


Abstract
RNA helicases of the DEAD box family are involved in almost all cellular processes involving RNA molecules. Here we describe functional characterization of the yeast RNA helicase Dbp8p (YHR169w). Our results show that Dbp8p is an essential nucleolar protein required for biogenesis of the small ribosomal subunit. In vivo depletion of Dbp8p resulted in a ribosomal subunit imbalance due to a deficit in 40S ribosomal subunits. Subsequent analyses of pre-rRNA processing by pulse–chase labeling, northern hybridization and primer extension revealed that the early steps of cleavage of the 35S precursor at sites A1 and A2 are inhibited and delayed at site A0. Synthesis of 18S rRNA, the RNA moiety of the 40S subunit, is thereby blocked in the absence of Dbp8p. The involvement of Dbp8p as a bona fide RNA helicase in ribosome biogenesis is strongly supported by the loss of Dbp8p in vivo function obtained by site-directed mutagenesis of some conserved motifs carrying the enzymatic properties of the protein family.
INTRODUCTION
RNA helicases of the DEAD box protein family are present in almost all organisms and over 100 such proteins from a variety of species are found in the sequence databases. The genome of the yeast Saccharomyces cerevisiae by itself contains 26 DEAD box proteins. They are characterized by the presence of eight conserved motifs (1,2) grouped in a central core domain and flanked by poorly conserved N- and C-terminal extensions. Based on sequence variations within the conserved motifs, related proteins are grouped in the DEAH box family, the Ski2p family, the Snf2p family or the Upf1p family. Several DEAD box and DEAH box proteins have been shown to possess in vitro helicase activity, i.e. they can unwind short duplex RNAs in an ATP-dependent fashion (3–8). Furthermore, site-directed mutagenesis combined with in vitro enzymatic assays allowed the assignment of specific functions, like binding of ATP, ATP hydrolysis and RNA helicase activity, to some of the conserved motifs (9–11). Regarding the highly variable N- and C-terminal extensions, they may confer substrate specificity (12), direct the protein to its subcellular localization (13) or bind accessory proteins.
So far, DEAD box proteins and their relatives have been shown to be involved in pre-mRNA splicing, ribosome biogenesis, RNA export from the nucleus, translation initiation and RNA decay, but in most cases their substrates and thus their precise functions have still not been elucidated. Ribosome biogenesis is the cellular process that employs by far the largest number of putative RNA helicases. Indeed, 15 of these proteins from yeast have been reported to be involved in this process [Dbp3p (13), Fal1p (14), Rrp3p (15), Rok1p (16), Drs1p (17), Dbp10p (18), Dbp4p (19), Spb4p (20), Mak5p (21), Dbp6p (22), Dbp7p (23), Dob1p (24), Dhr1p (25), Dhr2p (25) and Sen1p (26)]. Synthesis of ribosomes represents a major metabolic activity in the eukaryotic cell (27). It has been calculated that an exponentially growing yeast cell contains ∼200 000 ribosomes and that up to 50% of the RNA polymerase II activity is devoted to transcription of ribosomal protein (r-protein) encoding genes. In addition, rRNA is encoded by 120–200 repeats on chromosome XII, representing ∼10% of the yeast genome. These repeats are actively transcribed by RNA polymerase I to give rise to a large pre-rRNA precursor, which contains, in addition to the mature 18S, 5.8S and 25S rRNAs, external and internal spacer sequences that have to be accurately removed by a series of ordered endo- and exonucleolytic cleavages during pre-rRNA processing (Fig. 1). Furthermore, very early in ribosome biogenesis rRNA sequences within the pre-rRNA are modified by pseudo-uridinylation as well as by base and ribose methylation. These modifications are guided by approximately 100 small nucleolar RNAs (snoRNAs) that hybridize to specific regions of the pre-rRNA. In addition, pre-rRNA processing and ribosome assembly are tightly linked and during transcription of the large pre-rRNA precursor, trans-acting factors and ribosomal proteins are already associated with the nascent transcript. Over 65 trans-acting factors involved in ribosome biogenesis have been characterized in S.cerevisiae and the mature ribosome contains 32 and 46 r-proteins in the small and large ribosomal subunits, respectively (28,29).
Figure 1.

Pre-rRNA processing in S.cerevisiae. (A) Structure and processing sites of the 35S pre-rRNA. This precursor contains the sequences for the mature 18S, 5.8S and 25S rRNAs, which are separated by the two internal transcribed spacers ITS1 and ITS2 and flanked by the two external spacers 5′-ETS and 3′-ETS. The location of the various probes (labeled B–I) used in this study are indicated. (B) Pre-rRNA processing pathway. The 35S pre-rRNA is cleaved at site A0 by the endonuclease Rnt1p generating the 33S pre-rRNA. This molecule is subsequently processed at sites A1 and A2 to give rise to the 20S and 27SA2 precursors, resulting in separation of the pre-rRNAs destined for the small and large ribosomal subunits. It is thought that the early pre-rRNA cleavages A0–A2 are carried out by a large snoRNP complex, which is likely to be assisted by the putative ATP-dependent RNA helicases Dbp4p, Fal1p, Rok1p, Rrp3p, Dhr1p, Dhr2p and Dbp8p. Final maturation of the 20S precursor takes place in the cytoplasm, where an endonucleolytic cleavage at site D yields the mature 18S rRNA. The 27SA2 precursor is processed by two alternative pathways that both lead to the formation of mature 5.8S and 25S rRNAs. In the major pathway the 27SA2 precursor is cleaved at site A3 by RNase MRP. The putative ATP-dependent RNA helicase Dbp3p assists in this processing step. The 27SA3 precursor is 5′→3′ exonucleolytically digested up to site B1S to yield the 27SBS precursor, a reaction requiring the exonucleases Xrn1p and Rat1p. A minor pathway processes the 27SA2 molecule at site B1L, producing the 27SBL pre-rRNA. While processing at site B1 is completed, the 3′-end of mature 25S rRNA is generated by processing at site B2. The subsequent ITS2 processing of both 27SB species appears to be identical. Cleavage at sites C1 and C2 releases the mature 25S rRNA and the 7S pre-rRNA. The latter undergoes exosome-dependent 3′→5′ exonuclease digestion to the 3′-end of the mature 5.8S rRNA; this reaction also requires the putative ATP-dependent RNA helicase Dob1p. Assembly of the 60S subunit and the associated pre-rRNA processing reactions requires four other putative ATP-dependent RNA helicases: Dbp6p, Dbp7p, Dbp10p and Spb4p. The roles of the Drs1p and Mak5p proteins have not yet been defined. Only trans-acting factors belonging to the class of putative RNA helicases are indicated. For reviews on pre-rRNA processing and trans-acting factors see Kressler et al. (28) and Venema and Tollervey (29).
Thus, ribosome biogenesis is a very complex process that requires a multitude of RNA–RNA and RNA–protein interactions and involves numerous structural rearrangements. Furthermore, due to the high demand for ribosomes in the cell the process of ribosome biogenesis needs to be precisely coordinated. One can assume then that putative RNA helicases play a crucial role both in the structural rearrangements and as directional driving forces during this process. They are probably required for melting local rRNA secondary structures to allow coordinated binding of r-proteins or trans-acting factors and appropriate formation of mutually exclusive RNA–RNA interactions. Moreover, RNA helicases may be involved in pairing and dissociation of the guide snoRNAs. Indeed, Dbp4p has been isolated in a screen for suppressors of a snoRNA (U14) mutant (19) and Dhr1p has been shown to be associated with the snoRNA U3 (25). They may also act as cofactors of nucleolytic activities, as has been shown for Dob1p and the exosome (24). Finally, it cannot be excluded that some of the putative RNA helicases are involved in rearrangements of RNA–protein interactions.
To better understand the process of ribosome biogenesis and to fully characterize the DEAD box protein family of putative RNA helicases we analyzed the gene YHR169w, which we rename DBP8 for DEAD box protein 8 encoding gene.
MATERIALS AND METHODS
Strains, plasmids, oligonucleotides, media and microbiological methods
Saccharomyces cerevisiae strains and plasmids used in this study are described in Table 1 and in the following sections. The oligonucleotides used in this study are listed in Table 2. Genetic manipulations and preparation of media were according to established procedures (31,32). Yeast cells were transformed using the lithium acetate method (33). Escherichia coli DH10B was used for all recombinant DNA techniques (34). Tetrad dissections were performed using a Singer MS manual micromanipulator.
Table 1. Yeast strains and plasmids used in this study.
| Yeast straina |
Relevant genotype |
Reference |
| W303 | MATa/MATα ura3-1/ura3-1 ade2-1/ade2-1 his3-11,15/his-11,15 leu2-3,112/leu2-3,112 trp1-1/trp1-1 | (30) |
| MCD8H | DBP8/Δdbp8::HIS3MX6 | This study |
| MCD8H-3Ab | MATα DBP8 | This study |
| MCD8H-3Cc |
MATα Δdbp8::HIS3MX6 [pMCD8-1] |
This study |
| Plasmidd |
Plasmid description |
Auxotrophic marker |
| pMCD8-1 | YCplac33-DBP83 | URA3 |
| pMCD8-2 | YCplac111-GAL::HADBP8 (pAS24-DBP8) | LEU2 |
| pMCD8-4 | YCplac33-HADBP8 | URA3 |
| pMCD8-6 | YCplac111-HADBP8 | LEU2 |
| pMCD8-7 | YCplac111-DBP8 l | LEU2 |
| PMCD8-8 | YCplac111-ProtA DBP8 | LEU2 |
| pMCD8-9A | YCplac111-dbp8-1 | LEU2 |
| pMCD8-9B | YCplac111-ProtA dbp8-1 | LEU2 |
| pMCD8-10A | YCplac111-dbp8-2 | LEU2 |
| pMCD8-10B | YCplac111-ProtA dbp8-2 | LEU2 |
| pMCD8-11A | YCplac111-dbp8-3 | LEU2 |
| pMCD8-11B | YCplac111-ProtA dbp8-3 | LEU2 |
| pMCD8-12A | YCplac111-dbp8-4 | LEU2 |
| pMCD8-12B | YCplac111-ProtA dbp8-4 | LEU2 |
aAll strains are derivatives of W303.
bWild-type meiotic segregant of MCD8H.
cΔdbp8 meiotic segregant of MCD8H requiring a plasmid-borne DBP8 allele to support growth. Depending on the experimental conditions and as indicated in the text, the original plasmid pMCD8-1 was replaced by other DBP8-containing plasmids.
dAll plasmids were derivatives of the centromeric YCplac series of plasmids (36).
Table 2. Oligonucleotides used in this study.
| Name |
Sequence (5′→3′) |
Remarks |
| DBP8-P1 | GCTTTAGATCTCCGTAGCATCATATCAGAAACTTTTCAAGATGCGTACGCTGCAGGTCGAC | SFH–PCR disruption of DBP8 |
| DBP8-P2 | GTACGATATGTACGTCTTTAAATCTTATTTAGA ATTTTTCAATCGATGAATTCGAGCTCG | SFH–PCR disruption of DBP8 |
| DBP8-G1 | GGGGCCCCGTCGACATGGCAGACTTTAAATCTT | GAL::DBP8 |
| DBP8-G2 | GGGGCCCCAAGCTTGTGTTATGTACCAGGCG | GAL::DBP8 |
| DBP8-P3 | ATCCTTGGGAGGTAGAGCACT | HA-DBP8 |
| DBP8-P4 | GCGGGCTATCCCTATGACGTCCCGGACTATGCAGCAGACTTTAAATCTTTAGGTC | HA-DBP8 |
| DBP8-P5 | GTCATAGGGATAGCCCGCATAGTCAGGAACATCGTATGGGTATCTAGATGCCATCTTGAAAAGTTTCTGATATG | HA-DBP8 |
| DBP8-P6 | GGCTCTAGATCCGGATCCGCAGACTTTAAATCTTTAGGTCTT | pMCD8-7 |
| DBP8-A1 | GCCAAGACTGGTTCTGCAGCAACTATTGCATTTGCAGGG | dbp8-1 |
| DBP8-A2 | CCCTGCAAATGCAATAGTTGCTGCAGAACCAGTCTTGGC | dbp8-1 |
| DBP8-B1 | GCTAAATATTTAGTTCTAGCTGCAGCCGATATTTTGCTAACTAGC | dbp8-2 |
| DBP8-B2 | GCTAGTTAGCAAAATATCGGCTGCAGCTAGAAC TAAATATTTAGC | dbp8-2 |
| DBP8-C1 | GACAAACACTTCTATTCGCTGCAGCCATAACG GACCAAGTAAAG | dbp8-3 |
| DBP8-C2 | CTTTACTTGGTCCGTTATGGCTGCAGCGAATAGAAGTGTTTGTC | dbp8-3 |
| DBP8-D1 | CCAGATGTATTCATCCATGCTGCAGGTCGTACGGCCCG | dbp8-4 |
| DBP8-D2 | CGGGCCGTACGACCTGCAGCATGGATGAATACATCTGG | dbp8-4 |
| I | CCA GAT AAC TAT CTT AAA AGA AGA AGC | Primer extension |
Disruption of the DBP8 gene
A HIS3MX6 marker cassette with short flanking DBP8 homology regions was synthesised by PCR (SFH–PCR) using Pfu polymerase (Stratagene), the plasmid pFA6a-HIS3MX6 as DNA template and the primer couple DBP8-P1 and DBP8-P2 (35). After concentration by ethanol precipitation, the SFH–PCR dbp8::HIS3MX6 product was transformed into the diploid strain W303 to substitute for the entire DBP8 coding sequence by homologous recombination. Transformants were selected on SD-His plates. Correct integration of the HIS3MX6 marker at the DBP8 genomic locus resulted in the heterozygous diploid strain MCD8H (DBP8/Δdbp8::HIS3MX6). MCD8H was further subjected to sporulation and tetrad analysis.
Cloning of the DBP8 gene
The DBP8 gene was cloned from cosmid 9986 (chromosome VII, coordinates 423778–459166) purchased from ATCC. Briefly, after complete digestion of the cosmid with HindIII, a restriction fragment of 8.3 kb was purified from an agarose gel (Gene Clean; BIO101 Inc.) and further digested with SphI and EcoRI. A SphI–EcoRI DNA fragment of 3 kb containing the DBP8 gene was purified from an agarose gel and subcloned into SphI and EcoRI-restricted YCplac33 (36), yielding plasmid pMCD8-1. The haploid strain MCD8H-3C [pMCD8-1] (Δdbp8), which was obtained after sporulation and tetrad dissection of MCD8H transformed with pMCD8-1, exhibited a wild-type growth behavior under all the conditions tested, confirming that the plasmid-borne DBP8 allele fully complemented the dbp8-null allele.
Construction of the GAL::DBP8 allele and in vivo depletion of Dbp8p
DBP8 was amplified by PCR using a high fidelity DNA polymerase (Pfu Turbo DNA polymerase; Stratagene), plasmid pMCD8-1 as the DNA template and the primer couple DBP8-G1 and DBP8-G2 to introduce SalI and HindIII restriction sites. The PCR product was digested with SalI and HindIII and cloned into SalI and HindIII-restricted plasmid pAS24 (37), yielding plasmid pMCD8-2 in which expression of HA-DBP8 is under control of the GAL1-10 promoter.
The GAL::DBP8 strain was obtained after transformation of MCD8H-3C [pMCD8-1] with pMCD8-2 followed by plasmid shuffling on SGal-Leu plates containing 5-fluoroorotic acid (5-FOA). Growth behavior on galactose and glucose media was further studied to test for faithful complementation and shut off under non-permissive conditions.
For in vivo depletion the GAL::DBP8 strain was grown in YPGal medium at 30°C until mid exponential phase (OD600 = 1) and then inoculated in YPD medium. Cell growth was monitored over a period of 36 h during which the culture was regularly diluted into fresh YPD medium to maintain exponential growth. At different time points samples were taken to perform protein and RNA extractions and polysome analysis.
Sucrose gradient analyses
Polysome analysis and ribosome subunit quantification were performed as previously described (14).
RNA analysis
Pulse–chase labeling of pre-rRNA and RNA preparations were performed as described previously (22). Steady-state levels of pre-rRNAs were assessed by northern and primer extension analyses according to Venema et al. (38). For the primer extension, performed with primer I, a 10-fold molar excess of cold primer was mixed with the radiolabeled primer immediately before annealing. The oligonucleotides used for northern analysis and primer extension were as previously described (18), except for oligonucleotide I, which is described in Table 2. For localization of the oligonucleotides see Figure 1A.
In situ localization of HA–Dbp8p
A plasmid-borne, HA-epitope-tagged DBP8 allele was constructed by fusion PCR (39). Briefly, two DNA fragments (A and B) with sequence overlap were generated in a first PCR series using Pfu polymerase, plasmid pMCD8-1 as the DNA template and the two primer couples described in Table 2: DBP8-P3 and DBP8-P4, DBP8-P5 and a 24mer reverse sequencing primer (–48; New England Biolabs). Fusion PCR was performed using gel-purified fragments A and B as the DNA template and DBP8-P3 and the reverse sequencing primer as primers. An AflII DNA fragment of 1.26 kb from the fusion PCR product and containing the DBP8 promoter region, the HA epitope and the 15 first codons of the DBP8 ORF was subcloned into AflII-restricted plasmid pMCD8-1, yielding plasmid pMCD8-4. A SphI–EcoRI DNA fragment of 3 kb from pMCD8-4 was further subcloned into SphI and EcoRI-restricted plasmid YCplac111, yielding pMCD8-6.
The HA-DBP8 strain was obtained after transformation of MCD8-3H [pMCD8-1] with pMCD8-6 followed by plasmid shuffling on SD-Leu plates containing 5-FOA. The plasmid-borne HA-DBP8 allele complemented the dbp8-null allele to the wild-type extent under all tested conditions. Western blot analysis with monoclonal mouse anti-HA antibodies detected a single protein, migrating at the expected apparent molecular mass of 50 kDa, in whole cell lysates of the HA-DBP8 strain but not of the control DBP8 strain.
Indirect immunofluorescence analyses were performed with the HA-DBP8 strain as previously described (22), except that the secondary antibodies were goat anti-mouse fluorescein-conjugated antibodies and goat anti-rabbit rhodamine-conjugated antibodies (dilution 1/200) (Pierce).
Contruction of a plasmid-borne ProtA-DBP8 allele and site-directed mutagenesis of DBP8
A XbaI site blocked by dam methylation followed by an in-frame BamHI site were created by PCR at the 5′-end of the DBP8 ORF. PCR was performed using pMCD8-1 as the DNA template and primers DBP8-P3 and DPB8-P6. The XbaI fragment was subcloned into XbaI-restricted plasmid pMCD8-6, yielding pMCD8-7. The resulting plasmid-borne DBP8 allele harbors in-frame XbaI and BamHI restriction sites following the ATG and drives expression of a Dbp8p with six additional amino acids in its N-teminal part (SRSGSA). It is a fully functional allele that complemented the dbp8-null allele to the wild-type extent under all conditions tested. A 390 bp XbaI–BamHI fragment from plasmid pRAT1, encoding two IgG-binding domains of Staphylococcus aureus protein A (ProtA), and the 461 bp BamHI–XbaI insert from pMCD8-7 were co-ligated into XbaI-restricted plasmid pMCD8-7, yielding plasmid pMCD8-8. This resulted in fusion of the intact DBP8 coding sequence in-frame to the ProtA IgG-binding domain under control of the cognate DBP8 promoter. The ProtA-DBP8 strain was obtained by transformation of MCD8H-3C [pMCD8-1] followed by plasmid shuffling. The plasmid-borne ProtA-DBP8 allele complemented the dbp8-null allele to the wild-type extent under all conditions tested. Western blot analysis with rabbit serum detected a single protein migrating at the expected apparent molecular mass of 63 kDa in whole cell lysates of the ProtA-DBP8 strain but not of the control DBP8 strain.
Site-directed mutagenesis of DBP8 was performed using a QuickChange Site-Directed Mutagenesis kit (Stratagene) according to the procedure provided by the manufacturer. Briefly, a couple of complementary primers (described in Table 2) carrying the mutation to be introduced were used in a PCR reaction using Pfu polymerase and plasmid pMCD8-7 as the DNA template. After digestion with DpnI restriction enzyme, the PCR product was transformed in Escherichia coli and plasmids were recovered. DNA restriction fragments carrying the mutations were checked by sequencing and subcloned into plasmids pMCD8-7 and pMCD8-8. The resulting plasmid-borne dbp8 mutant alleles and plasmid-borne ProtA-dbp8 mutant alleles were constructed as follows. The dbp8-1 allele corresponds to the change AX4GKT→AX4AAT. A 461 bp BamHI–XbaI insert carrying the mutation was subcloned into BamHI and XbaI-restricted plasmid pMCD8-7, yielding plasmid pMCD8-9A. The 1.3 kb BamHI–SphI insert from pMCD8-9A was subcloned into BamHI and SphI-restricted plasmid pMCD8-8, yielding plasmid pMCD8-9B (see Table 1).
The dbp8-2 allele corresponds to the change DEAD →AAAD. A 799 bp BamHI–SacII insert carrying the mutation was subcloned into BamHI and SacII-restricted plasmids pMCD8-7 and pMCD8-8, yielding plasmids pMCD8-10A and pMCD8-10B, respectively.
The dbp8-3 allele corresponds to the change TAT→AAA. A 338 bp XbaI–SacII insert carrying the mutation was subcloned into XbaI and SacII-restricted plasmid pMCD8-7, yielding plasmid pMCD9-11A. The 1.7 kb SphI–BamHI insert from pMCD8-8 was subcloned into SphI and BamHI-restricted plasmid pMCD8-11A, yielding plasmid pMCD8-11B.
The dbp8-4 allele corresponds to the change HRSGR→HAAGR. A 233 bp SacII–BsiWI insert carrying the mutation was subcloned into SacII and BsiWI-restricted plasmids pMCD8-7 and pMCD8-8, yielding plasmids pMCD8-12A and pMCD8-12B, respectively.
The plasmid-borne dbp8 mutant alleles were transformed into strain MCD8-3H [pMCD8-1] and tested by plasmid shuffling on 5-FOA plates. Whole cell lysates of strains carrying the ProtA-dbp8 mutant alleles were analyzed by western blot using rabbit serum and goat anti-rabbit alkaline phosphatase-conjugated antibodies.
RESULTS
Dbp8p is a putative RNA helicase essential for vegetative growth
DBP8 (YHR169W) has been identified during an ‘in silico’ screen for new members of the S.cerevisiae family of putative ATP-dependent RNA helicases (23). It is located on the right arm of chromosome VIII and encodes a basic protein of 431 amino acids with a predicted pI of 9.78 and a predicted mass of 47.8 kDa. Dbp8p contains the eight conserved motifs constituting the so-called helicase core region characteristic of the DEAD box proteins (Fig. 2). In addition to this core region, Dbp8p has relatively short N- and C-terminal extensions that do not contain any obvious structural features. BLAST alignments for Dbp8p versus the SwissProt database identified proteins from Homo sapiens (Q9Y6V7), Arabidopsis thaliana (Q9SA27) and Drosophila melanogaster (Q9V528) as potential orthologs of Dbp8p. The human and yeast proteins are both relatively short in size for RNA helicases (483 and 431 amino acids, respectively) and their homology (48% identity, 65% similarity) is not restricted to the helicase core region but also covers the N- and C-terminal domains. The homologies for the plant and fly proteins are 44% identity/63% similarity and 42% identity/58% similarity, respectively.
Figure 2.

Alignment of the yeast and human Dbp8p proteins. Identical residues are marked * below the sequence. The eight highly conserved motifs are indicated by a bar in between the sequences. It is important to note that identical residues are found throughout the two proteins including the N- and C-terminal extension of the helicase core domain. The last 15 residues of the human protein are not shown. The alignment was performed using ClustalW at EBI (www.ebi.ac).
In order to gain an insight into the cellular functions of Dbp8p, one of the DBP8 ORFs of the diploid strain W303 was deleted and replaced by the heterologous marker HIS3MX6 using a one-step, PCR-based strategy. Upon sporulation of the resulting heterozygous diploid strain MCD8H (Δdbp8/DBP8) a 2:2 segregation for viability was observed and no His+ spores were recovered, indicating that DBP8 is an essential gene. After transformation with pMCD8-1, a URA3 centromeric plasmid harboring the cognate DBP8 allele, and sporulation MCD8H yielded mostly tetrads of four viable spores with all the His+ spores harboring plasmid pMCD8-1. The complete lack of growth of this progeny on 5-FOA-containing medium further confirmed the absolute requirement for DBP8 for vegetative growth. Under all tested conditions the Δdbp8 haploid strain MCD8H-3C harboring plasmid pMCD8-1 exhibited the same growth behavior as an otherwise isogenic wild-type strain, indicating that the DBP8 plasmid-borne allele fully complemented the dbp8-null allele.
A conditional system to study the cellular functions of Dbp8p, a GAL::DBP8 strain
In order to further study the essential cellular functions of Dbp8p, we established a conditional system based on depletion of Dbp8p by transcriptional repression of a plasmid-borne GAL::DBP8 fusion gene. The galactose promoter drives expression of an N-terminally HA-tagged Dbp8p in galactose-based medium but this is repressed in glucose-containing medium. The GAL::DBP8 strain was obtained after transformation of the MCD8H-3C [pMCD8-1] strain with plasmid pMCD8-2 followed by plasmid shuffling on galactose plates containing 5-FOA. In YPGal medium the GAL::DBP8 strain had the same growth behavior as an otherwise isogenic wild-type strain, indicating that the GAL::DBP8 allele fully complemented the dbp8-null allele. As expected, only residual growth of the GAL::DBP8 strain was observed on YPD plates (Fig. 3A). After a shift from YPGal to YPD liquid medium the growth rate of the GAL::DBP8 strain remained similar to that of the wild-type strain for the first 12 h, then progressively decreased to reach a doubling time of ∼10 h after 36 h in YPD medium (Fig. 3B). As shown by western blot analysis, cellular depletion of Dbp8p was concomitant with the decrease in growth rate (Fig. 3C).
Figure 3.

Depletion of Dbp8p impairs growth of yeast cells. (A) Growth comparison of MCD8H-3A (wt), MCD8H-3C [pMCD8-1] (DBP8) and MCD8H-3C [pMCD8-2] (GAL::DBP8) strains. Freshly grown cells were streaked on YPGal (galactose) and YPD (glucose) plates and incubated at 30°C for 3 days. (B) Growth curves of MCD8H-3C [pMCD8-1] (DBP8; closed circles) and MCD8-3H [pMCD8-2] (GAL::DBP8; open circles) at 30°C after logarithmic cultures were shifted from YPGal to YPD medium for up to 36 h. Data are given as estimated doubling time values at different time points in YPD. (C) Western blot analysis of depletion of Dbp8p. Whole cell lysates of the GAL::DBP8 strain were prepared from samples harvested at the indicated times. Equal amounts of proteins were separated by 12.5% SDS–PAGE and HA–Dbp8p (indicated by an arrow) was detected by western blot using monoclonal mouse anti-HA antibody 16B12 and goat anti-mouse alkaline phosphatase-conjugated antibodies. Prestained broad range molecular weight markers (Bio-Rad) were used as standards for molecular mass determination.
In vivo depletion of Dbp8p leads to a 40S ribosomal subunit shortage
Amongst the DEAD box proteins that have already been characterized, several have been implicated in translation initiation and ribosome biogenesis (2). We first investigated by polysome profile analysis whether Dbp8p was involved in one of these two processes. The GAL::DBP8 and the wild-type strains were grown in parallel in YPGal medium at 30°C, then shifted to YPD medium and polysome extracts were prepared from cells harvested at different times. After 6 h in YPD medium the polysome profile of the GAL::DBP8 strain is very similar to that of the wild-type strain (Fig. 4A and B). In perfect correlation with the depletion of Dbp8p and the decrease in growth rate described before, differences in the GAL::DBP8 polysome profiles appeared after 12 h and were even more pronounced after 18 h in YPD medium. The size and number of polyribosomes dropped and the peak corresponding to the free 40S ribosomal subunit pool was nearly undetectable, whereas there was a strong increase in the free 60S ribosomal subunit pool (Fig. 4C and D). This reflected a ribosomal subunit imbalance due to a deficit in 40S ribosomal subunits, which was confirmed by quantifying total ribosomal subunits in polysome run-off and low Mg2+ cell extracts. The A254 60S:40S ratio measured for the GAL::DBP8 strain increased from 1.6 to 5.9 after 18 h in YPD medium, whereas it was around 1.6 and remained constant for the wild-type strain. These results suggest that Dbp8p plays a role in 40S ribosomal subunit metabolism.
Figure 4.

Depletion of Dbp8p results in a deficit in 40S ribosomal subunits and a decrease in polysomes. The DBP8 strain MCD8H-3C [pMCD8-1] (A) was grown in YPD at 30°C. The GAL::DBP8 strain MCD8H-3C [pMCD8-2] was grown at 30°C in YPGal and shifted to YPD for 6 (B), 12 (C) and 18 h (D). Cells were harvested at an OD600 of 0.8 and similar amounts of cell extracts (12 A260 units) were resolved in 7–50% (w/v) sucrose gradients. Gradient analysis was performed with an ISCO UV-6 gradient collector with continuous monitoring at A254. Sedimentation is from left to right. The peaks of free 40S and 60S ribosomal subunits, 80S free couples/monosomes and polysomes are indicated. Due to a strong increase in the free 60S ribosomal subunit, the corresponding peak overlaps the free 80S/monosome peak in (D).
Formation of the 18S rRNA is impaired upon Dbp8p in vivo depletion
The biogenesis of ribosomal subunits is tightly linked to the formation of the rRNAs. Therefore, to further investigate the 40S ribosomal subunit shortage phenotype of the GAL::DBP8 strain we analyzed the effects of depletion of Dbp8p on synthesis and processing of pre-rRNA. For that purpose, [5,6-3H]uracil pulse–chase labeling experiments were performed on wild-type and GAL::DBP8 strains after ∼20 h depletion in glucose-containing medium.
In the wild-type strain the 35S precursor was rapidly converted into the 32S pre-rRNA and then into the 27S and 20S pre-rRNAs, which were subsequently processed to the mature 25S and 18S rRNAs, respectively (Fig. 5, lanes 1–5). Within the first minutes of chase the radioactive 35S pre-rRNA was entirely processed and the labeled mature rRNAs reached their maximum levels after 5 min, when most of the labeled precursors had disappeared. In the GAL::DBP8 strain processing was clearly delayed, since a significant level of labeled 35S precursor persisted after >30 min of chase. The radioactive 35S pre-rRNA was converted into labeled 27SB precursor and an aberrant 23S molecule, but neither 27SA nor 20S labeled precursors were detectable. The 27SB precursor was processed following the same kinetics as in the wild-type strain and resulted in similar levels of labeled mature 25S rRNA. In contrast, only very little labeled 18S mature rRNA was formed. Similar experiments performed with [methyl-3H]methionine revealed that the overall methylation of the pre-rRNAs was unaffected upon Dbp8p depletion (data not shown). The formation of low molecular weight RNAs (5S and 5.8S rRNAs and tRNAs) was also unaffected (data not shown).
Figure 5.
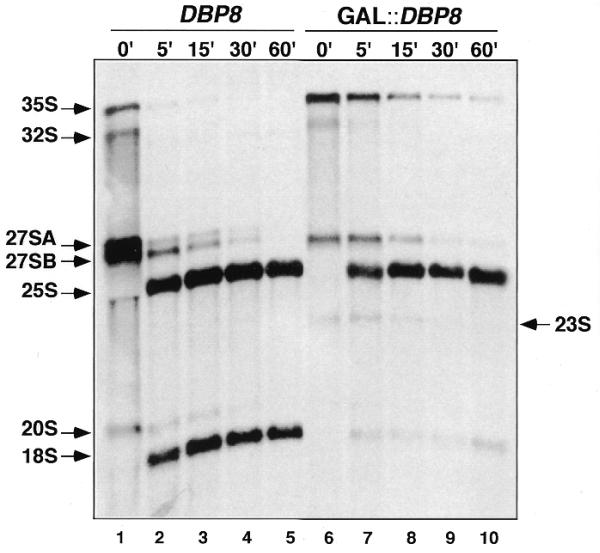
Synthesis of the mature 18S rRNA is strongly reduced in Dbp8p-depleted cells. Strains MCD8H-3C [pMCD8-1] (DBP8) and MCD8H-3C [pMCD8-2, YCplac33] (GAL::DBP8) were grown at 30°C in SGal-Ura then shifted to SD-Ura for 20 h. Cells were pulse labeled with [5,6-3H]uracil for 2 min and then chased with an excess of cold uracil. Total RNA was extracted from cell samples harvested at the indicated chase time points, resolved on a 1.2% agarose–6% formaldehyde gel, transferred to nylon membrane and visualized by fluorography. Approximately 25 000 c.p.m. was loaded in each lane. The positions of the different pre-rRNAs and mature rRNAs are indicated.
These results show that the deficit of 40S ribosomal subunits observed upon Dbp8p depletion is due to impaired production of mature 18S rRNA and strongly suggest that Dbp8p is involved in 40S ribosomal subunit biogenesis.
Dbp8p is required for pre-rRNA processing at A0, A1 and A2
To further define the pre-rRNA processing steps that are affected upon Dbp8p depletion, steady-state levels of mature rRNAs and pre-rRNA intermediates were assessed by northern blot and primer extension analyses. Different oligonucleotides hybridizing to defined regions of the 35S pre-rRNA transcript (Fig. 6A) were used to monitor specific processing intermediates in the GAL::DBP8 strain during the time course of depletion of Dbp8p and in an isogenic wild-type strain as a control. As seen in Figure 6A, depletion of Dbp8p resulted in a drastic decrease in mature 18S rRNA steady-state level, whereas that of mature 25S rRNA was not affected. Probing with oligonucleotide B, which hybridizes 5′ upstream of site A0, revealed that with ongoing Dbp8p depletion the 35S pre-rRNA and an aberrant 23S processing product accumulated (Fig. 6B). The increase in 35S pre-rRNA steady-state level, detected by all the other precursor-specific probes used (Fig. 6C–F), indicated that cleavage at site A0 is impaired or at least delayed in the absence of Dbp8p. The aberrant 23S processing product could still be detected by oligonucleotides C (Fig. 6C) and D (Fig. 6D) but not with oligonucleotide E (Fig. 6E). Thus, it extends from the 5′-end of the 5′-external transcribed spacers (ETS) to the A3 site and originates from direct cleavage of the 35S precursor at site A3 within internal transcribed spacer 1 (ITS), which occurs in the absence of cleavages at sites A0, A1 and A2 (29). Consistent with generation of the 23S processing product, we observed that the steady-state levels of the 27SA2 pre-rRNA and 20S pre-rRNA, which is the direct precursor of mature 18S rRNA, were strongly reduced upon Dbp8p depletion (Fig. 6C–E). Only a slight decrease in 32S pre-rRNA levels was observed upon Dbp8p depletion, probably due to the faint detection of this species in our northern hybridization. Depletion of these pre-rRNA species, products of cleavages at sites A1 and A2, indicated that these cleavage steps were impaired in the absence of Dbp8p. In contrast, as shown by hybridization with oligonucleotide F (Fig. 6F) and G (data not shown), the steady-state level of the 27SB processing intermediates remained unchanged during depletion, indicating that the major subsequent processing step, cleavage at site A3, was unaffected. Moreover, northern blot analyses of low molecular weight RNAs using oligonucleotides G and H revealed that the steady-state levels of 7S pre-rRNA and mature 5.8S rRNA were similar in wild-type and Dbp8p-depleted cells, indicating that processing in ITS2 was also unaffected (data not shown).
Figure 6.

Depletion of Dbp8p affects the steady-state levels of pre-rRNA and mature rRNA species. Strains MCD8H-3C [pMCD8-1] (DBP8) and MCD8H-3C [pMCD8-2] (GAL::DBP8) were grown in YPGal then shifted to YPD. Cells were harvested at the indicated times and total RNAs were extracted. Equal amounts (5 µg) of total RNA were resolved on a 1.2% agarose–6% formaldehyde gel and transferred to a nylon membrane. The same membrane was stained with methylene blue (A) and then consecutively hybridized with the different probes B–F indicated in Figure 1A (B–F, respectively). The positions of the different pre-rRNAs and mature rRNAs are indicated.
Detection of the 33S and 27SA3 pre-rRNA species, discrimination between the 27SBL and 27SBS precursors and confirmation of the accuracy of cleavage, which are not amenable to northern hybridization, were performed by primer extension analyses. The 33S pre-rRNA steady-state level, as shown by the primer extension stop at site A0, seems to rise after 12 h of Dbp8p depletion and then returns progressively to initial levels. However, as described above, the appearance of the 23S processing intermediate showed that cleavage at site A0 was clearly delayed in the Dbp8p-depleted strain. The levels of 27SA3, 27SBL and 27SBS pre-rRNAs, shown by primer extension stops A3, B1L and B1S, respectively, remained unchanged during Dbp8p depletion (Fig. 7B and C). In contrast, and consistent with the results of northern hybridization, the steady-state level of 27SA2 pre-rRNA, shown by the primer extension stop at site A2, dropped after 12 h of depletion (Fig. 7B). The accuracy of cleavage at sites A0, A1, A2, A3, B1L and B1S was conserved upon Dbp8p depletion (Fig. 7 and data not shown).
Figure 7.
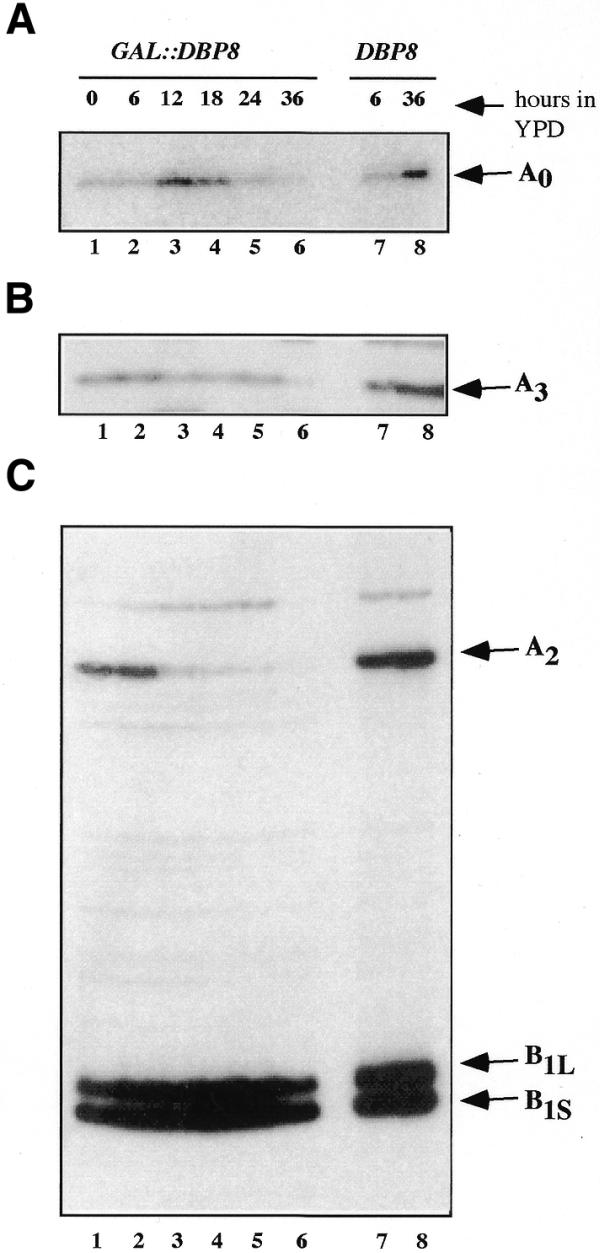
Primer extension analyses of the 27S precursors and the 33S pre-rRNA. The same samples of total RNA analyzed by northern blot in Figure 6 were used for primer extension analyses. (A) Primer extension through 5′-ETS was performed using oligonucleotide I and allows detection of the 33S pre-rRNA (site A0). (B) Primer extension using oligonucleotide E in ITS1 allows detection of the 27SA3 pre-rRNA (site A3). (C) Primer extension using oligonucleotide G in ITS2 reveals processing sites A2, B1L and B1S, allowing detection of 27SA2, 27SBL and 27SBL pre-rRNA species, respectively. The elevated signals in lane 8 (wt 36 h) are due to erroneously elevated RNA levels loaded on the gel.
Taken together, the results indicate that Dbp8p is essential for the processing cleavages at sites A1 and A2 and is required for optimal cleavage at site A0. Subsequent processing reactions in ITS1 and ITS2 leading to the production of 25S and 5.8S rRNAs do not require Dbp8p. Furthermore, the steady-state levels of the snoRNAs U3, U14, snR10 and snR30, which are amongst the trans-acting factors involved in the A0–A2 processing steps, were not affected by depletion of Dbp8p (data not shown), which strengthens a direct involvement of Dbp8p in these pre-rRNA processing steps.
Dbp8p is localized in the nucleolus
To determine the subcellular localization of Dbp8p, an epitope-tagged HA-DBP8 allele under the control of its cognate promoter was constructed. This allele was cloned into a LEU2 centromeric plasmid yielding pMCD8-6. The HA-DBP8 strain was obtained after transformation of the MCD8H-3C [pMCD8-1] strain with pMCD8-6 followed by plasmid shuffling. Under all conditions tested the HA-DBP8 strain exhibited the same growth behavior as an otherwise isogenic wild-type strain, demonstrating that the HA–Dbp8p protein was fully functional. Western blot analysis performed with primary monoclonal mouse anti-HA antibodies and secondary goat anti-mouse alkaline phosphatase-conjugated antibodies detected a single protein with an estimated molecular mass of 55 kDa in a whole cell lysate of the HA-DBP8 strain but not from the control strain (data not shown). Indirect immunofluorescence was performed with the HA-DBP8 strain following growth in YPD medium to an OD600 of 0.6. Detection of the HA–Dbp8p protein was performed with monoclonal mouse anti-HA antibodies and goat anti-mouse fluorescein-conjugated antibodies (Fig. 8C). The DNA was stained with DAPI to visualize the nucleus (Fig. 8A) and the nucleolus was visualized by detection of Nop1p with polyclonal rabbit anti-Nop1 and goat anti-rabbit rhodamine-conjugated antibodies (Fig. 8B). Superimposition of the signals obtained from detection of HA–Dbp8p and Nop1p, respectively, is shown in Figure 8D. A typical crescent-shaped staining mostly excluded from DAPI stained areas and characteristic of the nucleolar structure was observed with anti-Nop1p antibodies (Fig. 8B). A highly similar staining was obtained using anti-HA antibodies and was confirmed by the perfect superimposition of both signals, demonstrating that Dbp8p is exclusively localized in the nucleolus (Fig. 8C and D). Only weak background staining was obtained with anti-HA antibodies on the control strain containing untagged Dbp8p (data not shown).
Figure 8.
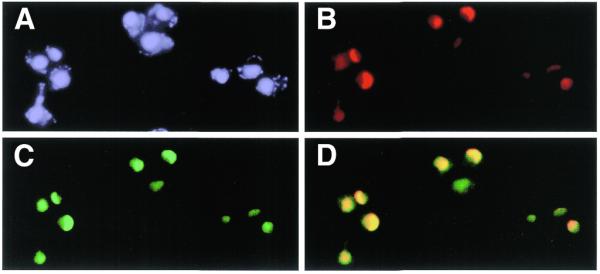
HA-Dbp8p localizes to the nucleolus. Indirect immunofluorescence was performed with cells expressing HA–Dbp8p from the DBP8 promoter (MCD8-3C [pMCD8-6]). (A) DNA was stained with DAPI. (B) The nucleolar protein Nop1 was detected with polyclonal rabbit anti-Nop1p antibodies, followed by decoration with goat anti-rabbit rhodamine-conjugated antibodies. (C) HA–Dbp8p was detected with monoclonal mouse anti-HA antibody 16B12, followed by decoration with goat anti-mouse fluorescein-conjugated antibodies. (D) Superimposition of the signals obtained from Nop1p and HA–Dbp8p.
The nucleolar localization of Dbp8p is again consistent with a direct involvement of the protein in pre-rRNA processing and ribosome biogenesis.
Genetic evidence that Dbp8p acts as an RNA helicase
As a member of the DEAD box protein family, Dbp8p contains all the sequence motifs of an ATP-dependent RNA helicase. Furthermore, for this class of enzymes specific activities like ATP and RNA binding, ATP hydrolysis and RNA unwinding have been assigned to some of the highly conserved motifs (2). To determine whether these motifs play a role in in vivo Dbp8p function we modified them by site-directed mutagenesis (Fig. 9A). Walker motifs A (AX4GKT) and B (DEAD), involved in ATP binding and hydrolysis, were modified to AX4AAT (dbp8-1 allele) and DAAD (dbp8-2 allele), respectively. Motif III (TAT), important for RNA unwinding, was converted into AAA (dbp8-3 allele) and motif VI (HRSGR), implicated in ATP hydrolysis-dependent RNA interaction (11), was replaced by HAAGR (dbp8-4 allele). These four plasmid-borne mutant alleles were then introduced into the MCD8H-3C [pMCD8-1] strain and their functionality was tested through their capability to allow loss of the plasmid-borne DBP8 allele monitored on 5-FOA-containing medium. The dbp8-1 and dbp8-2 mutant alleles were unable to support growth at all the tested temperatures. The dbp8-3 mutant allele supported growth, but with a 2-fold increase in doubling time (∼3 versus 1.5 h for the wild-type strain at 30°C). Furthermore, it exhibited an altered polysome profile reflecting a 40S ribosomal subunit deficit similar to those observed during depletion of Dbp8p (Fig. 9B). In contrast, the dbp8-4 mutant allele behaved like the wild-type allele.
Figure 9.
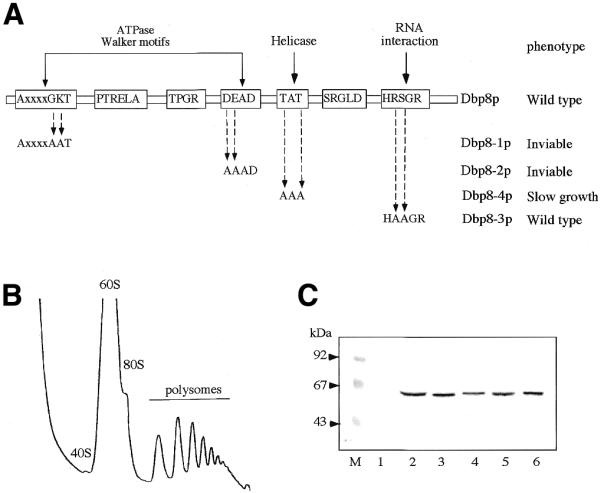
Site-directed mutagenesis of Dbp8p. (A) Four of the RNA helicase conserved motifs were individually altered by site-directed mutagenesis. The name of the mutant allele, the nature of the mutation and the associated growth phenotype are indicated. (B) Polysome analysis of MCD8-3C [pMCD8-11A] (dbp8-3) grown in YPD at 30°C. Twelve A260 units of cellular extract were resolved on a 7–50% sucrose gradient. Sedimentation is from left to right. The peaks of free 40S and 60S ribosomal subunits, 80S free couples/monosomes and polysomes are indicated. (C) Western blot analysis of the mutant proteins. Equal amounts of total protein extracts from MCD8H-3C [pMCD8-1] carrying, respectively, YCplac111 (lane 1), pMCD8-8 (ProtA–Dbp8p; lane 2), pMCD8-9B (ProtA–Dbp8-1; lane 3), pMCD9-10B (ProtA–Dbp8-2; lane 4), pMCD8-11B (ProtA–Dbp8-3; lane 5) and pMCD8-12A (ProtA–Dbp8-4; lane 6) were separated by 10% SDS–PAGE. The ProtA-tagged proteins were detected by western blot using rabbit immune serum and goat anti-rabbit alkaline phosphatase-conjugated antibodies. Pre-stained low range standard (Bio-Rad) was used as the standard for molecular mass determination.
To verify the level of expression of the mutant proteins, N-terminally ProtA-tagged versions of wild-type and mutant alleles were constructed, tested for growth under the same conditions as the untagged versions and fusion proteins were visualized by western blot. All four mutant proteins were stable and expressed to wild-type level (Fig. 9C).
These results reveal that at least two of the four conserved motifs studied are essential for in vivo Dbp8p function and strengthen the postulate that Dbp8p acts as an RNA helicase in the ribosome biogenesis process. The wild-type behavior of the dbp8-4 mutant indicates that the first arginine residue of motif VI is dispensable for Dbp8p function, however, it does not preclude the potential importance of other residues of the motif.
DISCUSSION
DEAD box proteins are involved in a variety of processes involving RNA molecules. To completely characterize the DEAD box family of RNA helicases from the yeast S.cerevisiae we analyzed the role of the DBP8 gene. The Dbp8p protein contains the eight conserved motifs of the DEAD box protein family. However, motif III has a conservative replacement of one of the amino acids (TAT instead of SAT) and motif VI is HRSGRxGR, whereas most helicases have a isoleucine or valine instead of the serine (except for Mak5p, Prp5p and Spb4p). The TAT sequence is also present in the Prp28p and Dbp3p proteins, involved in pre-mRNA splicing and ribosome biogenesis. Thus, the Dbp8p protein is a typical DEAD box protein despite these alterations. Comparison with sequence databases show significant homologies with proteins from H.sapiens, A.thaliana and D.melanogaster. This high evolutionary conservation between the yeast protein and its putative orthologs in several higher eukaryotes argues in favor of Dbp8p playing an essential role in a fundamental cellular process. Indeed, Dbp8p is involved in ribosome biogenesis, a process largely conserved in eukaryotes.
Since DBP8 is an essential gene, phenotypic analyses were done with a conditional system based on Dbp8p depletion by transcriptional repression of a plasmid-borne GAL::DBP8 gene. Our data clearly indicate that Dbp8p is involved in biogenesis of the 40S small ribosomal subunit. The first indication was from polysome analyses and quantification of the overall ribosomal subunits, which revealed that ongoing Dbp8p depletion was accompanied by a deficit in the 40S subunit pool leading to a ribosomal subunit imbalance, i.e. an increase in 60S relative to 40S subunits. Subsequent analysis of pre-rRNA processing by pulse–chase labeling showed that synthesis of 18S rRNA is impaired whereas 25S, 5.8S and 5S rRNAs are normally produced. A more detailed analysis of the different processing intermediates by northern hybridization and primer extension indicated that endonucleolytic cleavage of the 35S pre-rRNA at sites A1 and A2 is inhibited and at least delayed at site A0. This was reflected in the decrease in 32S, 20S and 27SA2 pre-rRNAs associated with accumulation of the 35S precursor and appearance of the aberrant 23S intermediate. As has been described for other proteins required for these early cleavages, such an inhibition results in direct cleavage of the 35S pre-rRNA precursor at site A3, producing a 23S aberrant intermediate and 27SA3 pre-rRNA. Whereas the latter follows the normal processing pathway resulting in production of the 25S and 5.8S mature rRNAs, the 23S species is not further processed to yield mature 18S rRNA. Therefore, depletion of Dbp8p results in a deficit of 18S rRNA compared to the other mature rRNAs. Furthermore, HA epitope-tagged Dbp8p was found to localize exclusively in the nucleolus, the cellular compartment where ribosome biogenesis mainly takes place, arguing for a direct role of Dbp8p in formation of 18S rRNA and the 40S ribosomal subunit.
Several other RNA helicases have been described as required for early cleavage of pre-rRNA. Depletion of Fal1p, Rok1p, Rrp3p and Dhr2p results in a more or less strong delay in cleavage at site A0 and inhibition of cleavage at sites A1 and A2, characterized by accumulation of the 35S pre-rRNA precursor and the 23S intermediate, as upon depletion of Dbp8p. So far, neither substrate nor interacting partner has been identified for any of these RNA helicases and their precise functions in ribosome biogenesis are still not elucidated. Upon depletion of Dhr1p, a DEAH box protein, cleavage at sites A1 and A2 is predominantly affected and the protein has been shown to be physically associated with U3 snoRNA (25). On the basis of these observations, a more precise function has been proposed for Dhr1p in pre-rRNA processing. Rok1p and Dbp4p interact genetically with the snoRNAs snR10 and U14, respectively, however, in both cases no physical interaction has been detected. Furthermore, a detailed characterization of the pre-rRNA processing defects associated with Dbp4p impairment is still lacking. As for the other RNA helicases affecting early cleavages, except Dhr1p, we cannot so far assign a precise function to Dbp8p in these early processing steps. We could, however, tell that accumulation of the four snoRNAs U3, U14, snR30 and snR10 involved in these processing steps is not impaired upon Dbp8p depletion (data not shown).
Although a helicase activity has not been demonstrated for any of these putative RNA helicases involved in early processing reactions, we strongly believe that Dbp8p has such an activity and that this activity is absolutely required for ribosome biogenesis. Two motifs have been shown to be critical for ATP binding and hydrolysis, motif I (AxxGxGKT) and motif II (DEAD). Mutations in motifs I and II completely abolish the in vivo function of Dbp8p. Based on mutations made in RAD3 and eIF4A (10,40,41), the mutation in motif I is expected to abolish ATPase activity. Mutations in the DEAD motif have been shown to affect ATPase activity in Ded1p, eIF4A and many other putative helicases (3,10,40). Thus, these mutant phenotypes are in agreement with the requirement for ATPase activity for in vivo function of Dbp8p. The modification of motif III (TAT→AAA) within Dbp8p, although viable, severely affects growth. Furthermore, analysis of polysomes and ribosomal subunits from these mutant cells revealed a deficit of 40S ribosomal subunits similar to that observed upon Dbp8p depletion (Fig. 9B). Certain motif III mutations result in vivo in a dominant negative phenotype for Prp2p and Prp43p (42,43) and in a cold-sensitive phenotype for Prp28p (44). Motif III mutations in mouse eIF4A and Nph-II from vaccinia virus result in loss of their RNA helicase activity in vitro whereas their ATPase activity is poorly affected or even enhanced (10,45). This motif is thus considered as critical for RNA helicase activity. A mutation in motif VI (HRSGR→HAAGR) of Dbp8p has no effect on growth under the test conditions. Substitutions of R with E, G, I, S or T (but not K) in Tif1p (yeast eIF4A) completely abolished the in vivo activity of the protein and, in addition, resulted in a strong dominant negative phenotype when overexpressed (9,46). Similar results have been reported for mammalian eIF4A protein, where in vitro activity of the eIF4A protein is affected (11). In contrast, mutations of the first R in motif VI to K, I or T in the ROK1 gene did not reveal a phenotype (47). Thus, mutation of the first R in this motif may have drastic effects or may be silent. Indeed, other residues within this motif have been shown to be important, especially the second arginine residue. It was originally proposed that motif VI is involved in RNA binding. Although mutational analysis and in vitro assays using mammalian eIF4A were in accord with this postulate, similar analyses using the vaccinia NPH-II helicase protein showed an involvement in ATP hydrolysis but not in RNA binding (48). The structural data indicate that motif VI is required to hold the ATP in place (49–51). This nicely explains the results obtained from eIF4A and NPH-II, since the bound nucleotide influences the affinity for the RNA substrate (3,52,53). In the case of Dbp8p it is possible that the nucleotide can be held in place even after mutating the HRSGRxGR motif, due to compensatory structural elements present in this protein. On the other hand, affinity for the RNA substrate may not be regulated in the same way by the bound nucleotide, allowing the protein to function to a sufficient extent to allow almost wild-type growth.
Dbp8p is the seventh putative RNA helicase required for biogenesis of the 40S subunit and the sixteenth helicase required for ribosome biogenesis in general (Fig. 1). The question thus arises, why are so many RNA helicases required for ribosome biogenesis? In view of the massive production of ribosomes in the exponentially growing cell, it is not surprising that this process requires an optimal machinery. In this sense, RNA helicases may help to dissociate intra- and intermolecular RNA–RNA interactions for efficient rearrangements to take place. The use of highly specific helicases certainly contributes to optimization of the process. Moreover, recognition of specific substrate structures avoids backward reactions or inhibiting product–catalyst interactions. Thus, Dbp8p is a putative RNA helicase presumably involved in a very specific step of ribosome biogenesis. Further work is required to elucidate interacting proteins and its substrate.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dieter Kressler and Jesús de la Cruz for critical reading of the manuscript. We are grateful to E.C.Hurt for the kind gift of anti-Nop1 antibodies. This work was supported by a grant from the Swiss National Science Foundation (to P.L.).
References
- 1.Linder P., Lasko,P.F., Ashburner,M., Leroy,P., Nielsen,P.J., Nishi,K., Schnier,J. and Slonimski,P.P. (1989) Birth of the D-E-A-D box. Nature, 337, 121–122. [DOI] [PubMed] [Google Scholar]
- 2.de la Cruz J., Kressler,D. and Linder,P. (1999) Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci., 24, 192–198. [DOI] [PubMed] [Google Scholar]
- 3.Iost I., Dreyfus,M. and Linder,P. (1999) Ded1p, a DEAD-box protein required for translation initation in Saccharomyces cerevisiae, is an RNA helicase. J. Biol. Chem., 274, 17677–17683. [DOI] [PubMed] [Google Scholar]
- 4.Tseng S.S.-I., Weaver,P.L., Liu,Y., Hitomi,M., Tartakoff,A.M. and Chang,T.-H. (1998) Dbp5p, a cytosolic RNA helicase, is required for poly(A)+ RNA export. EMBO J., 17, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwer B. and Gross,C.H. (1998) Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J., 17, 2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., Wagner,J.D.O. and Guthrie,C. (1998) The DEAH-box splicing factor Prp16 unwinds RNA duplexes in vitro. Curr. Biol., 8, 441–451. [DOI] [PubMed] [Google Scholar]
- 7.Rozen F., Edery,I., Meerovitch,K., Dever,T.E., Merrick,W.C. and Sonenberg,N. (1990) Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol. Cell. Biol., 10, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogers G.W.J., Richter,N.J. and Merrick,W.C. (1999) Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem., 274, 12236–12244. [DOI] [PubMed] [Google Scholar]
- 9.Schmid S.R. and Linder,P. (1991) Translation initiation factor 4A from Saccharomyces cerevisiae: analysis of residues conserved in the D-E-A-D family of RNA helicases. Mol. Cell. Biol., 11, 3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pause A. and Sonenberg,N. (1992) Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J., 11, 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pause A., Méthot,N. and Sonenberg,N. (1993) The HRIGRXXR region of the DEAD box RNA helicase eukaryotic translation initiation factor 4A is required for RNA binding and ATP hydrolysis. Mol. Cell. Biol., 13, 6789–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y. and Guthrie,C. (1998) PRP16, a DEAH-box RNA helicase, is recruited to the spliceosome primarily via its nonconserved N-terminal domain. RNA, 4, 1216–1229. [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver P.L., Sun,C. and Chang,T.-H. (1997) Dbp3p, a putative RNA helicase in Saccharomyces cerevisiae, is required for efficient pre-rRNA processing predominantly at site A3. Mol. Cell. Biol., 17, 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kressler D., de la Cruz,J., Rojo,M. and Linder,P. (1997) Fal1p is an essential DEAD-box protein involved in 40S-ribosomal-subunit biogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol., 17, 7283–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Day C.L., Chavanikamannil,F. and Abelson,J. (1996) 18S rRNA processing requires the RNA helicase-like protein Rrp3. Nucleic Acids Res., 24, 3201–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venema J., Bousquet-Antonelli,C., Gelugne,J.-P., Caizergues-Ferrer,M. and Tollervey,D. (1997) Rok1p is a putative RNA helicase required for rRNA processing. Mol. Cell. Biol., 17, 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ripmaster T.L., Vaughn,G.P. and Woolford,J.L.,Jr (1992) A putative ATP-dependent RNA helicase involved in Saccharomyces cerevisiae ribosome assembly. Proc. Natl Acad. Sci. USA, 89, 11131–11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger F., Daugeron,M.-C. and Linder,P. (2000) Dbp10p, a putative RNA helicase from Saccharomyces cerevisiae, is required for ribosome biogenesis. Nucleic Acids Res., 28, 2315–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang W.-Q., Clark,J.A. and Fournier,M.J. (1997) The rRNA-processing function of the yeast U14 small nucleolar RNA can be rescued by a conserved RNA helicase-like protein. Mol. Cell. Biol., 17, 4124–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de la Cruz J., Kressler,D., Rojo,M., Tollervey,D. and Linder,P. (1998) Spb4p, an essential putative RNA helicase, is required for a late step in the assembly of 60S ribosomal subunits in Saccharomyces cerevisiae. RNA, 4, 1268–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohtake Y. and Wickner,R.B. (1995) Yeast virus propagation depends critically on free 60S ribosomal subunit concentration. Mol. Cell. Biol., 15, 2772–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kressler D., de la Cruz,J., Rojo,M. and Linder,P. (1998) Dbp6p is an essential putative ATP-dependent RNA helicase required for 60S-ribosomal-subunit assembly in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daugeron M.C. and Linder,P. (1998) Dbp7p, a putative ATP-dependent RNA helicase of Saccharomyces cerevisiae is required for 60S ribosomal subunit assembly. RNA, 4, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Cruz J., Kressler,D., Tollervey,D. and Linder,P. (1998) Dob1p (Mtr4p) is a putative ATP-dependent RNA helicase required for the 3′ end formation of 5.8S rRNA in Saccharomyces cerevisiae. EMBO J., 17, 1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colley A., Beggs,J., Tollervey,D. and Lafontaine,D.L.J. (2000) Dhr1p, a putative DEAH-box RNA helicase is associated with the box C+D snoRNA U3. Mol. Cell. Biol., 20, 7238–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasmussen T.P. and Culbertson,M.R. (1998) The putative nucleic acid helicase Sen1p is required for formation and stability of termini and for maximal rates of synthesis and levels of accumulation of small nucleolar RNAs in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 6885–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warner J.R. (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci., 24, 437–440. [DOI] [PubMed] [Google Scholar]
- 28.Kressler D., Linder,P. and de la Cruz,J. (1999) Protein trans-acting factors involved in ribosome biogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 7897–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venema J. and Tollervey,D. (1999) Ribosome synthesis in Saccharomyces cerevisiaeAnnu. Rev. Genet., 33, 261–331. [DOI] [PubMed] [Google Scholar]
- 30.Thomas B.J. and Rothstein,R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]
- 31.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) In Current Protocols in Molecular Biology, Ch. 13. John Wiley & Sons, New York, NY.
- 32.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 33.Gietz D., St Jean,A., Woods,R.A. and Schiestl,R.H. (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res., 20, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Wach A., Brachat,A., Pöhlmann,R. and Philippsen,P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- 36.Gietz R.D. and Sugino,A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt A., Bickle,M., Beck,T. and Hall,M.N. (1997) The yeast phosphatidylinositol kinase homolog TOR2 activates RHO1 and RHO2 via the exchange factor ROM2. Cell, 88, 531–542. [DOI] [PubMed] [Google Scholar]
- 38.Venema J., Planta,R.J. and Raué,H.A. (1998) In vivo mutational analysis of ribosomal RNA in Saccharomyces cerevisiae. In Martin,R. (ed.) Protein Synthesis: Methods and Protocols. Humana Press, Totowa, NJ, pp. 257–270. [DOI] [PubMed]
- 39.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 40.Blum S., Schmid,S.R., Pause,A., Buser,P., Linder,P., Sonenberg,N. and Trachsel,H. (1992) ATP hydrolysis by initiation factor 4A is required for translation initiation in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 89, 7664–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sung P., Higgins,D., Prakash,L. and Prakash,S. (1988) Mutation of lysine-48 to arginine in the yeast RAD3 protein abolishes its ATPase and DNA helicase activities but not the ability to bind ATP. EMBO J., 7, 3263–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plumpton M., McGarvey,M. and Beggs,J.D. (1994) A dominant negative mutation in the conserved RNA helicase motif ‘SAT’ causes splicing factor PRP2 to stall in spliceosomes. EMBO J., 13, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arenas J.E. and Abelson,J.N. (1997) Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl Acad. Sci. USA, 94, 11798–11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang T.-H., Latus,L.J., Liu,Z. and Abbott,J.M. (1998) Genetic interactions of conserved regions in the DEAD-box protein Prp28p. Nucleic Acids Res., 25, 5033–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gross C.H. and Shuman,S. (1998) The nucleoside triphosphate and helicase activities of vaccinia virus NPH-II are essential for virus replication. J. Virol., 72, 4729–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmid S.R., Buser,P., Coppolecchia,R., Fischli,A. and Linder,P. (1993) Analysis of the genes encoding eIF4A. In Brown,A.J.P., Tuite,M.F. and McCarthy,J.E.G. (eds) Protein Synthesis and Targeting in Yeast. Springer-Verlag, Heidelberg, Germany, pp. 131–142.
- 47.Oh J.Y. and Kim,J. (1999) ATP hydrolysis of the DEAD box protein Rok1p is required for in vivo ROK1 function. Nucleic Acids Res., 27, 2753–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gross C.H. and Shuman,S. (1996) The QRxGRxGRxxxG motif of the vaccinia virus DExH box RNA helicase NPH-II is required for ATP hydrolysis and RNA unwinding but not for RNA binding. J. Virol., 70, 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao N., Hesson,T., Cable,M., Hong,Z., Kwong,A.D., Le,H.V. and Weber,P.C. (1997) Structure of the hepatitis C virus RNA helicase domain. Nature Struct. Biol., 4, 463–467. [DOI] [PubMed] [Google Scholar]
- 50.Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- 51.Cho H.-S., Ha,N.-C., Kang,L.-W., Chung,K.M., Back,S.H., Jang,S.K. and Oh,B.-H. (1998) Crystal structure of RNA helicase from genotype 1b hepatitis C virus: a feasible mechanism of unwinding duplex RNA. J. Biol. Chem., 273, 15045–15052. [DOI] [PubMed] [Google Scholar]
- 52.Lorsch J.R. and Herschlag,D. (1998) The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry, 37, 2180–2193. [DOI] [PubMed] [Google Scholar]
- 53.Lorsch J.R. and Herschlag,D. (1998) The DEAD box protein eIF4A. 2. A cycle of nucleotide and RNA-dependent conformational changes. Biochemistry, 37, 2194–2206. [DOI] [PubMed] [Google Scholar]