

Abstract
The title molecule, C8H6BrN, is almost planar (r.m.s. deviation for the non-H atoms = 0.008 Å). In the crystal, weak π–π stacking interactions [centroid–centroid separations = 3.782 (2) and 3.919 (2) Å] generate [100] columns of molecules.
Related literature
For the synthesis, see: Johnson & Sandborn (1941 ▶). 2-Bromo-4-methylbenzonitrile derivatives are used as intermediates in the synthesis of phthalocyanine dyes. For applications of phthalocyanine dyes in photo redox reactions and photodynamic cancer therapy, see: Simon & Sirlin (1989 ▶); Simon et al. (1989 ▶).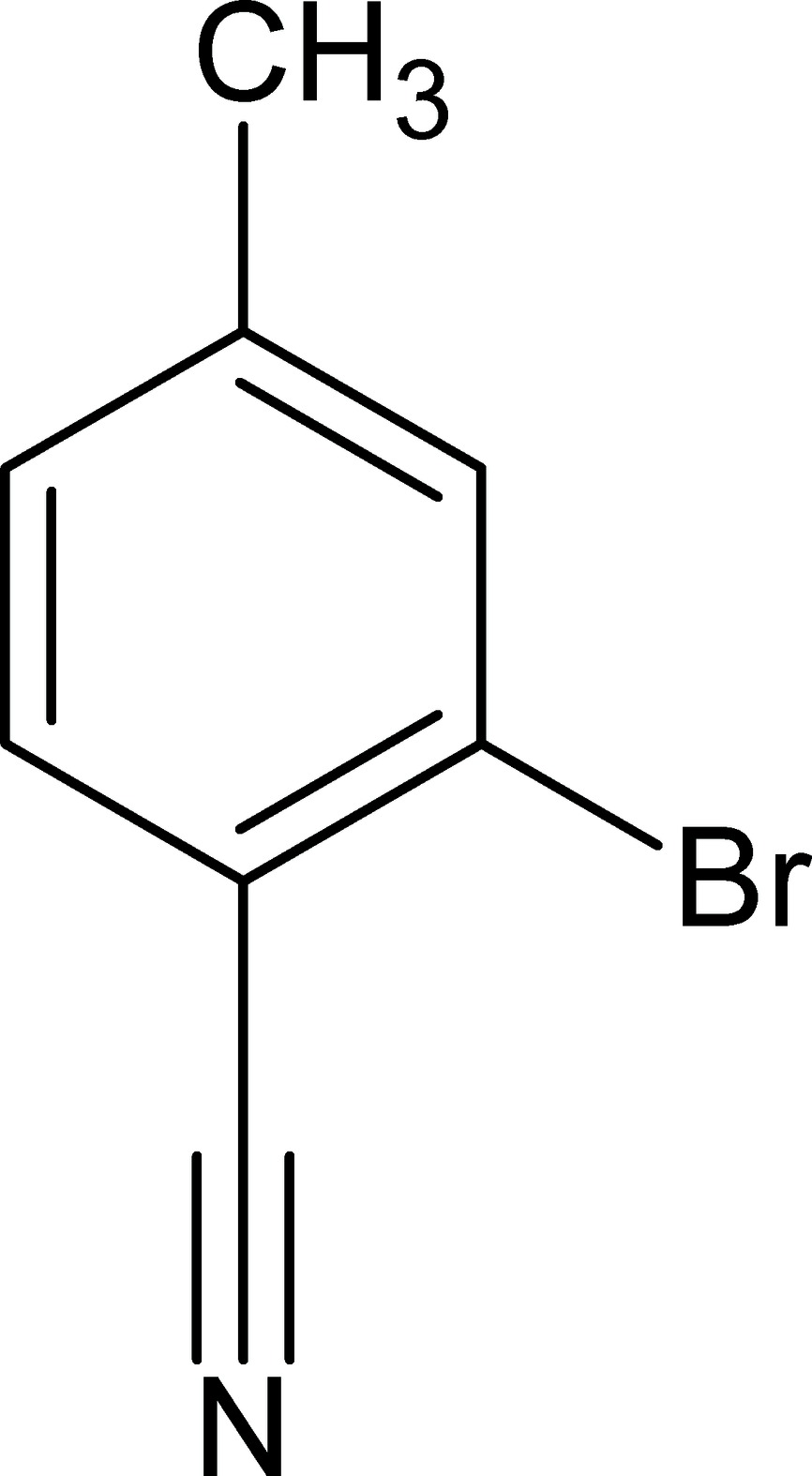
Experimental
Crystal data
C8H6BrN
M r = 196.05
Triclinic,
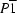
a = 7.5168 (11) Å
b = 7.8383 (11) Å
c = 7.9428 (11) Å
α = 69.243 (7)°
β = 64.375 (8)°
γ = 87.567 (8)°
V = 391.14 (10) Å3
Z = 2
Mo Kα radiation
μ = 5.17 mm−1
T = 296 K
0.41 × 0.28 × 0.19 mm
Data collection
Bruker Kappa APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2007 ▶) T min = 0.226, T max = 0.440
8084 measured reflections
1921 independent reflections
1244 reflections with I > 2σ(I)
R int = 0.025
Refinement
R[F 2 > 2σ(F 2)] = 0.033
wR(F 2) = 0.084
S = 1.01
1921 reflections
92 parameters
H-atom parameters constrained
Δρmax = 0.44 e Å−3
Δρmin = −0.49 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and PLATON (Spek, 2009 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶) and PLATON.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536809048983/hb5232sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809048983/hb5232Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
Muhammad Shahid acknowledges the Higher Education Commission of Pakistan for providing funds, the Institute of Chemistry, University of the Punjab, for providing research facilities and the Education Department, Government of the Punjab, for their co-operation.
supplementary crystallographic information
Comment
Synthesis of 2-bromo-4-methylbenzonitrile derivatives are important compounds due to their use as intermediates in the synthesis of phthalocyanine dyes. The substituted phthalocyanine dyes have been used for photo redox reactions (Simon & Sirlin, 1989) and photodynamic cancer therapy (Simon et al.. 1989).
The title compound(I) is almost planar. The cyano plane (C4/C8/N1) is oriented at a dihedral angle of 79.7 (3)° with respect to aromatic ring (C1/C2/C3/C4/C5/C6). The dihedral angle between the plane containing the methyl carbon (C1/C2/C6/C7) and aromatic ring plane is 0.22 (0.18)°. No significant intermolecular or intramolecular hydrogen bonding interaction has been observed in the molecule.
Experimental
3-Bromo-4-amino toluene (10 g, 54 mmol) (Johnson & Sandborn, 1941) was dissolved in HCl (30 ml, 17%). The mixture was cooled to 273 K in an ice-salt mixture. Over 5 min, an aqueous solution (9 ml) of NaNO2 (4.3 g) was added to the above mixture. The temperature was maintained at 273-278 K. A mixture of aqueous solution (6%) of Cu(I)cyanide and KCN (40%) was heated to 333 K and added to the above cold neutralized diazonium salt solution. After work up of reaction, colourless blocks of (I) were obtained by the slow evaporation of water.
Refinement
The H atoms were geometrically placed (C—H = 0.93–0.96Å) and refined as riding with Uiso(H) = 1.2Ueq(C) or 1.5Ueq(methyl C).
Figures
Fig. 1.

The molecular structure of (I) with 50% displacement ellipsoids.
Fig. 2.

Unit cell packing diagram.
Crystal data
| C8H6BrN | Z = 2 |
| Mr = 196.05 | F(000) = 192 |
| Triclinic, P1 | Dx = 1.665 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 7.5168 (11) Å | Cell parameters from 3073 reflections |
| b = 7.8383 (11) Å | θ = 2.2–21.2° |
| c = 7.9428 (11) Å | µ = 5.17 mm−1 |
| α = 69.243 (7)° | T = 296 K |
| β = 64.375 (8)° | Block, colourless |
| γ = 87.567 (8)° | 0.41 × 0.28 × 0.19 mm |
| V = 391.14 (10) Å3 |
Data collection
| Bruker Kappa APEXII CCD diffractometer | 1921 independent reflections |
| Radiation source: fine-focus sealed tube | 1244 reflections with I > 2σ(I) |
| graphite | Rint = 0.025 |
| φ and ω scans | θmax = 28.3°, θmin = 2.8° |
| Absorption correction: multi-scan (SADABS; Bruker, 2007) | h = −9→9 |
| Tmin = 0.226, Tmax = 0.440 | k = −10→10 |
| 8084 measured reflections | l = −10→10 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.033 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.084 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.0326P)2 + 0.2649P] where P = (Fo2 + 2Fc2)/3 |
| 1921 reflections | (Δ/σ)max < 0.001 |
| 92 parameters | Δρmax = 0.44 e Å−3 |
| 0 restraints | Δρmin = −0.49 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.32613 (7) | 0.89370 (4) | 0.35907 (6) | 0.07874 (19) | |
| C1 | 0.2019 (4) | 0.3498 (4) | 0.7427 (4) | 0.0525 (7) | |
| C2 | 0.2395 (4) | 0.5395 (4) | 0.6606 (4) | 0.0512 (7) | |
| H2 | 0.2419 | 0.6033 | 0.7387 | 0.061* | |
| C3 | 0.2732 (4) | 0.6352 (4) | 0.4660 (4) | 0.0457 (6) | |
| C4 | 0.2711 (4) | 0.5439 (4) | 0.3461 (4) | 0.0445 (6) | |
| C5 | 0.2349 (5) | 0.3537 (4) | 0.4270 (5) | 0.0532 (7) | |
| H5 | 0.2337 | 0.2899 | 0.3485 | 0.064* | |
| C6 | 0.2008 (5) | 0.2588 (4) | 0.6222 (5) | 0.0582 (8) | |
| H6 | 0.1765 | 0.1310 | 0.6747 | 0.070* | |
| C7 | 0.1639 (6) | 0.2446 (5) | 0.9569 (5) | 0.0764 (10) | |
| H7A | 0.2191 | 0.3191 | 1.0006 | 0.115* | |
| H7B | 0.2253 | 0.1339 | 0.9654 | 0.115* | |
| H7C | 0.0231 | 0.2136 | 1.0415 | 0.115* | |
| C8 | 0.3088 (5) | 0.6413 (4) | 0.1400 (5) | 0.0540 (7) | |
| N1 | 0.3389 (5) | 0.7126 (4) | −0.0230 (5) | 0.0771 (9) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.1226 (4) | 0.0413 (2) | 0.0893 (3) | 0.01752 (18) | −0.0609 (3) | −0.02676 (18) |
| C1 | 0.0514 (18) | 0.0578 (18) | 0.0451 (17) | 0.0061 (14) | −0.0231 (14) | −0.0136 (14) |
| C2 | 0.0571 (18) | 0.0587 (18) | 0.0515 (18) | 0.0159 (14) | −0.0288 (15) | −0.0308 (15) |
| C3 | 0.0525 (17) | 0.0403 (14) | 0.0491 (17) | 0.0096 (12) | −0.0251 (14) | −0.0193 (13) |
| C4 | 0.0449 (16) | 0.0480 (16) | 0.0410 (16) | 0.0047 (12) | −0.0188 (13) | −0.0174 (13) |
| C5 | 0.0641 (19) | 0.0478 (16) | 0.0525 (18) | 0.0029 (14) | −0.0249 (15) | −0.0249 (14) |
| C6 | 0.069 (2) | 0.0437 (16) | 0.056 (2) | 0.0005 (14) | −0.0258 (16) | −0.0151 (15) |
| C7 | 0.086 (3) | 0.085 (3) | 0.050 (2) | 0.007 (2) | −0.0321 (19) | −0.0132 (18) |
| C8 | 0.0586 (19) | 0.0560 (18) | 0.0478 (19) | −0.0003 (14) | −0.0225 (15) | −0.0205 (15) |
| N1 | 0.098 (2) | 0.077 (2) | 0.0512 (18) | −0.0056 (17) | −0.0332 (17) | −0.0171 (16) |
Geometric parameters (Å, °)
| Br1—C3 | 1.882 (3) | C4—C8 | 1.440 (4) |
| C1—C2 | 1.380 (4) | C5—C6 | 1.371 (4) |
| C1—C6 | 1.384 (4) | C5—H5 | 0.9300 |
| C1—C7 | 1.503 (4) | C6—H6 | 0.9300 |
| C2—C3 | 1.368 (4) | C7—H7A | 0.9600 |
| C2—H2 | 0.9300 | C7—H7B | 0.9600 |
| C3—C4 | 1.384 (4) | C7—H7C | 0.9600 |
| C4—C5 | 1.383 (4) | C8—N1 | 1.133 (4) |
| C2—C1—C6 | 118.2 (3) | C6—C5—H5 | 119.9 |
| C2—C1—C7 | 121.0 (3) | C4—C5—H5 | 119.9 |
| C6—C1—C7 | 120.8 (3) | C5—C6—C1 | 121.2 (3) |
| C3—C2—C1 | 121.0 (3) | C5—C6—H6 | 119.4 |
| C3—C2—H2 | 119.5 | C1—C6—H6 | 119.4 |
| C1—C2—H2 | 119.5 | C1—C7—H7A | 109.5 |
| C2—C3—C4 | 120.8 (3) | C1—C7—H7B | 109.5 |
| C2—C3—Br1 | 119.6 (2) | H7A—C7—H7B | 109.5 |
| C4—C3—Br1 | 119.6 (2) | C1—C7—H7C | 109.5 |
| C5—C4—C3 | 118.6 (3) | H7A—C7—H7C | 109.5 |
| C5—C4—C8 | 119.6 (3) | H7B—C7—H7C | 109.5 |
| C3—C4—C8 | 121.8 (3) | N1—C8—C4 | 177.7 (3) |
| C6—C5—C4 | 120.3 (3) |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB5232).
References
- Bruker (2007). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst. 32, 837–838.
- Johnson, J. R. & Sandborn, L. T. (1941). Org. Synth. Coll. 1, 111–116.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Simon, J., Bassoul, P. & Norvez, S. (1989). New. J. Chem 13, 13–31.
- Simon, J. & Sirlin, C. (1989). Pure Appl. Chem. 61, 1625–1629.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536809048983/hb5232sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536809048983/hb5232Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report