

Abstract
In the molecule of the title compound, C8H7NO4, the nitro group is approximately coplanar with the benzene ring [dihedral angle = 0.6 (1)°], while the dihedral angle between the methoxycarbonyl group and the benzene ring is 8.8 (1)°. In the crystal structure, weak intermolecular aromatic C—H⋯Ocarboxyl and C—H⋯Onitro hydrogen-bonding interactions are present.
Related literature
For related literature on benzoates, see: Zhang (1992 ▶); Zhang et al. (1990 ▶); Zhang et al. (1995 ▶).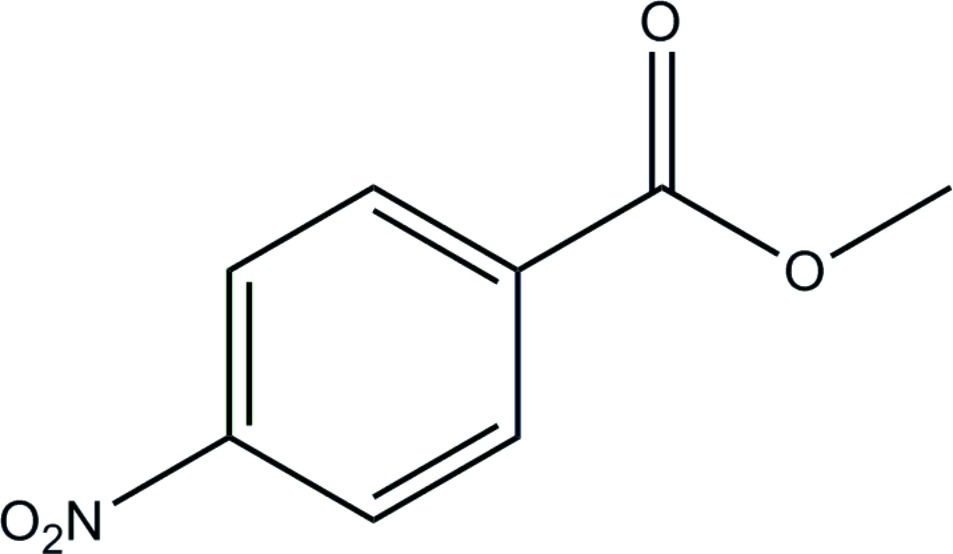
Experimental
Crystal data
C8H7NO4
M r = 181.15
Monoclinic,
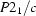
a = 7.109 (3) Å
b = 17.092 (6) Å
c = 7.193 (3) Å
β = 116.292 (4)°
V = 783.6 (5) Å3
Z = 4
Mo Kα radiation
μ = 0.13 mm−1
T = 93 K
0.43 × 0.40 × 0.10 mm
Data collection
Rigaku SPIDER CCD-detector diffractometer
Absorption correction: ψ scan (North et al., 1968 ▶) T min = 0.948, T max = 0.988
6176 measured reflections
1787 independent reflections
1445 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.034
wR(F 2) = 0.080
S = 1.00
1787 reflections
119 parameters
H-atom parameters constrained
Δρmax = 0.31 e Å−3
Δρmin = −0.18 e Å−3
Data collection: RAPID-AUTO (Rigaku, 2004 ▶); cell refinement: RAPID-AUTO; data reduction: RAPID-AUTO; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053680904745X/zs2018sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680904745X/zs2018Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯O2i | 0.95 | 2.59 | 3.384 (2) | 141 |
| C5—H5⋯O4ii | 0.95 | 2.58 | 3.378 (2) | 142 |
Symmetry codes: (i) 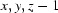 ; (ii)
; (ii) 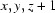 .
.
Acknowledgments
The authors acknowledge financial support from Jiangsu Institute of Nuclear Medicine.
supplementary crystallographic information
Comment
Benzoates are important intermediates in the chemistry of pigments and pharmaceuticals, which are used worldwide (Zhang, 1992; Zhang et al., 1990; Zhang et al., 1995). We report here the crystal structure of methyl 4-nitrobenzoate, C8H7NO4 (I). In the structure of the title compound (Fig. 1) the bond lengths and angles are within expected ranges. The nitro substituent group is nearly coplanar with the benzene ring [dihedral angle, 0.6 (1)°], while the methoxycarbonyl group forms a dihedral angle of 8.8 (1)° with the benzene ring. In the crystal structure, adjacent molecules are linked by weak intermolecular aromatic C—H···Ocarboxyl and Onitro hydrogen bonds (Table 1).
Experimental
4-Nitrobenzoic acid (5.0 g, 30 mmol) was dissolved in hot methanol (10 ml), then six drops of concentrated sulfuric acid were added. The mixture was stirred at 353 K for 4 h, poured into ice water and stirred for 3 min. After filtering, washing with water and drying in vacuum, a white powder was then obtained (yield: 73%). The crude product was purified by recrystallization from methanol (yield: 51%). Colourless plate-shaped crystals [m.p. 369 (2) K] were obtained after several days, by slow evaporation of a 1:1 (v/v) methanol-water solution. .
Refinement
Positional parameters of all H atoms were calculated geometrically and were allowed to ride on the C atoms to which they are bonded, with Caryl—H = 0.95 Å, and Cmethyl—H = 0.98 Å, and with Uiso(H) = 1.2Ueq(C)
Figures
Fig. 1.

Atom numbering scheme for the title compound (I) with the displacement ellipsoids drawn at the 30% probability level.
Crystal data
| C8H7NO4 | F(000) = 376 |
| Mr = 181.15 | Dx = 1.536 Mg m−3 |
| Monoclinic, P21/c | Melting point: 369(2) K |
| Hall symbol: -P 2ybc | Mo Kα radiation, λ = 0.71073 Å |
| a = 7.109 (3) Å | Cell parameters from 2257 reflections |
| b = 17.092 (6) Å | θ = 3.2–27.4° |
| c = 7.193 (3) Å | µ = 0.13 mm−1 |
| β = 116.292 (4)° | T = 93 K |
| V = 783.6 (5) Å3 | Plate, colorless |
| Z = 4 | 0.43 × 0.40 × 0.10 mm |
Data collection
| Rigaku SPIDER CCD-detector diffractometer | 1787 independent reflections |
| Radiation source: rotating anode | 1445 reflections with I > 2σ(I) |
| graphite | Rint = 0.023 |
| ω scans | θmax = 27.5°, θmin = 3.2° |
| Absorption correction: ψ scan (North et al., 1968) | h = −9→9 |
| Tmin = 0.948, Tmax = 0.988 | k = −20→22 |
| 6176 measured reflections | l = −9→9 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.034 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.080 | H-atom parameters constrained |
| S = 1.00 | w = 1/[σ2(Fo2) + (0.0303P)2 + 0.336P] where P = (Fo2 + 2Fc2)/3 |
| 1787 reflections | (Δ/σ)max < 0.001 |
| 119 parameters | Δρmax = 0.31 e Å−3 |
| 0 restraints | Δρmin = −0.18 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.35100 (15) | 0.66883 (5) | 0.67509 (14) | 0.0205 (2) | |
| O2 | 0.30933 (15) | 0.59510 (5) | 0.91414 (14) | 0.0216 (2) | |
| O3 | 0.14128 (16) | 0.28185 (5) | 0.23700 (16) | 0.0265 (2) | |
| O4 | 0.14076 (17) | 0.36321 (6) | 0.00524 (15) | 0.0275 (2) | |
| N1 | 0.15702 (17) | 0.34819 (6) | 0.17881 (17) | 0.0185 (2) | |
| C1 | 0.25319 (19) | 0.54972 (7) | 0.39808 (19) | 0.0155 (3) | |
| H1 | 0.2638 | 0.6016 | 0.3563 | 0.019* | |
| C2 | 0.21513 (19) | 0.48826 (7) | 0.2601 (2) | 0.0160 (3) | |
| H2 | 0.2007 | 0.4971 | 0.1240 | 0.019* | |
| C3 | 0.19872 (19) | 0.41347 (7) | 0.3264 (2) | 0.0155 (3) | |
| C4 | 0.21927 (19) | 0.39748 (7) | 0.5240 (2) | 0.0167 (3) | |
| H4 | 0.2065 | 0.3456 | 0.5643 | 0.020* | |
| C5 | 0.25907 (19) | 0.45958 (7) | 0.6606 (2) | 0.0160 (3) | |
| H5 | 0.2749 | 0.4503 | 0.7970 | 0.019* | |
| C6 | 0.27592 (18) | 0.53560 (7) | 0.59852 (19) | 0.0149 (3) | |
| C7 | 0.31372 (19) | 0.60132 (7) | 0.7484 (2) | 0.0162 (3) | |
| C8 | 0.3760 (2) | 0.73724 (8) | 0.8036 (2) | 0.0239 (3) | |
| H8A | 0.2532 | 0.7425 | 0.8310 | 0.029* | |
| H8B | 0.3890 | 0.7840 | 0.7313 | 0.029* | |
| H8C | 0.5025 | 0.7314 | 0.9351 | 0.029* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0302 (5) | 0.0155 (5) | 0.0190 (5) | −0.0039 (4) | 0.0139 (4) | −0.0029 (4) |
| O2 | 0.0259 (5) | 0.0241 (5) | 0.0167 (5) | −0.0026 (4) | 0.0111 (4) | −0.0005 (4) |
| O3 | 0.0390 (6) | 0.0150 (5) | 0.0290 (6) | −0.0025 (4) | 0.0182 (5) | 0.0006 (4) |
| O4 | 0.0437 (6) | 0.0225 (5) | 0.0202 (5) | −0.0009 (5) | 0.0177 (5) | −0.0016 (4) |
| N1 | 0.0196 (5) | 0.0163 (5) | 0.0211 (6) | 0.0005 (4) | 0.0102 (5) | 0.0002 (4) |
| C1 | 0.0151 (6) | 0.0144 (6) | 0.0172 (7) | −0.0002 (5) | 0.0075 (5) | 0.0022 (5) |
| C2 | 0.0152 (6) | 0.0189 (6) | 0.0142 (6) | 0.0007 (5) | 0.0067 (5) | 0.0024 (5) |
| C3 | 0.0132 (6) | 0.0160 (6) | 0.0172 (6) | 0.0011 (5) | 0.0066 (5) | −0.0008 (5) |
| C4 | 0.0152 (6) | 0.0153 (6) | 0.0204 (7) | 0.0008 (5) | 0.0088 (5) | 0.0033 (5) |
| C5 | 0.0143 (6) | 0.0196 (6) | 0.0150 (6) | 0.0012 (5) | 0.0071 (5) | 0.0036 (5) |
| C6 | 0.0115 (5) | 0.0175 (6) | 0.0152 (6) | 0.0003 (5) | 0.0055 (5) | −0.0002 (5) |
| C7 | 0.0134 (6) | 0.0181 (6) | 0.0162 (6) | 0.0005 (5) | 0.0059 (5) | 0.0017 (5) |
| C8 | 0.0339 (8) | 0.0176 (6) | 0.0243 (7) | −0.0055 (6) | 0.0167 (6) | −0.0060 (5) |
Geometric parameters (Å, °)
| O1—C7 | 1.3429 (15) | C2—H2 | 0.9500 |
| O1—C8 | 1.4517 (15) | C3—C4 | 1.3901 (18) |
| O2—C7 | 1.2111 (16) | C4—C5 | 1.3880 (18) |
| O3—N1 | 1.2308 (14) | C4—H4 | 0.9500 |
| O4—N1 | 1.2290 (15) | C5—C6 | 1.3962 (18) |
| N1—C3 | 1.4770 (16) | C5—H5 | 0.9500 |
| C1—C2 | 1.3872 (18) | C6—C7 | 1.4965 (18) |
| C1—C6 | 1.3988 (18) | C8—H8A | 0.9800 |
| C1—H1 | 0.9500 | C8—H8B | 0.9800 |
| C2—C3 | 1.3873 (17) | C8—H8C | 0.9800 |
| C7—O1—C8 | 115.59 (10) | C4—C5—C6 | 120.30 (12) |
| O4—N1—O3 | 123.79 (11) | C4—C5—H5 | 119.8 |
| O4—N1—C3 | 118.11 (10) | C6—C5—H5 | 119.8 |
| O3—N1—C3 | 118.10 (11) | C5—C6—C1 | 120.22 (12) |
| C2—C1—C6 | 120.25 (12) | C5—C6—C7 | 118.78 (11) |
| C2—C1—H1 | 119.9 | C1—C6—C7 | 120.98 (11) |
| C6—C1—H1 | 119.9 | O2—C7—O1 | 123.89 (12) |
| C1—C2—C3 | 118.10 (12) | O2—C7—C6 | 124.65 (11) |
| C1—C2—H2 | 121.0 | O1—C7—C6 | 111.46 (11) |
| C3—C2—H2 | 121.0 | O1—C8—H8A | 109.5 |
| C2—C3—C4 | 123.11 (12) | O1—C8—H8B | 109.5 |
| C2—C3—N1 | 118.00 (11) | H8A—C8—H8B | 109.5 |
| C4—C3—N1 | 118.89 (11) | O1—C8—H8C | 109.5 |
| C5—C4—C3 | 118.01 (12) | H8A—C8—H8C | 109.5 |
| C5—C4—H4 | 121.0 | H8B—C8—H8C | 109.5 |
| C3—C4—H4 | 121.0 | ||
| C6—C1—C2—C3 | 0.59 (18) | C4—C5—C6—C1 | −0.12 (18) |
| C1—C2—C3—C4 | −0.22 (18) | C4—C5—C6—C7 | 178.53 (11) |
| C1—C2—C3—N1 | 179.66 (11) | C2—C1—C6—C5 | −0.43 (18) |
| O4—N1—C3—C2 | 0.62 (17) | C2—C1—C6—C7 | −179.05 (11) |
| O3—N1—C3—C2 | −179.62 (11) | C8—O1—C7—O2 | −3.17 (18) |
| O4—N1—C3—C4 | −179.49 (12) | C8—O1—C7—C6 | 176.11 (10) |
| O3—N1—C3—C4 | 0.27 (17) | C5—C6—C7—O2 | −8.15 (19) |
| C2—C3—C4—C5 | −0.31 (18) | C1—C6—C7—O2 | 170.48 (12) |
| N1—C3—C4—C5 | 179.80 (11) | C5—C6—C7—O1 | 172.58 (11) |
| C3—C4—C5—C6 | 0.48 (18) | C1—C6—C7—O1 | −8.78 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C1—H1···O1 | 0.95 | 2.39 | 2.7149 (19) | 100 |
| C2—H2···O2i | 0.95 | 2.59 | 3.384 (2) | 141 |
| C5—H5···O4ii | 0.95 | 2.58 | 3.378 (2) | 142 |
Symmetry codes: (i) x, y, z−1; (ii) x, y, z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: ZS2018).
References
- North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968). Acta Cryst. A24, 351–359.
- Rigaku (2004). RAPID-AUTO. Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Zhang, S. G. (1992). Technical Book of Fine Chemicals. Beijing: Science Publishing.
- Zhang, A. Y., Qian, B., Min, J. & Fang, Q. X. (1995). J. Shanxi Normal Univ. (Nat. Sci. Ed.), 23, 44–47.
- Zhang, Z. S., Wu, J. G. & Deng, R. W. (1990). J. Lanzhou Univ. (Nat. Sci. Ed.), 26, 69–75.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053680904745X/zs2018sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053680904745X/zs2018Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report