

Summary
The p110δ isoform of class I phosphoinositide 3-kinase (PI3Ks) plays a major role in B cell receptor signaling, while its p110γ counterpart is thought to predominate in leukocyte chemotaxis. Consequently, emphasis has been placed on developing PI3Kγ selective inhibitors to treat disease states that result from inappropriate tissue accumulation of leukocytes. We now demonstrate that PI3Kδ blockade is effective in treating an autoimmune disorder in which neutrophil infiltration is required for tissue injury. Using the K/BxN serum transfer model of arthritis, in which neutrophils and leukotriene B4 (LTB4) participate, we show that genetic deletion or selective inhibition of PI3Kδ diminishes joint erosion to a level comparable to its PI3Kγ counterpart. Moreover, the induction and progression of joint destruction was profoundly reduced in the absence of both PI3K isoforms and correlated with a limited ability of neutrophils to migrate into tissue in response to LTB4. However, the dynamic interplay between these isoforms is not pervasive, as fMLP-induced neutrophil extravasation was primarily reliant on PI3Kγ. Our results not only demonstrate that blockade of PI3Kδ has potential therapeutic value in the treatment of chronic inflammatory conditions, but also provide evidence that dual inhibition of these lipid kinases may yield superior clinical results.
Keywords: Cell trafficking, neutrophils, kinases, autoimmunity
Introduction
Class I PI3Ks are a family of intracellular signaling proteins that are essential components of migratory, proliferative, and differentiation pathways in many cell types including those that are involved in innate and adaptive immunity. The holoenzymes consist of a regulatory (designated p50, p55, p85, or p101) and a catalytic subunit (designated p110α, p110β, p110γ, or p110δ) that are essential for their recruitment to the plasma membrane and subsequent generation of the key lipid second messenger phosphatidylinositiol (3,4,5)-trisphosphate (PIP3). [1] Activation of this signaling pathway is believed to occur through two distinct receptor types, receptor tyrosine kinases (RTK) or G-protein-coupled receptors (GPCR), each of which utilizes specific p110 isoforms. For instance, the former is thought to primarily activate α, β, and δ isoforms of p110 (designated class Ia) while the latter only p110γ (designated class Ib). However, this dichotomy may not be so straightforward as previous reports not only demonstrate that PI3Kγ in macrophages can be activated indirectly through the tyrosine kinase receptors such as CSF-1R/c-fms, but that PI3Kδ contributes to PIP3 production in neutrophils in response to GPCR activation by lipid mediators of inflammation (i.e. LTB4), or bacterial products (i.e. fMLP). [2-4] Such observations would suggest that the function of these two classes of PI3Ks may overlap in particular subsets of leukocytes and that activation of these signaling pathways may not be restricted to a particular type of receptor.
Although the therapeutic ramifications associated with this redundancy in activation is unclear, evidence is mounting that selective targeting of either p110γ or p110δ catalytic domains may prove beneficial in treating specific inflammatory disease states. For instance, a major, but not unanticipated, phenotype associated with mice lacking PI3Kγ activity is a reduction in neutrophil chemotaxis to fMLP and LTB4 as well as a partial impairment in antigen receptor signaling in T cells but not B cells. [5-8] Validation of p110γ as a therapeutic target is suggested by the ability of an orally active small-molecule inhibitor of this catalytic domain to partially reduce joint destruction in an animal model of inflammatory arthritis, which correlated with a defect in neutrophil migration. [9] By contrast to its gamma counterpart, genetically inactivated or p110δ-deficient mice have a significant reduction in B cell antigen receptor (BCR) signaling, a substantial decline in immunoglobulin levels, and diminished numbers of immature and mature B cells. [8,10,11] They do, however, have a partial impairment in neutrophil chemotaxis in response to acute inflammatory insults or exposure to a chemotactic agent. [4,12-14] Importantly, such studies have also demonstrated that selective targeting of the p110δ catalytic domain with an orally active small-molecule inhibitor is achievable, yielding similar results to that observed for genetically altered animals. Despite the accumulated evidence thus far, it is unclear whether blockade of p110γ, p110δ, or perhaps both catalytic domains would yield superior results in the potential treatment of inflammatory disease states such as rheumatoid arthritis (RA) where neutrophils are known to contribute to tissue injury. [15]
Rodent models of arthritis that permit assessment of therapies directed against the effector phase of the immune response lend themselves to just such a comparison as they more closely portray the clinical situation: the need to treat individuals with established disease. In this regard, the murine K/BxN serum transfer model of inflammatory arthritis has proven useful in broadening our understanding of the contribution that particular leukocyte subsets and inflammatory cytokines play in joint destruction. [16] These animals express a transgene encoded TCR that confers reactivity to a self-peptide derived from glucose-6-phosphate isomerase, a ubiquitously expressed glycolytic enzyme, that when presented in the context of the MHC class II molecule Ag7 leads to the generation of arthritogenic immunoglobulins. [17] Importantly, transfer of serum from K/BxN mice into healthy animals results in polyarthritis within days even in the absence of lymphocytes or mast cells. [18,19] Moreover, it mimics all the classic histological features associated with RA in humans including a predominance of infiltrating neutrophils, pannus formation, and a cartilage and bone erosive synovitis. Considerable insight has been gained into the pathogenesis of joint destruction associated with this animal model including the indispensable role of neutrophils and leukotriene B4 (LTB4) in the initiation and perpetuation of arthritis. [20-22]
Here, we evaluated the extent to which PI3Kδ contributes to initiation and progression of inflammation associated with the K/BxN serum transfer model of arthritis. Moreover, we explored its possible overlapping role with PI3Kγ in this model as well as in intravital studies that assessed neutrophil migration in response to exogenous chemoattractants. We show that genetic deletion or pharmacological blockade of p110δ is as effective in protecting against and in reducing the extent of disease associated with autoantibody-induced arthritis as observed for its gamma counterpart. That said, our results clearly indicate that the combined activities of these class Ia and Ib PI3Ks are absolutely critical for the development of inflammation in response to K/BxN serum transfer as well as in supporting LTB4-mediated neutrophil accumulation in tissue. Finally, we provide direct evidence that this redundancy in function may be restricted to specific GPCRs, as PI3Kγ primarily orchestrates the extravascular accumulation of neutrophils in response to the bacterial product fMLP.
Results
p110δ-deficiency reduces joint injury in serum-induced arthritis
Previously, it has been established that p110δ plays a role in supporting the acute influx of leukocytes into tissues in response to the application of bacterial products, cytokines, or chemoattractants. To better understand the potential therapeutic utility that inhibition of this signaling pathway may have in reducing tissue injury that results from an inappropriate or uncontrolled immune response as occurs in autoimmune disease states, we evaluated the contribution of PI3Kδ versus its gamma counterpart in mediating joint inflammation in the K/BxN-serum transfer model of arthritis. Administration of arthritogenic serum to p110δ−/− mice resulted in a significant and similar reduction in the extent of paw edema as compared to p110γ−/− animals (ranging from 45% to 53%) from days seven to fourteen (Figure 1A). Histological scoring of afflicted joints also revealed a diminution in synovial inflammation as well as bone and cartilage erosion, suggesting that a deficiency in the p110δ catalytic domain impairs the onset and perhaps the progression of disease (Figure 1, B-G, J). Given the recent observation that these two class I PI3K isoforms may have overlapping, but temporally distinct functions in promoting leukocyte accumulation in tissue in response to exogenous administration of chemoattractants, we next evaluated the effect that a deficiency in both p110δ and p110γ would have on the development of inflammatory arthritis. [13] By contrast to singly deficient mice, p110γδ−/− animals develop minimal paw swelling. Moreover, histological evaluation of joints from these animals showed relatively normal articular surfaces, intact joint spaces, and absence of significant periarticular inflammation 14 days post administration of autoreactive antibodies (Figure 1, H-J). Results suggest that the combined activities of these lipid kinases are critical for induction of inflammatory arthritis in this animal model.
Figure 1.
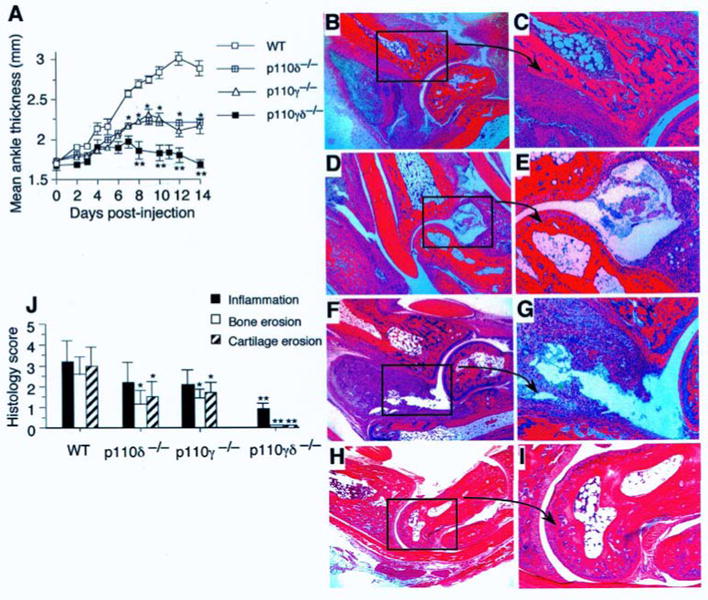
Hind paw swelling and histopathology of p110-deficient mice post-induction of K/BxN serum transfer arthritis. (A) Ankle thickness was measured from the day of injection of arthritogenic serum (D0) to completion of the study (D14). Data are representative of two independent experiments (total 10 mice; mean ± SEM). *, P <0.001 (WT vs. p110 deficient cells). (B-I) Representative histopathology of ankle joints from WT (B, C), p110δ−/− (D, E), p110γ−/− (F, G), p110γδ−/− (H, I) at experimental day 14 (insert 200× total magnification). (J) Histopathogical scoring (mean ± SD) of ankles from the same mice 14 days post-administration of arthritogenic serum. *, P <0.01 (WT vs. p110δ or p110γ deficient); **, P <0.001 (p110γδ−/− vs. p110δ or p110γ deficient).
Treatment of arthritis by blockade of p110δ activity
As genetic deletion of p110δ alone or in combination with p110γ results in several developmental defects in a variety of immunocompetent cells, it is important to determine whether inhibition of this catalytic domain in non-genetically altered animals would have a similar effect on the progression of arthritis, that is, after the onset of clinically significant disease (day 5). [8,23-25] In this “therapeutic” treatment model, WT mice received the p110δ inhibitor IC87114 at 20 mg/kg orally (three times per day) commencing on the fifth day post-injection of arthritogenic serum. This yielded plasma concentrations of drug (peak of 9 ± 2.6 μM and trough of 3.2 ± 1.3 μM; mean ± SD) known to selectively block the activity of p110δ, but not p110α, p110β, or p110γ.[4] This resulted in a significant reduction in the overall extent of inflammation and associated bone and cartilage erosion (Figure 2, A,F). However, a substantially greater therapeutic impact was achieved when IC87114 was given to p110γ−/− animals as compared to WT or p110δ−/− mice. Not only was there a complete resolution in hind paw swelling, but histological evaluation of joints revealed no evidence of cartilage or bone destruction. (Figure 2, A, D-F). Peak and trough plasma levels of IC87114 (20 mg/kg) were 14.3 ± 3 μM and 6.3 ± 3.2 μM for p110δ−/− animals, and 12.4 ± 2.7 μM and 5.5 ± 2.1 μM for p110γ−/− mice (mean ± SD). Clearly, it is the combined activities of these class Ia and class Ib PI3K isoforms and not developmental abnormalities in immunocompetent cells that account for these observations. Interestingly, neutrophils not only rely on PI3K activity for effector function, but these cells have been shown to play an essential role in the initiation and progression of joint injury in the K/BxN serum transfer model of arthritis. [22] Consistent with the latter observations is the reduction in neutrophil infiltrates in afflicted joints of WT and p110γ−/− mice treated with IC87114 for 10 days (24.3 ± 3% versus 92 ± 5%, respectively; mean ± SEM) as well as the ability of PMN-depleting antibodies to prevent the inflammatory effects of arthritogenic serum when pre-administered to WT mice (Figure 2, G and H).
Figure 2.
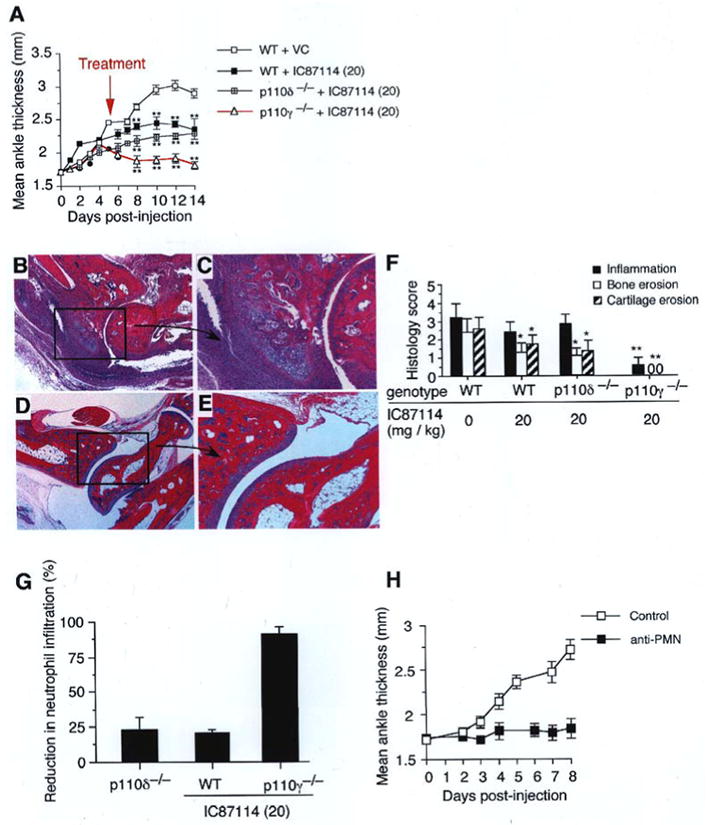
Treatment of autoantibody-induced arthritis via PI3K inhibition. (A) Ankle thickness measurements in WT mice treated with VC or 20 mg/kg (20) of IC87114 or p110-deficient mice treated with 20 mg/kg (20) of drug commencing on day 5 post-injection of arthritogenic serum (n=5 animals per group). Data are representative of two independent experiments (mean ± SEM). **, P <0.001 (WT + VC vs. WT + IC87114 (20) and p110γ−/− + VC vs. p110γ−/− + IC87114 (20)). (B-E) Representative histopathology of ankle joints from WT + VC (B, C) or p110γ−/− + IC87114 (20) (D, E) mice at experimental day 14. (F) Histopathogical scoring (mean ± SD) of ankles from the same mice 14 days post-administration of arthritogenic serum. *, P <0.01 and **, P <0.001 compared to WT + VC. (G) Reduction in neutrophil infiltration in joint sections as compared to WT (mean ± SEM). Results are from two experiments performed in duplicate (mean ± SEM). *, P < 0.01 (WT vs. p110 deficient or WT + IC87114); VC = vehicle control. (H) Effect of antibody-induced depletion of neutrophils on the induction of arthritis in WT mice (n=4 animals per group). Data are representative of one experiment (mean ± SEM).
PI3Kδ activity is required for LTB4-mediated neutrophil tissue accumulation in vivo
In addition to the requirement for neutrophils, the induction and progression of joint injury associated with autoantibody-driven erosive synovitis is also critically reliant on the LTB4. [20,21] As a deficiency in either p110δ or p110γ yielded a similar reduction in joint swelling and tissue damage, it is reasonable to assume that both class I PI3K isoforms may be required for maximal influx of neutrophils into inflamed tissues in response to this chemoattractant. To address this issue, we evaluated the ability of p110-deficient cells to undergo transendothelial migration in response to superfusion of TNFα-stimulated cremaster muscle with LTB4. Extravasation of neutrophils singly deficient in PI3Ks was reduced by a similar extent in response to this chemoattractant (∼33%) as compared to WT (Figure 3, A and B). By contrast, genetic deletion of both catalytic domains or treatment of p110γ-deficient mice with IC87114 diminished tissue accumulation of these cells by >70%. Similar results were obtained in transwell chemotaxis assays (Figure 3, C and D). These findings suggest that PI3Kδ and its gamma counterpart work in concert to promote maximal neutrophil migration into inflamed tissues in response to a chemoattractant known to contribute to autoantibody-mediated arthritis in mice.
Figure 3.
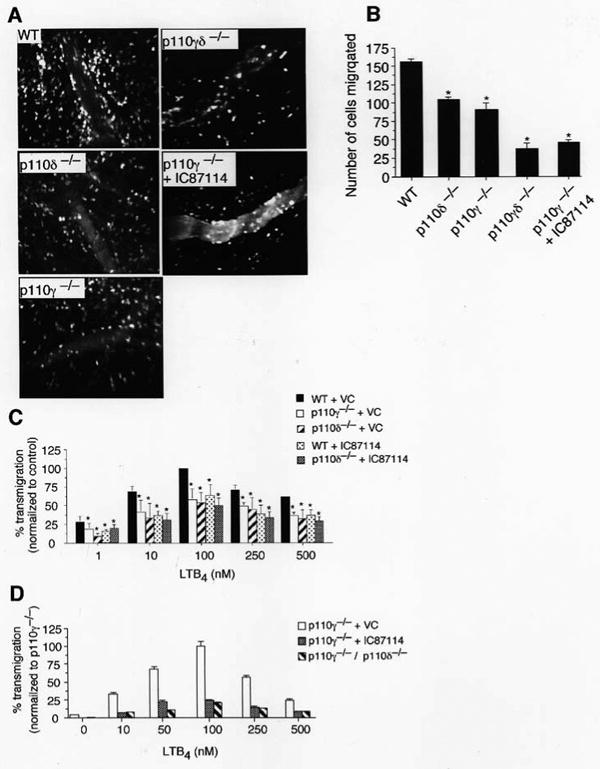
LTB4-mediated neutrophil transendothelial migration in WT or p110-deficient mice. (A) Representative intravital photomicrographs depicting the extent of LTB4-induced neutrophil extravasation into TNFα-inflamed cremaster muscle (CM) of p110 deficient and IC87114 treated mice. High molecular weight FITC-dextran was administered intravenously to distinguish intra-from extravascular cells. (B) Quantitation of the number of the migrated cells at 1.5 h post-application of LTB4 (n=3 mice per genotype, minimum of 4 vessels per animal). (C) LTB4-mediated migration of WT, p110δ−/−, p110γ−/− across bare transwell inserts in the presence or absence of IC87114. Data represent the mean ± SEM. *, P < 0.001
p110δ-deficiency does not impair tissue accumulation of neutrophils in response to fMLP
Neutrophils play a central role in our ability to mount an effective innate immune response to infectious agents such as bacteria. Although a desirable attribute of pharmacological blockade of PI3Kδ activity is reduced autoantibody-induced erosive synovitis, it is important to determine whether such inhibition would also potentially limit host defense to specific pathogens by curtailing the influx of cells into tissues in response to bacterial products such as fMLP. Thus, we evaluated the ability of p110δ−/− neutrophils to undergo transendothelial migration in TNFα-stimulated venules upon application of a concentration of fMLP (10 μM) shown to induce significant accumulation of these cells in extravascular tissues. [26] Interestingly, a deficiency in p110δ did not impair neutrophil migration as compared to WT cells (99 ± 3 cells vs. 81 ± 4 cells; mean ± SEM, P = 0.002) (Figure 4, A and B). Similar results were obtained in IC87114-treated WT mice. By contrast, genetic deletion of p110γ alone or in combination with p110δ reduced fMLP-mediated neutrophil chemotaxis by 63% and 74%, respectively. Thus, therapeutic blockade of PI3Kδ does not appear to impair neutrophil accumulation in inflamed tissues in response to the bacterial product fMLP.
Figure 4.
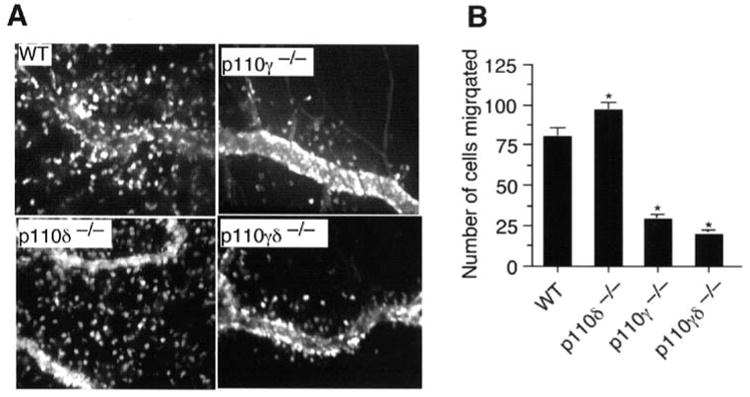
fMLP-mediated neutrophil transendothelial migration in WT or p110-deficient mice. (A) Representative intravital photomicrographs depicting the extent of neutrophil extravasation into TNFα-inflamed cremaster muscle (CM) of p110 deficient and IC87114 treated mice. High molecular weight FITC-dextran was administered intravenously to distinguish intra- from extravascular cells. (B) Quantitation of the number of the migrated cells at 1.5 h post-application of fMLP (n=3 mice per genotype, minimum of 8 vessels per animal). Data represent the mean ± SEM. *, P < 0.001
Role of PI3Kδ in supporting neutrophil locomotion in inflamed tissues
Once extravasated, neutrophils maintain a high degree of motility that is essential for reaching end targets, whether that is an invading pathogen or autoantibody coated tissue. Previously, it has been suggested that neutrophil locomotion in tissues may rely solely on the activity of PI3Kγ. [27] Whether PI3Kδ activity also regulates the speed at which neutrophils move in response to a chemoattractant and if these in vitro observations are also relevant in a living animal remains to be determined. To address these issues, we studied the extravascular behavior of p110δ-deficient neutrophils in TNFα-treated cremaster muscle superfused with either LTB4 or fMLP. In the former case, migration velocities of p110δ−/− neutrophils were comparable to WT (0.12 ± 0.005 μm/s versus 0.11 ± 0.004 μm/s; mean ± SEM), but slightly reduced (1.3 fold slower) in cells lacking only p110γ (Figure 5A). There was, however, a modest and similar reduction (∼1.5-fold) in distance traveled by p110 singly deficient cells from the point of origin over a 7 min observation period (Figure 5, B and C). By contrast, p110γδ−/− deficient neutrophils exhibited limited movement, traveling a distance of only 12 ± 2 μm (mean ± SEM) from the initial point of origin as compared to 42 ± 3 μm for WT. Migration velocities were also severely curtailed (2.3-fold slower) under identical experimental conditions, suggesting that the combined activities of the PI3Kγ and PI3Kδ are also required for effective neutrophil locomotion in extravascular tissues. Similar results were obtained by administering IC87114 to p110γ−/− mice. Interestingly, the velocity of migrating p110δ−/− neutrophils in response to fMLP was slightly higher than for its WT counterpart (0.17 ± 0.006 μm/s vs. 0.13 ± 0.004 μm/s, respectively; P<0.0001) as well as overall distance traversed in tissues (47 ± 2 μm vs. 37 ± 1 μm, respectively; P<0.0001) (Figure 6, A-C). Clearly, a lack of PI3Kδ activity alone does not appear to significantly interfere with neutrophil movement in tissues in response to LTB4 or fMLP.
Figure 5.
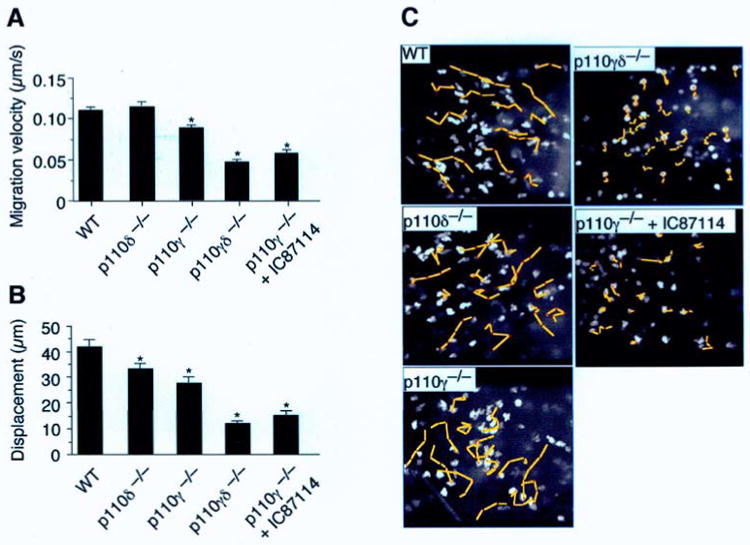
Role of class I PI3Ks in supporting neutrophil motility in tissue. (A) Mean velocity of neutrophils in LTB4-superfused CM over a 7 min time interval (n=4 individual experiments per genotype, total 40 cells per genotype). (B) Distance (μm) traveled from point of origin and (C) photomicrographs depicting the movement of neutrophils (yellow lines; total 7 min observation) in extravascular tissue superfused with LTB4 (1.5 h). *, P < 0.001.
Figure 6.
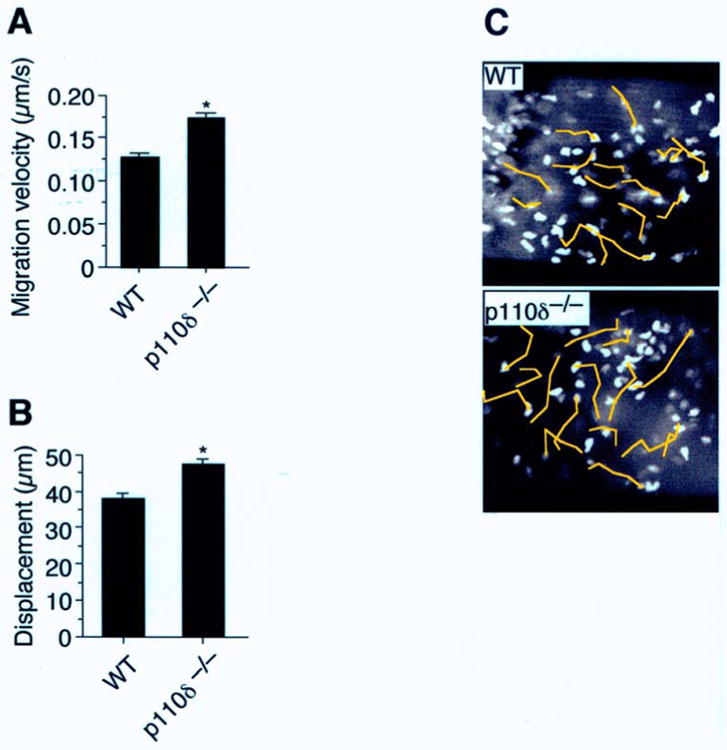
Role of class I PI3Ks in supporting fMLP-induced neutrophil motility in tissue. (A) Mean velocity of neutrophils in fMLP-superfused CM (1.5h) during a 7 min time interval (n=3 individual experiments per genotype, total 40 cells per genotype). (B) Distance (μm) traveled from point of origin and (C) photomicrographs depicting the movement of neutrophils (yellow lines; total 7 min observation) in extravascular tissue. *, P < 0.001.
Discussion
Multiple studies utilizing either direct application of cytokines and/or chemoattractants as well as acute tissue injury models have been performed in an attempt to not only understand the role(s) that PI3Kδ or PI3Kγ play in modulating various immune responses, but also to determine whether selective inhibitors of either p110 catalytic domain may prove to be effective anti-inflammatory agents. Clearly, PI3Kγ is a key participant in GCPR-mediated processes in neutrophils and that pharmacological blockade can limit the ability of this leukocyte subset to inflict tissue injury in murine models of autoimmunity. Although signaling through PI3Kδ is directly linked to RTK, it does contribute to PIP3 production as well as neutrophil migration through GPCRs such as BLT1, the primary receptor for LTB4. The current study explored whether selective inhibition of p110δ would yield similar therapeutic results to that reported for PI3Kγ in mitigating tissue injury.
To achieve this goal, we utilized the K/BxN serum transfer model of inflammatory arthritis. This model not only bypasses development defects in adaptive immunity associated with a deficiency in either p110δ (primarily B cell) alone or in combination with p110γ (B, T and NK cell) that would potentially impair mechanisms that initiate disease, but more accurately simulates the clinical situation in which therapies would be administered to patients with established disease. Moreover, neutrophils and the endogenously produced chemoattractant LTB4 are known to be key mediators of inflammation induced by administration of arthritogenic antibodies, the former confirmed in our PMN-depletion study. Our results clearly show that 1) p110δ-deficient mice have a similar reduction in joint swelling and bone and cartilage erosion as their p110γ−/− counterparts, and 2) that pharmacological inhibition of its catalytic domain after onset of clinical symptoms can limit disease progression. Importantly, inactivation of both p110 isoforms, either by genetic deletion or treatment of p110γ−/− mice with IC87114, yielded almost no evidence of the destructive consequences of inflammatory synovitis. Conceivably, these results may reflect interdependence between PI3Kγ and PI3Kδ to initiate and amplify PIP3 production in immunocompetent cells. This is supported by studies demonstrating that effective production of reactive oxygen species in human neutrophils and efficient antigen-receptor signaling in T cells requires the participation of both p110δ and p110γ. [23,25,28]
To provide direct evidence that not only does PI3Kδ play an equal role in supporting GPCR-mediated neutrophil extravasation in response to LTB4, we utilized confocal intravital microscopy to measure the in vivo migration of GFP-expressing neutrophils. In this model of inflammation, PI3Kδ and its gamma counter do not appear to serve temporally distinct roles as previously reported, but rather work in concert to promote effective neutrophil accumulation into tissue. [13] This is supported by the equivalent reduction in number of LTB4-migrated cells (∼1.5-fold) in the absence of either p110δ or p110γ, and the profound impairment in this process (∼3-fold) when both catalytic domains are genetically deleted. Moreover, in the context of TNFα-induced inflammation, the velocity of migration and the degree of cell displacement that occurred in extravascular tissue in response to LTB4 was also severely curtailed when both PI3Kδ and PI3Kγ were inactivated. In fact, the movement of p110γδ−/− neutrophils appeared stochastic rather than chemoattractant directed (see Supplementary video). Based on these results and the significant role that neutrophils and LTB4 play in the K/BxN mouse model of arthritis, it is not unreasonable to assume that the combined activities of both class Ia and Ib PI3Ks are required for efficient trafficking of these cells into inflamed joint tissue.
It is interesting to note that although both PI3Kδ and PI3Kγ are essential for LTB4-mediated neutrophil chemotaxis in our intravital studies, it is the activity of the latter that appears to be primarily accountable for their migration in response to fMLP. This is in contrast to a previous report in which transfection of a neutrophil-like cell line with a dominant negative mutant of p85 but not p110γ dramatically reduced fMLP-mediated chemotaxis. [29] However, our results are consistent with multiple studies demonstrating impaired migration of p110γ−/− neutrophils to this bacterial product, and are the first to directly rule out a significant contribution from its delta counterpart in supporting tissue accumulation of these cells.
In conclusion, our series of in vivo studies demonstrate that pharmacological inhibition of PI3Kδ may be an effective strategy in the treatment of inflammatory disorders such as RA, and that the efficacy of such therapy can be enhanced in the absence of PI3Kγ. Moreover, it appears that the involvement of PI3Kδ in GPCR signaling is more restricted than its gamma counterpart. However, it is precisely this signaling diversity within class I PI3Ks that may provide an opportunity to preferentially block neutrophil activity implicated in the pathogenic responses rather than in support of host defense. Clearly, broadening our understanding of the role that class Ia and class Ib PI3Ks play in innate and adaptive immunity will be essential for tailoring therapies to specific disease processes.
Materials and methods
Animals
p110γ−/−, p110δ−/−, p110γδ−/−, and WT littermate controls (backcrossed a minimum of 8 generations on to B6) have been described and were used between 6 and 8 weeks of age. [7] KRN transgenic mice (C. Benoist, Harvard Medical School, Boston, MA) were mated with NOD/LtJ mice (The Jackson Laboratory) to generate arthritogenic serum. [18] All procedures were approved by the Columbia University Animal Care and Use Committee and were in accordance with National Institutes of Health policies.
Arthritis studies
For antibody-transfer arthritis studies, 200 μl of serum pooled from 8 week old arthritic K/BxN mice was injected i.p. on days 0 and 2. Ankle thickness was measured using a dial thickness gage (Mitutoyo America) and mean ± SEM values determined for a minimum of 5 animals in each control and experimental group. All experiments were performed in duplicate, except those involving the p110γδ−/− mice. For studies involving pharmacological blockade of PI3K activity, mice received an oral dose of either the small molecule p110δ inhibitor IC87114 (20 mg/kg) or vehicle control (PEG400) every 8h starting on day 5 post administration of arthritogenic serum and ending on day 14. Plasma levels of this compound were determined by liquid chromatography/mass spectroscopy. Progression of inflammation was assessed by caliper measurement of hind limb ankle thickness and mean ± SEM values determined for 10 animals in each group.
Depletion of neutrophils
To evaluate the role of neutrophils in K/BxN serum-induced arthritis, WT mice received intraperitoneal injections of a rabbit anti-mouse PMN polyclonal antibody (200 μl, Accurate Chemical and Scientific) every other day for a total of 3 doses beginning the day prior to serum transfer. [30] This resulted in a sustained decrease in circulating GR-1+ cells (<15%) as determined by flow cytometry. Control mice received a polyclonal antibody that does not interact with murine leukocytes or cause neutropenia.
Histological scoring
At necropsy (day 14), hind paws were fixed in 10% neutral buffered formalin, decalcified, and cut into 5 μm sections (Histology Consultant Services, INC., Everson, WA). Specimens were then stained with hematoxylin and eosin for general evaluation of joint structure. Four components of the arthritic process were assessed in the distal tibia and the tarsal bones as well as in the surrounding fascia using a “blinded” analytical paradigm: inflammation score (in peri-articular soft tissues and bone marrow): 0 = normal, 1 = few inflammatory cells, 2 = mild inflammation (a few small focal aggregates, with diffuse minimal perisynovial infiltration), 3 = moderate inflammation (many small focal aggregates, with diffuse and extensive perisynovial infiltration), 4 = marked inflammation (many large aggregates and intra-articular fibrin, as well as diffuse and extensive perisynovial infiltration); bone score (assessing the periosteal proliferative bone reaction): 0 = normal, 1 = minimal repair (a few small periosteal osteophytes), 2 = mild repair (many small periosteal osteophytes), 3 = moderate repair (many small to medium periosteal osteophytes), 4 = marked repair (many medium to large periosteal osteophytes), 5 = extensive repair (osteophytes along essentially entire lengths of periosteal surface); erosion score (examining bone and cartilage destruction): 0 = normal, 1 = minimal bone erosion (1 to 2 small, shallow foci), 2 = mild bone erosion (1 to 4 foci of medium size and depth), 3 = moderate bone erosion (5 or more foci of medium size that extend partially through the cortical bone), 4 = marked bone erosion (multiple small to medium foci extending partly or completely through the cortical bone, 5 = extensive bone erosion (cortical penetration exceeds > 25% of the entire bone length at multiple sites); cartilage: 0 = normal, 1 = minimal (a few scalloped depressions in peripheral articular cartilage of major joints), 2 = mild (scalloping extends one third of way across major affected joints), 3 = moderate (scalloping extends across 75% of affected joints), 4 = marked (scalloping involves entire articular surfaces of major affected joints). Arthritic lesions were graded separately for each hind paw of every animal using tiered, semi-quantitative grading metrics. [31]
Neutrophil quantification in joint tissues was performed in 4 adjacent sections. For each section, 5 different optic fields were examined and total number of cells and neutrophils were counted per optic field. The reduction in neutrophil infiltration associated with p110 deficiency was calculated as a percentage of WT and is expressed as the mean ± SEM.
Neutrophil purification and in vitro transmigration assay
Mouse bone marrow polymorphonuclear cells were isolated by discontinuous Percoll gradient and LTB4-induced neutrophil chemotaxis was assessed using a transwell assay system (1.5 h, 37°C) as previously described. [4,7]
Intravital microscopy studies
Surgical preparation of the cremaster muscle in mice lacking class I PI3Ks, but expressing GFP under the control of the LysM promoter, has been previously described. [4,7] One exception to this procedure was that the muscle was cut with a fine scissor and not cautery to minimize tissue injury. An inflammatory reaction was induced by administration of an intrascrotal injection of murine TNFα (PeproTech Inc) consisting of 50 ng in 200 μl of normal saline for a total of 3 h. Subsequently, the cremaster muscle was surgically exposed and the tissue continuously bathed in either LTB4 (BIOMOL) or fMLP (Sigma) at a concentration of 100 nM and 10 μM, respectively, for 1.5 h. The number of GFP-expressing cells that extravasated into the tissue in response to these chemoattractants was determined by fluorescence microscopy using a system equipped with a Yokogawa CSU-22 spinning disk confocal scanner and 488 nm laser line (Revolution XD, Andor™ Technology). A 20× water-immersion Olympus objective (LUMPlanFl, 0.5 NA) coupled to a piezo driver enabled viewing in the z-axis (∼1μm sequential sections; total 75 μm). To distinguished intra- vs. extra-vessel cells, FITC-dextran (FD250S, Sigma) was administered intravenously. Data sets were flattened along the z-axis as maximum intensity projections so to enable determination of the total number of cells that had migrated into a 150 × 200 μm region on either side of the vessel wall. High-resolution time-lapse imaging (every 2 s for 7 min) of GFP+ cells was used to track their movement once migrated into the surrounding tissue. Off-line analysis was performed using a cell tracking software (Andor™ tracker). Four consecutive velocities per cell were measured for a total of 20 cells per experiment (n=4).
Statistical analyses
Continuous variables: Clinical scores (joint swelling) were expressed as mean ± SEM. The ANOVA test was performed on the histological data using commercial statistical software (SigmaStat v2.0; Systat Software, Richmond, CA). Ordinal variables: Histopathologic results were expressed as the mean ± SD. A chi-square test was performed on these data using different commercial statistical software (JMP v5.0; SAS, Cary, NC). A Student t test was also used for intravital studies. P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the Institut de Genetique et de Biologie Moleculaire et Cellulaire for providing KRN mice, Deborah Novak at Washington University School of Medicine for technical advice, and Natalie Diacovo for data analysis. This work is supported by the NHLBI grant HL075805-01A2 (T.G. Diacovo).
Abbreviations
- GPCR
G-protein-coupled receptor
- LTB4
leukotriene B4
- PIP3
phosphatidylinositiol (3,4,5)-trisphosphate
- RA
rheumatoid arthritis
- RTK
receptor tyrosine kinase
Footnotes
Conflict of Interest: One author (KP) has declared a financial interest in a company whose potential product was studied in the present work.
References
- 1.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 2.Jones GE, Prigmore E, Calvez R, Hogan C, Dunn GA, Hirsch E, Wymann MP, Ridley AJ. Requirement for PI 3-kinase gamma in macrophage migration to MCP-1 and CSF-1. Exp Cell Res. 2003;290:120–131. doi: 10.1016/s0014-4827(03)00318-5. [DOI] [PubMed] [Google Scholar]
- 3.Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. Essential role of Phosphoinositide 3-kinase δ in neutrophil directional movement. J Immunol. 2003;170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- 4.Puri KD, Doggett TA, Douangpanya J, Hou Y, Tino WT, Wilson T, Graf T, et al. Mechanisms and implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue. Blood. 2004;103:3448–3456. doi: 10.1182/blood-2003-05-1667. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 7.Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG. The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood. 2005;106:150–157. doi: 10.1182/blood-2005-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bilancio A, Okkenhaug K, Camps M, Emery JL, Ruckle T, Rommel C, Vanhaesebroeck B. Key role of the p110delta isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110delta function in B cells. Blood. 2006;107:642–650. doi: 10.1182/blood-2005-07-3041. [DOI] [PubMed] [Google Scholar]
- 9.Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 10.Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Carpino N, Wang D, Ihle JN. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22:8580–8591. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, Humphries LA, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196:753–763. doi: 10.1084/jem.20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Puri KD, Penninger JM, Kubes P. Leukocyte PI3Kgamma and PI3Kdelta have temporally distinct roles for leukocyte recruitment in vivo. Blood. 2007;110:1191–1198. doi: 10.1182/blood-2006-11-060103. [DOI] [PubMed] [Google Scholar]
- 14.Pinho V, de Castro Russo R, Amaral FA, de Sousa LP, Barsante MM, de Souza DG, Alves-Filho JC, et al. Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. J Immunol. 2007;179:7891–7898. doi: 10.4049/jimmunol.179.11.7891. [DOI] [PubMed] [Google Scholar]
- 15.Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond. Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 16.Kyburz D, Corr M. The KRN mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25:79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 17.Maccioni M, Zeder-Lutz G, Huang H, Ebel C, Gerber P, Hergueux J, Marchal P, et al. Arthritogenic monoclonal antibodies from K/BxN mice. J Exp Med. 2002;195:1071–1077. doi: 10.1084/jem.20011941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–61. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 19.Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med. 2007;204:2797–2802. doi: 10.1084/jem.20071391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203:837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J Exp Med. 2006;203:829–835. doi: 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 23.Swat W, Montgrain V, Doggett TA, Douangpanya J, Puri K, Vermi W, Diacovo TG. Essential role of PI3Kdelta and PI3Kgamma in thymocyte survival. Blood. 2006;107:2415–2422. doi: 10.1182/blood-2005-08-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, Colonna M. p110gamma and p110delta Phosphoinositide 3-Kinase Signaling Pathways Synergize to Control Development and Functions of Murine NK Cells. Immunity. 2007;27:214–227. doi: 10.1016/j.immuni.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Webb LM, Vigorito E, Wymann MP, Hirsch E, Turner M. Cutting edge: T cell development requires the combined activities of the p110gamma and p110delta catalytic isoforms of phosphatidylinositol 3-kinase. J Immunol. 2005;175:2783–2787. doi: 10.4049/jimmunol.175.5.2783. [DOI] [PubMed] [Google Scholar]
- 26.Foy DS, Ley K. Intercellular adhesion molecule-1 is required for chemoattractant-induced leukocyte adhesion in resting, but not inflamed, venules in vivo. Microvasc Res. 2000;60:249–260. doi: 10.1006/mvre.2000.2272. [DOI] [PubMed] [Google Scholar]
- 27.Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, Crabbe T, et al. PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol. 2007;9:86–91. doi: 10.1038/ncb1517. [DOI] [PubMed] [Google Scholar]
- 28.Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- 29.Boulven I, Levasseur S, Marois S, Paré G, Rollet-Labelle E, Naccache PH. Class IA phosphatidylinositide 3-kinases, rather than p110 gamma, regulate formyl-methionyl-leucyl-phenylalanine-stimulated chemotaxis and superoxide production in differentiated neutrophil-like PLB-985 cells. J Immunol. 2006;176:7621–7627. doi: 10.4049/jimmunol.176.12.7621. [DOI] [PubMed] [Google Scholar]
- 30.Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, et al. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation. 2005;112:232–240. doi: 10.1161/CIRCULATIONAHA.104.517391. [DOI] [PubMed] [Google Scholar]
- 31.Bolon B, Morony S, Cheng Y, Hu YL, Feige U. Osteoclast numbers in Lewis rats with adjuvant-induced arthritis: identification of preferred sites and parameters for rapid quantitative analysis. Vet Pathol. 2004;41:30–36. doi: 10.1354/vp.41-1-30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.