) Linkage-disequilibrium mapping without genotyping. Nature Genet., 18, 225–230. [DOI] [PubMed] [Google Scholar]
- 25.Nelson S.F. (1995) Genomic mismatch scanning: current progress and potential applications. Electrophoresis, 16, 279–285. [DOI] [PubMed] [Google Scholar]
- 26.Welford S.M., Gregg,J., Chen,E., Garrison,D., Sorensen,P.H., Denny,C.T. and Nelson,S.F. (1998) Detection of differentially expressed genes in primary tumor tissues using representational differences analysis coupled to microarray hybridization. Nucleic Acids Res., 26, 3059–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahue R.S., Au,K.G. and Modrich,P. (1989) DNA mismatch correction in a defined system. Science, 245, 160–164. [DOI] [PubMed] [Google Scholar]
- 28.Parker B.O. and Marinus,M.G. (1992) Repair of DNA heteroduplexes containing small heterologous sequences in Escherichia coli. Proc. Natl Acad. Sci. USA, 89, 1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Au K.G., Welsh,K. and Modrich,P. (1992) Initiation of methyl-directed mismatch repair. J. Biol. Chem., 267, 12142–12148. [PubMed] [Google Scholar]
- 30.Yamaguchi M., Dao,V. and Modrich,P. (1998) MutS and MutL activate DNA helicase II in a mismatch-dependent manner. J. Biol. Chem., 273, 9197–9201. [DOI] [PubMed] [Google Scholar]
- 31.Dao V. and Modrich,P. (1998) Mismatch-, MutS-, MutL- and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J. Biol. Chem., 273, 9202–9207. [DOI] [PubMed] [Google Scholar]
- 32.Smith J. and Modrich,P. (1996) Mutation detection with MutH, MutL and MutS mismatch repair proteins. Proc. Natl Acad. Sci. USA, 93, 4374–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lishanski A., Ostrander,E.A. and Rine,J. (1994) Mutation detection by mismatch binding protein, MutS, in amplified DNA: application to the cystic fibrosis gene. Proc. Natl Acad. Sci. USA, 91, 2674–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cobb B.D. and Clarkson,J.M. (1994) A simple procedure for optimising the polymerase chain reaction (PCR) using modified Taguchi methods. Nucleic Acids Res., 22, 3801–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng G. and Winkler,M.E. (1995) Single-step purifications of His6-MutH, His6-MutL and His6-MutS repair proteins of Escherichia coli K-12. Biotechniques, 19, 956–965. [PubMed] [Google Scholar]
- 36.Law J.C. and Deka,A. (1995) Identification of a PstI polymorphism in the p21Cip1/Waf1 cyclin-dependent kinase inhibitor gene. Hum. Genet., 95, 459–460. [DOI] [PubMed] [Google Scholar]
- 37.Eckert K.A. and Kunkel,T.A. (1991) DNA polymerase fidelity and the polymerase chain reaction. PCR Methods Appl., 1, 17–24. [DOI] [PubMed] [Google Scholar]
- 38.Tindall K.R. and Kunkel,T.A. (1988) Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry, 27, 6008–6013. [DOI] [PubMed] [Google Scholar]
- 39.Barnes W.M. (1994) PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc. Natl Acad. Sci. USA, 91, 2216–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackwell L.J., Bjornson,K.P. and Modrich,P. (1998) DNA-dependent activation of the hMutSalpha ATPase. J. Biol. Chem., 273, 32049–32054. [DOI] [PubMed] [Google Scholar]
- 41.Young M.C., Kuhl,S.B. and von Hippel,P.H. (1994) Kinetic theory of ATP-driven translocases on one-dimensional polymer lattices. J. Mol. Biol., 235, 1436–1446. [DOI] [PubMed] [Google Scholar]
- 42.Young M.C., Schultz,D.E., Ring,D. and von Hippel,P.H. (1994) Kinetic parameters of the translocation of bacteriophage T4 gene 41 protein helicase on single-stranded DNA. J. Mol. Biol., 235, 1447–1458. [DOI] [PubMed] [Google Scholar]
- 43.Blackwell L.J., Martik,D., Bjornson,K.P., Bjornson,E.S. and Modrich,P. (1998) Nucleotide-promoted release of hMutSalpha from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. J. Biol. Chem., 273, 32055–32062. [DOI] [PubMed] [Google Scholar]
- 44.Gradia S., Acharya,S. and Fishel,R. (1997) The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell, 91, 995–1005. [DOI] [PubMed] [Google Scholar]
- 45.Ban C. and Yang,W. (1998) Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell, 95, 541–552. [DOI] [PubMed] [Google Scholar]
- 46.Hall M.C. and Matson,S.W. (1999) The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J. Biol. Chem., 274, 1306–1312. [DOI] [PubMed] [Google Scholar]
- 47.Krawczak M., Ball,E.V. and Cooper,D.N. (1998) Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am. J. Hum. Genet., 63, 474–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hawkins G.A. and Hoffman,L.M. (1997) Base excision sequence scanning. Nature Biotechnol., 15, 803–804. [DOI] [PubMed] [Google Scholar]