

Abstract
ATF1 (activating transcription factor 1), a stimulus-induced CREB family transcription factor, plays important roles in cell survival and proliferation. Phosphorylation of ATF1 at Ser63 by PKA (cAMP-dependent protein kinase) and related kinases was the only known post-translational regulatory mechanism of ATF1. Here, we found that HIPK2 (homeodomain-interacting protein kinase 2), a DNA-damage-responsive nuclear kinase, is a new ATF1 kinase that phosphorylates Ser198 but not Ser63. ATF1 phosphorylation by HIPK2 activated ATF1 transcription function in the GAL4-reporter system. ATF1 is a transcriptional repressor of ferritin H, the major intracellular iron storage gene, through an ARE (antioxidant-responsive element). HIPK2 overrode the ATF1-mediated ARE repression in a kinase-activity-dependent manner in HepG2 cells. Furthermore, DNA-damage-inducing agents doxorubicin, etoposide and sodium arsenite induced ferritin H mRNA expression in HIPK2+/+ MEF cells, whereas it was significantly impaired in HIPK2−/− MEF cells. Induction of other ARE-regulated detoxification genes such as NQO1 (NADPH quinone oxidoreductase 1), GST (glutathione S-transferase) and HO1 (heme oxygenase 1) by genotoxic stress was also decreased in HIPK2-deficient cells. Taken together, these results suggest that HIPK2 is a new ATF1 kinase involved in the regulation of ferritin H and other antioxidant detoxification genes in genotoxic stress conditions.
Keywords: HIPK2, ATF1, Phosphorylation, Ferritin, Genotoxic stress, Transcription
Introduction
ATF1 (activating transcription factor 1) belongs to the CREB transcription factor family that contains a basic-leucine zipper (b-zip) motif for DNA binding and dimerization (Rehfuss et al., 1991). ATF1 and CREB control transcription of many target genes through homo- or heterodimerization within the family or with other b-zip transcription factors on the sequences related to an AP1 or cAMP-response element (Mayr and Montminy, 2001). ATF1 plays pivotal roles in cell survival and proliferation. Increased ATF1 expression in transformed lymphocytes and metastatic melanoma cells appears to enhance the growth potential of these tumor cells (Hsueh and Lai, 1995; Jean et al., 2000). In addition, a gene fusion between Ewing's sarcoma gene EWS and ATF1 by t(12;22) chromosomal translocation was found in clear-cell sarcoma (Zucman et al., 1993), and the EWS-ATF1 chimeric transcription factor plays a vital role in maintaining viability, tumorigenicity and metastatic potential of these cells (Bosilevac et al., 1999; Jean et al., 2000). Stimulus-coupled activation of ATF1 is induced by growth factors as well as stress-inducing agents, in which ATF1 is phosphorylated at Ser63 located in the kinase-inducible (KID) domain by PKA (cAMP-dependent protein kinase) and several other serine-threonine (Ser-Thr) kinases (Mayr and Montminy, 2001).
ATF1 regulates cell survival and proliferation by acting either as a transcriptional activator (Atlas et al., 2001; Belmonte et al., 2001; Kingsley-Kallesen et al., 1999; Lee and Pedersen, 2003; Lu and Sack, 2008; Rolli et al., 1999; Zaman et al., 1999; Zhang et al., 2004) or a repressor (Dong et al., 2002; Okuyama et al., 1996; Salnikow et al., 1997). We recently found that ATF1 is a transcriptional repressor of the ferritin H gene (Iwasaki et al., 2007). Ferritin is the major intracellular iron storage protein comprising 24 subunits of H and L forms (Theil, 2003). Ferritin H contains the ferroxidase activity that oxidizes ferrous iron to ferric iron for iron storage, whereas ferritin L stabilizes the multimeric ferritin shell (Arosio and Levi, 2002). Ferritin synthesis is subject to both transcriptional and translational regulation (MacKenzie et al., 2008a). Iron-mediated translational regulation via interactions of iron regulatory proteins with iron-responsive elements of ferritin H and L mRNA was extensively and elegantly characterized (Hentze et al., 2004; Rouault, 2006). Iron-independent regulation of ferritin H and L genes was observed under such conditions as oxidative stress, inflammation and cell differentiation (MacKenzie et al., 2008a; MacKenzie and Tsuji, 2008). We demonstrated that both mouse and human ferritin H genes are transcriptionally activated under oxidative stress via a well-conserved far-upstream enhancer element, antioxidant-responsive element (ARE) (Iwasaki et al., 2006; Tsuji, 2005; Tsuji et al., 1995; Tsuji et al., 2000). The induction of ferritin during oxidative stress is an important cell defence mechanism against oxidative cell damage (Kaur et al., 2003; MacKenzie et al., 2008b; Pham et al., 2004; Sakamoto et al., 2009).
The molecular mechanism through which ATF1 transcriptional function is regulated in oxidative and genotoxic stress conditions remains incompletely understood. To better understand the ATF1-mediated ARE regulation, in this study we attempted to find ATF1-interacting proteins by yeast two-hybrid screening and have identified homeodomain-interacting protein kinase 2 (HIPK2) as an ATF1 binding protein. HIPK2 belongs to an emerging family of nuclear Ser-Thr kinases that share significant homologies with the DYRK (MNB) dual-specificity kinases (Hofmann et al., 2000; Kim et al., 1998). These protein kinases play important roles in gene regulation, cell growth, differentiation and survival (Calzado et al., 2007; Rinaldo et al., 2007); however, molecular mechanisms behind their functions and diversities remain largely unknown. Among these kinases, HIPK2 has received much attention to its key regulatory role in DNA damage response. HIPK2 was initially identified as a binding protein and pivotal regulator of a homeodomain transcription factor, NKX1 (Kim et al., 1998). HIPK2 is involved in both cell survival and apoptosis (Sombroek and Hofmann, 2009). HIPK2 was shown to play a pivotal role in TGF-β-dependent cell survival of midbrain dopamine neurons via Smad interaction and subsequent activation of TGF-β target genes (Zhang et al., 2007). HIPK2 was also shown to be proapoptotic in response to ultraviolet (UV) exposure through p53 phosphorylation at Ser46, which inhibits MDM2-mediated degradation of p53 and ultimately activates p53-dependent apoptosis (D'Orazi et al., 2002; Hofmann et al., 2002). Furthermore, HIPK2 was shown to induce phosphorylation of anti-apoptotic transcriptional co-repressor CtBP (C-terminal binding protein) at Ser422, which in turn triggers CtBP ubiquitination and degradation (Zhang et al., 2005; Zhang et al., 2003).
In this study, we demonstrate that HIPK2 is a novel ATF1 kinase that phosphorylates a new phosphorylation site, Ser198, but not Ser63, and that HIPK2 appears to counteract the repressor function of ATF1 on the ferritin H ARE, resulting in induction of ferritin H and several other ARE-regulated antioxidant detoxification genes. These results suggest that HIPK2 is an important nuclear Ser-Thr kinase involved in cellular response to genotoxic and oxidative stress through post-translational regulation of the ATF1 transcription factor and expression of the ferritin H and other ARE-regulated antioxidant genes.
Results
HIPK2 phosphorylates ATF1
ATF1 serves as either a transcriptional activator or a repressor, including transcriptional repression of the ferritin H gene we observed (Iwasaki et al., 2007). To understand the regulatory mechanism of the ATF1 transcription factor and its effect on the ferritin H gene, yeast two-hybrid screening was employed to identify potential ATF1 binding proteins. A GAL4 DNA-binding domain fused to the human full-length ATF1 cDNA was used as bait for screening a murine B cell cDNA library, in which we identified HIPK2 as an ATF1 binding protein that allowed a transformed yeast clone to grow on His (−) dropout agar plates (Fig. 1A). To verify ATF1 and HIPK2 interaction in mammalian cells, HA-ATF1 and Flag-HIPK2 [either wild-type (wt) or kinase-dead (kd) HIPK2] were transiently transfected into HEK293 cells and cell lysates were subjected to immunoprecipitation with an anti-HIPK2 antibody followed by western blotting with anti-HA antibody. Only when HA-ATF1 and Flag-HIPK2 (either wt or kd) were co-expressed in the cells, did the HIPK2 antibody co-precipitate HA-ATF1 (Fig. 1B, top, lanes 5 and 6). In this experiment, we reproducibly detected a slower migrated ATF1 band in western blotting when co-expressed with wild-type HIPK2 (Fig. 1B, middle, lane 5) but not with kinase-dead HIPK2 (lane 6). Similar results of HIPK2-induced ATF1 retardation were observed in K562 and SH-SY5Y cells (B.-W. Huang and K. Sakamoto, unpublished observation). The fact that the retarded ATF1 migration was induced in a HIPK2 kinase-dependent manner suggests that ATF1 might be phosphorylated either directly or indirectly by HIPK2.
Fig. 1.

HIPK2 is an ATF1 binding protein. (A) PJ69-4A yeast clones transformed with indicated combinations of bait and prey (pGBDATF1 and pAct2HIPK2) were tested for their growth on a histidine-deficient agar plate. (B) Indicated combinations of pHA-ATF1 and pFlagHIPK2 [wild type (wt) or kinase dead (kd)] were transfected into HEK293 cells, and cell lysates were immunoprecipitated with anti-HIPK2 followed by western blotting with anti-HA antibody (top). The right-end lane was loaded with 10 μg of HEK293 whole-cell lysate transiently transfected with pHA-ATF1. The same set of cell lysates were subjected to western blotting with anti-HA (middle) or anti-HIPK2 (bottom). Of note is that wt HIPK2 (but not kd HIPK2) and HA-ATF1-transfected cell lysate exhibited a slower migrated ATF1 band (middle, lanes 5 and 6).
To further characterize the retarded ATF1, we expressed HA-ATF1 and HIPK2 (wt or kd) in HEK293 cells and whole-cell lysates prepared from duplicated transfection plates were independently analyzed by western blotting for detection of HA-ATF1. The results in Fig. 2A reproducibly showed that the retarded HA-ATF1 band was induced only when wild-type HIPK2 was co-expressed. To characterize whether the retarded ATF1 is phosphorylated, the whole-cell lysates were treated with phosphatase prior to the western blotting with anti-HA antibody. Indeed, the phosphatase treatment abolished the retarded band (Fig. 2B), indicating that the retarded ATF1 band is phosphorylated. The possibility of phosphorylation on the HA-tag is ruled out because there is no Ser-Thr in the tag sequence (Tyr-Pro-Tyr-Asp-Val-Pro-Asp-Tyr-Ala); a non-tagged ATF1 also showed the HIPK2-induced retardation (see below in Fig. 5).
Fig. 2.

HIPK2 phosphorylates ATF1. (A) Forty micrograms of HEK293 cell lysates transfected in duplicate with pCMVHAATF1 along with pCMVFlag (empty vector) or pCMVHIPK2-K221R kd or pCMVHIPK2 wt were independently separated on SDS-PAGE and analyzed by western blotting with anti-HA antibody. The HAATF1 and retarded HAATF1 bands were indicated by an arrowhead and arrow, respectively (top). The expression level of HIPK2 representing each duplicate was analyzed by western blotting with anti-HIPK2 antibody (bottom). (B) Forty micrograms of one of duplicated cell lysates in A were untreated or treated with λ-phosphatase for 37°C for 20 minutes prior to SDS-PAGE and western blotting with anti-HA antibody.
Fig. 5.

HIPK2 phosphorylates ATF1 at Ser198. (A) Wild-type Flag-HIPK2 and various ATF1 mutants (Ser63, Thr99, Ser164, Ser198 to Ala) were co-transfected into HEK293 cells and nuclear fractions were subjected to western blotting with anti-ATF1 antibody, anti-HIPK2 antibody or anti-lamin B antibody. ATF1 and retarded ATF1 are indicated with arrowheads. (B) Ten nanograms of His-tagged recombinant ATF1 wt, Ser198Ala (198A) or Ser63Ala (63A) was incubated with 4 ng recombinant HIPK2 (kinase domain aa 165–564) or PKA in the presence of 5 μCi γ-32P-ATP at 30°C for 20 minutes. Samples were loaded on 10% SDS-PAGE and phosphorylated bands were detected by autoradiograph. Comparable loading of the proteins was verified by gel Silver staining (bottom).
To test whether HIPK2 directly phosphorylates ATF1, we performed an in vitro kinase assay by incubating recombinant ATF1 and HIPK2 in the presence of γ-32P-ATP followed by SDS-PAGE and autoradiography. Recombinant PKA was used as a positive control for ATF1 phosphorylation at Ser63. ATF1 was 32P-labeled after incubation with 4 or 12 ng of HIPK2 in vitro in a dose-dependent manner (Fig. 3A). To our knowledge, Ser63 of ATF1 is the only phosphorylation site induced by PKA and related kinases (Rehfuss et al., 1991). Interestingly, HIPK2 phosphorylated wild-type ATF1 as well as Ser63Ala-mutant ATF1 (Fig. 3A). As expected, PKA, an ATF1 Ser63 kinase (Rehfuss et al., 1991), phosphorylated wild-type ATF1 but not Ser63Ala-mutant ATF1 (Fig. 3A). Thus, we concluded that HIPK2 directly phosphorylates ATF1 at new Ser-Thr site(s).
Fig. 3.

HIPK2 directly phosphorylates a novel ATF1 site, not Ser63. (A) Increasing amounts of recombinant HIPK2 (amino acids 165-564) was mixed with 2 μg of bacterially expressed His-ATF1 or His-ATF1 Ser63Ala mutant and incubated in the presence of γ-32P-ATP at 30°C for 20 minutes. Samples were separated on SDS-PAGE and visualized by autoradiography. PKA was used as a control. Asterisk indicates autophosphorylated HIPK2. Coomassie Blue staining of the same gel is shown (bottom). The stained band below the 76 kDa marker is BSA added to recombinant HIPK2 solution as a carrier protein. The molecular weight of the recombinant HIPK2 (a.a.165–564) is approximately 55 kDa. (B) Wt HIPK2 or kd HIPK2 (K221R) plasmid DNA were co-expressed with wt ATF1 or Ser63Ala-mutant ATF1 in HEK293 cells. Twenty-four hours after transfection, total cell lysates were collected and 40 μg of samples were separated on SDS-PAGE and then analyzed by western blotting using anti-phospho ATF1 Ser63 antibody (top), anti-ATF1 antibody (middle) or anti-HIPK2 antibody (bottom). Cell lysate with co-expression of PKA and ATF1 was used as a positive control of ATF1 Ser63 phosphorylation (lane 8).
These results do not exclude the possibility that HIPK2 also phosphorylates ATF1 at Ser63 in addition to the novel phosphorylation site. To test this possibility, Flag-HIPK2 (wt or kd) or PKA along with HA-ATF1 (wt or Ser63Ala) plasmids were expressed in HEK293 cells and the status of phospho-Ser63 ATF1 was determined by western blotting using an anti-phospho Ser63 ATF1 antibody. Expression of ATF1 alone showed a detectable amount of spontaneously phosphorylated ATF1 at Ser 63 (Fig. 3B, top, lane 2). By contrast, no phospho-Ser63 ATF1 band was detected when ATF1 Ser63Ala was expressed alone (lane 3). When wild-type HIPK2 was co-expressed with wild-type ATF1, no increase in ATF1 Ser63 phosphorylation was observed (Fig. 3B, top, lane 5, compare with lane 2). However, the anti-ATF1 western blot of the same sample (Fig. 3B, middle, lane 5) showed a clear retarded ATF1 band, suggesting that HIPK2 phosphorylated ATF1. Importantly, when wild-type HIPK2 was co-expressed with Ser63Ala-mutant ATF1, phospho-Ser63 ATF1 was undetectable (Fig. 3B, top, lane 6) but the retarded ATF1 band was similarly detected (middle, lane 6). Conversely, when wild-type ATF1 was co-expressed with kinase-dead HIPK2, the spontaneous basal level of phospho-Ser63 ATF1 was detected (top, lane 7) but no retarded ATF1 band was observed (middle, lane 7). When the catalytic subunit of PKA was co-expressed with wild-type ATF1, an increase in phosphorylation of Ser63 ATF1 was detected (Fig. 3B, top, lane 8), indicating that induced phosphorylation of ATF1 at Ser63 is detectable in our assays. Collectively, these results suggest that HIPK2 does not phosphorylate Ser63 but phosphorylates new Ser-Thr site(s) other than Ser63 of ATF1.
Phosphorylation at Ser198 of ATF1 and activation by HIPK2
To our knowledge, Ser63 in ATF1 is the only phosphorylation site so-far identified by PKA (Rehfuss et al., 1991), MSK1 (mitogen and stress activated kinase 1) (Wiggin et al., 2002) and several other kinases (Mayr and Montminy, 2001), leading to activation of the transcription function of ATF1. To examine the effect of HIPK2 on ATF1 transcription function, we employed GAL4-luciferase reporter assays by co-transfection of ATF1-GAL4-DNA binding-domain-fused plasmid (pFA-ATF1) and a GAL4-luciferase reporter along with HIPK2 into HEK293 cells. PKA was used as a positive control for ATF1 activation via Ser63 phosphorylation. In this assay, HIPK2 enhanced ATF1-dependent luciferase expression as efficiently as PKA (Fig. 4), suggesting that HIPK2 activates ATF1. When both HIPK2 and PKA were co-expressed, ATF1 was further activated, suggesting that the pathways of ATF1 activation by HIPK2 and PKA are independent.
Fig. 4.

HIPK2 activates ATF1 Transcription function. GAL4-DBD-luciferase plasmid was transfected into HEK293 cells together with GAL4 or GAL4-ATF1 (1–228) plus HIPK2 or PKA expression plasmid. The luciferase assay was carried out 48 hours after transfection. GAL4-ATF1 plus empty-vector-transfected sample was set as 1.0. DNA transfection was carried out in duplicate in each experiment and the result of four independent experiments and standard errors are shown.
Ser63 in ATF1 is flanked by Pro62. HIPK2 preferentially phosphorylates Ser-Thr sites adjacent to proline residues [such as Pro421-Ser422-Pro423 in CtBP (Zhang et al., 2003), Pro296-Ser297 in Grocho (Choi et al., 2005), Ser46-Pro47 in p53 (D'Orazi et al., 2002; Hofmann et al., 2002)]. Therefore, we predicted that Ser63 in ATF1 is one of potential HIPK2 phosphorylation sites. However, HIPK2 did not increase ATF1 Ser63 phosphorylation (Fig. 3B) and Ser63Ala-mutant ATF1 was equivalently phosphorylated as wild-type ATF1 by HIPK2 in our in vitro kinase assay (Fig. 3A), suggesting that HIPK2 is not an ATF1 Ser63 kinase but phosphorylates new Ser-Thr site(s). There are at least six other potential HIPK2 phosphorylation sites in human ATF1 at Pro98-Thr99-Pro100, Pro163-Ser164, Ser198-Pro199 (these are conserved in CREB), Ser122-Pro123 and Thr184-Pro185-Ser186 (these are not conserved in CREB). To determine new ATF1 phosphorylation site(s) by HIPK2, non-tagged ATF1 plasmids mutated at the conserved Ser-Thr between ATF1 and CREB proteins (Ser63, Thr99, Ser164 and Ser198 of ATF1) to Ala were co-expressed with HIPK2 and cell lysates were subjected to western blotting with anti-ATF1 antibody and examined for whether a particular ATF1 mutation abolishes the retarded band. As shown in Fig. 5A, only the Ser198Ala ATF1 mutant failed to show the retardation under the similar expression levels of transfected HIPK2. To verify that Ser198 is the ATF1 phosphorylation site by HIPK2, we conducted an in vitro kinase assay using recombinant wild-type, Ser198Ala and Ser63Ala ATF1 proteins. Indeed, HIPK2 phosphorylated wild-type and Ser63Ala ATF1 proteins but failed to phosphorylate Ser198Ala ATF1 (Fig. 5B). By contrast, PKA phosphorylated wild-type and Ser198Ala ATF1 proteins but failed to phosphorylate Ser63Ala ATF1 as expected (Fig. 5B). Collectively, we concluded that HIPK2 directly phosphorylates ATF1 at Ser198.
Transcriptional regulation of the human ferritin H gene by ATF1 and HIPK2
Ferritin and a battery of antioxidant genes are transcriptionally regulated via an ARE under oxidative and chemical stress conditions (MacKenzie et al., 2008a). We recently demonstrated by gel retardation and ChIP assays that ATF1 binds to the ferritin H ARE and represses ferritin H transcription in K562 human erythroleukemia cells (Iwasaki et al., 2007). ATF1 also repressed the ferritin H ARE-dependent transcription in HepG2 cells, in which the luciferase construct containing the ferritin H ARE (−4.5 kb) was repressed by ATF1, whereas the one lacking the ARE (−4.4 kb) showed lower basal luciferase expression and no further transcriptional repression by ATF1 (Fig. 6A). To assess the role of HIPK2 in the transcriptional regulation of the human ferritin H gene, −4.5 kb ARE (+) or −4.4 kb ARE (−)-luciferase was co-transfected with HIPK2 expression plasmid into HepG2 cells. As shown in Fig. 6B, HIPK2 induced expression of luciferase driven by the ARE (+) reporter, whereas there was no effect on the ARE (−) ferritin-H-luciferase expression. Then, we asked if HIPK2-mediated activation of the ferritin H transcription is dependent on the kinase activity. To address this question, wt or kd HIPK2 were expressed with the −4.5 kb ARE (+) ferritin-H-luciferase reporter into HepG2 cells and luciferase activity was measured. Varied amounts of wt HIPK2 transfection showed increased expression of luciferase, whereas kd HIPK2 had no effect on ferritin-H-luciferase expression (Fig. 7A).
Fig. 6.

Transcriptional regulation of the human ferritin H gene by ATF1 and HIPK2 via the ARE. (A) One microgram of −4.5 kb ferritin H luciferase [ARE (+)] or −4.4 kb ferritin H luciferase [ARE (−)] was co-transfected with indicated amounts of pcDNA3.1ATF1 into HepG2 cells. Total input of plasmid DNA was adjusted to 2 μg with pcDNA3.1 empty vector. Cells were harvested for luciferase assays 48–60 hours after DNA transfection (in duplicate with pRL-CMV as an internal transfection control). Luciferase expression after normalization with pRL-CMV from five independent experiments is shown with standard errors. The luciferase activity in cell lysates co-transfected with −4.5 kb ARE (+) and empty vector was defined as 100%. (B) One migrogram of −4.5 kb ARE (+)- or −4.4 kb ARE (−)-luciferase was co-transfected with indicated amounts of pFlagHIPK2 into HepG2 cells. Forty to forty-eight hours after transfection, cell lysates were prepared to run on the luciferase assay. Fold-induction in cell lysate obtained from cells transfected with −4.4 kb ARE (−) with empty vector was defined as 1.0. DNA transfection was carried out in duplicate in each experiment and the results of five independent experiments and standard errors are shown.
Fig. 7.
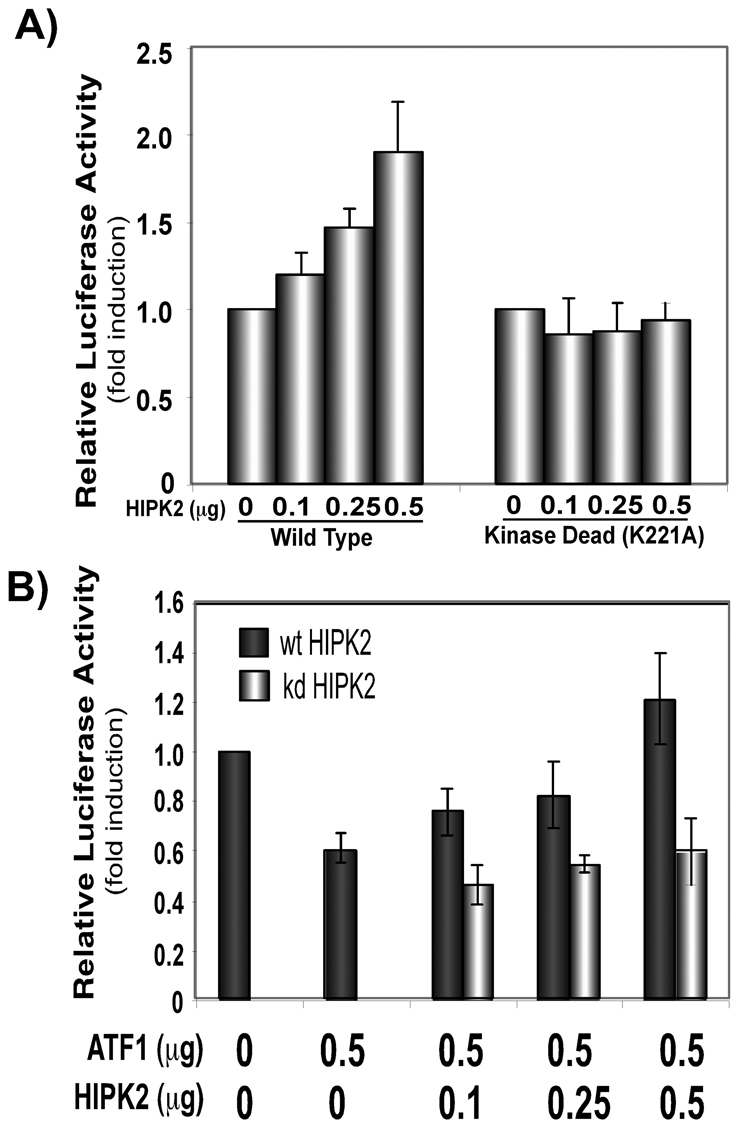
HIPK2 activates ferritin H transcription. (A) One migrogram of −4.5 kb human ferritin-H-luciferase reporter was co-transfected with indicated amounts of Flag-wtHIPK2 (wild type) or Flag-K221A-HIPK2 (kinase dead) into HepG2 cells. Fold-induction in cell lysate obtained from cells transfected with Flag empty vector was defined as 1.0. DNA transfection was carried out in duplicate in each experiment and the results of five independent experiments and standard errors are shown. (B) One migrogram of −4.5 kb human ferritin-H-luciferase reporter was co-transfected with indicated amounts of pCMVATF1 and pFlagHIPK2 (wt or kd) into HepG2 cells. Fold-induction in cell lysate obtained from cells transfected with empty vectors was defined as 1.0. DNA transfection was carried out in duplicate in each experiment and the results of four independent experiments and standard errors are shown.
We next examined whether HIPK2 affects the ATF1-mediated repression of ferritin H. To this end, ATF1 and HIPK2 plasmids were co-expressed in HepG2 cells along with −4.5 kb ARE (+) ferritin-H-luciferase and luciferase expression was measured. ATF1 reproducibly repressed ferritin-H-luciferase expression, and wt HIPK2 but not kd HIPK2 overrode the ATF1-mediated ferritin H repression (Fig. 7B). These results suggest that the reversal effect of HIPK2 on ATF1-mediated ARE repression is dependent on the HIPK2 kinase activity and that HIPK2-mediated phosphorylation of ATF1 might play a role in the reversal effect of HIPK2 on the ATF1-mediated ferritin H repression.
The role of endogenous HIPK2 in expression of ferritin and other antioxidant genes
To elucidate the role of endogenous HIPK2 in expression of ferritin in genotoxic stress, HIPK2+/+ and HIPK2−/− mouse embryonic fibroblasts (MEF) were treated with doxorubicin, etoposide and sodium arsenite for 20–24 hours, and ferritin H and L mRNA expression was measured. These genotoxic agents, known to activate HIPK2 (Hofmann et al., 2002; Rinaldo et al., 2007), induced ferritin H mRNA expression in HIPK2+/+ cells but not in HIPK2−/− MEF cells (Fig. 8A). The mouse ferritin L gene was shown to be regulated by an ARE (Wasserman and Fahl, 1997); however, these genotoxic agents showed only marginal effects on ferritin L mRNA expression even in HIPK2+/+ MEF cells (Fig. 8A). To examine whether the status of HIPK2 expression affects other well-characterized ARE-regulated antioxidant genes, 50 μM etoposide-treated MEF cells were analyzed in mRNA expression of NQO1, GSTα and HO1. Etoposide induced mRNA expression of NQO1 and GSTa in HIPK2+/+ MEF cells; however, the HIPK2 deficiency blunted the mRNA induction (Fig. 8B). To exclude the possibility of adaptation effects in MEF cells, similar experiments were performed in SH-SY5Y cells by knocking down HIPK2. As shown in Fig. 8C, etoposide treatment induced ferritin H and NQO1 expression and that was impaired by HIPK2 knockdown. These results suggest that HIPK2 plays an important role in upregulation of ferritin H and other ARE-regulated antioxidant genes in response to genotoxic stress.
Fig. 8.

HIPK2 deficiency affects expression of ferritin H and other ARE-regulated genes in genotoxic stress. (A) HIPK2+/+ or HIPK2−/− MEF cells were treated with 0.5 or 2.0 μg/ml doxorubicin (Dox), 10 or 50 μM etoposide (Eto) or 10 μM sodium arsenite (As). Twenty hours after the treatment, total RNA was isolated and subjected to real-time PCR for expression of ferritin H and L. (B) HIPK2+/+ or HIPK2−/− MEF cells were treated with 50 μM etoposide for 20 hours and expression of ferritin H (FH), ferritin L (FL), NAD(P)H quinone oxidoreductase-1 (NQO1), glutathione S-transferase-α (GSTa), or heme oxygenase-1 (HO1) mRNA was measured by real-time PCR. (C) SH-SY5Y cells were transfected with non-targeting (siControl) or HIPK2-targeting (siHIPK2) siRNA and incubated for 24 hours. Cells were then treated with 2 μM or 10 μM Etoposide for 48 hours and ferritin H and NQO1 mRNA expression was measured by real-time PCR. In B and C, mean and standard errors from three independent experiments were shown. *P<0.05, as determined by a Student's t-test.
Discussion
Receptor-mediated activation of the CREB family has been extensively studied, in which PKA and several other Ser-Thr kinases activate transactivation function of ATF1/CREB through phosphorylation at Ser63 in ATF1 and Ser133 in CREB that are located in the KID domain (Mayr and Montminy, 2001; Shaywitz and Greenberg, 1999). In this study, we demonstrate that HIPK2 phosphorylates ATF1 at Ser198, but not Ser63, and activates ATF1 transcription function. CREB and CREM (cAMP response element modulator) are closely related to ATF1 in their amino acid sequences and functional domains (Mayr and Montminy, 2001). In fact, the human ATF1 amino acid sequences containing the HIPK2 phosphorylation site are highly conserved in CREB and CREM transcription factors: ATF1, TVVMT-S(198)PV; CREB, GVVMASS(271)PA; CREM, GVVMAAS(226)PG. Therefore, this HIPK2-mediated ATF1 phosphorylation might be applicable to the entire CREB family. Indeed, we recently observed that HIPK2 phosphorylates CREB at Ser271, which activates CREB transcription function (Sakamoto et al., 2010).
CREB phosphorylation at Ser133 (equivalent to ATF1 Ser63) was shown to enhance subsequent recruitment of CREB-associated histone acetyltransferase CBP (CREB binding protein) (Arias et al., 1994; Chrivia et al., 1993; Kwok et al., 1994) via the KID, resulting in transcriptional activation of target genes. The HIPK2 phosphorylation site of ATF1 Ser198 is not in the KID but is localized adjacent to the second glutamine-rich region followed by the basic region of ATF1. We recently observed in ChIP assays that phosphorylation of CREB at Ser271 by HIPK2 showed no significant increase in DNA binding but increased recruitment of CBP on a BDNF promoter (Sakamoto et al., 2010). This suggests that HIPK2-mediated Ser198 phosphorylation might also stabilize the ATF1 and CBP interaction or facilitate the recruitment of CBP through phosphorylated Ser63. In addition, this new ATF1/CREB phosphorylation site by HIPK2 might recruit yet-unidentified coactivators to the vicinity of the binding site for transcriptional activation of target genes. The retarded migration of ATF1 with Ser198 phosphorylation but not Ser63 phosphorylation (Fig. 3B) implies that the impact of phosphorylation at these two sites on protein structure and activation mechanism might be different. Further investigation will be necessary for understanding the new regulatory mechanism of ATF1 and CREB family members by HIPK2.
HIPK2 has been characterized as a DNA-damage-responsive Ser-Thr kinase that functions either as a transcriptional co-repressor or co-activator (Rinaldo et al., 2007) and participates in cell death (Calzado et al., 2007) or survival (Zhang et al., 2007). HIPK2 protein levels are regulated by E3 ubiquitin ligases such as WSB1 (Choi et al., 2008) and SIAH1 (Winter et al., 2008). In addition, HIPK2 has recently been shown to regulate cellular hypoxic response, in which hypoxic conditions induce the interaction of HIPK2 and another ubiquitin E3 ligase SIAH2 that in turn facilitates HIPK2 polyubiquitination and proteasomal degradation (Calzado et al., 2009a). This HIPK2 degradation allows SIAH2 to induce degradation of prolyl hydroxylases, resulting in the stabilization of HIF1α (Calzado et al., 2009b). In accordance with the negative regulatory role of HIPK2 in HIF1α expression, HIPK2 was also shown to repress transcription of the HIF1a gene that causes sensitization of cells to doxorubicin-induced apoptosis under a hypoxia-mimicking condition (Nardinocchi et al., 2009). Thus, both at transcriptional and post-translational levels, HIPK2 appears to be a negative regulator of HIF1a expression and hypoxia-inducible gene expression. Conversely, ATF1 and CREB were reported to bind to the hypoxia response element together with HIF1α constitutively or an inducible manner under a hypoxia-mimicking condition (iron chelator or cobalt chloride treatment) (Ebert and Bunn, 1998; Kvietikova et al., 1995; Zaman et al., 1999) that upregulates hypoxia-responsive genes and protects cells from oxidative cell damage. Furthermore, ATF1 was identified as a hypoxia-responsive transcription factor, in which phosphorylation of ATF1 at Ser63 via a p38 MAP kinase pathway was involved in hypoxia-induced transcriptional activation of the uncoupling protein 3 gene in myotubes (Lu and Sack, 2008). The degradation of HIPK2 during hypoxia might cause the ratio to change towards more phospho-Ser63 ATF1, less phospho-Ser198 ATF1 along with HIF1α accumulation via the aforementioned mechanism. Our results suggest that phosphorylation of Ser63 (by PKA) and Ser198 (by HIPK2) activates ATF1 transcription function additively or independently (Fig. 4); however, the hypoxia response element might have the preference of phospho-Ser63 ATF1 in cooperation with HIF1α for expression of hypoxia-inducible genes. It remains to be determined in the future whether there is a positive or negative crosstalk between Ser63 and Ser198 phosphorylation of ATF1 in expression of its target genes.
Transcriptional repression of the ferritin H gene appears to be a strategy of oncogenes for enhancing cell proliferation because lower ferritin H expression levels ultimately increase the intracellular iron pool for cell proliferation and metabolism (Kakhlon et al., 2001). We previously demonstrated that the adenovirus E1A oncogene transcriptionally represses ferritin H in mouse NIH3T3 fibroblasts (Tsuji et al., 1993a), later it turned out to be regulated through the ARE (Tsuji et al., 1995; Tsuji et al., 2000). c-myc was shown to repress transcription of ferritin H along with an increased intracellular iron pool that was required for c-myc-induced cell transformation (Wu et al., 1999). Our results show that ATF1 is another transcriptional repressor of ferritin H in non-stress conditions (Fig. 6). Expression of ATF1 is upregulated in several cancer cells such as lymphomas (Hsueh and Lai, 1995) and metastatic melanoma cells (Jean et al., 2000). In addition, the EWS–ATF1-fused gene via t(12;22) chromosomal translocation is involved in proliferation of clear-cell sarcoma (Zucman et al., 1993). Thus, ATF1 could be another gene product that enhances cell proliferation through transcriptional repression of ferritin H and a subsequent increase in intracellular iron levels.
Iron is an essential element for various metabolic pathways and cell proliferation; however, excess iron is potentially detrimental because it catalyzes formation of a highly toxic hydroxyl radical in Fenton chemistry (Papanikolaou and Pantopoulos, 2005). Therefore, under oxidative and some chemical stress conditions, ferritin transcription is activated through the ARE and cells limit the availability of intracellular labile iron. In this study, under exposure to such genotoxic agents as doxorubicin and etoposide that are known to activate HIPK2 (Rinaldo et al., 2007), mRNA expression for ferritin H and other ARE-regulated antioxidant genes was upregulated in HIPK2+/+ cells but not HIPK2−/− or knockdown cells (Fig. 8). Thus, HIPK2 appears to be involved in ARE-regulated antioxidant gene transcription in these stress conditions. We observed that HIPK2−/− MEF cells proliferate much faster than HIPK2+/+ MEF cells as previously reported (Wei et al., 2007), and of note is that they are more susceptible to oxidative and genotoxic agents such as etoposide, doxorubicin and cisplatin (Sakamoto et al., 2010). The increased susceptibility to these agents could be, at least in part, due to the lack of induction of ARE-regulated ferritin H and other antioxidant genes in HIPK2-deficient cells.
Basic-leucine zipper transcription factors such as NRF2 (NFE2-related factor 2) and small MAF proteins (MAFK and MAFG) have been demonstrated to regulate various phase II detoxification genes via the ARE enhancer (Motohashi and Yamamoto, 2004). NRF2 and JUND are involved in the transcriptional activation of the ferritin H gene via the ARE during oxidative stress through their post-translational modifications such as redox and phosphorylation (Iwasaki et al., 2006; Tsuji, 2005). We were not able to detect endogenous HIPK2 protein by western blotting in HepG2 and other cell types, even after our trials with almost all commercially available anti-HIPK2 antibodies, perhaps because of generally low expression levels of HIPK2 as reported (Wang et al., 2001), and as noted by others that currently no commercially available antibody can efficiently detect endogenous HIPK2 protein (Boucher et al., 2009). Further investigation will be necessary to elucidate the HIPK2-induced ARE activation mechanism, specifically as to whether HIPK2 primarily activates, through phosphorylation, ATF1 and/or NRF2 or other ARE-binding proteins including coactivators such as p300 and CBP (Aikawa et al., 2006) and core histones.
Materials and Methods
Cell culture
The HepG2 human hepatocarcinoma cells and HEK293 human embryonic kidney cells (ATCC) were cultured in minimum essential medium (MEM) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids and 10% fetal bovine serum (FBS; Mediatech). SH-SY5Y cells were cultured in a 1:1 mixture of MEM and HAM's F12 medium supplemented with 0.1 mM non-essential amino acids and 10% FBS. HIPK2+/+ and HIPK2−/− mouse embryonic fibroblasts (MEF), kindly provided by Eric J. Huang at University of California San Francisco (Wei et al., 2007; Wiggins et al., 2004), were cultured in DMEM with 2 mM L-glutamine and 10% FBS. Cells were maintained and incubated during experiments at 37°C in a humidified 5% CO2 atmosphere.
Plasmids and antibodies
Cloning of the human ferritin H 5′ upstream enhancer and promoter region and construction of luciferase reporter plasmids were previously described (Tsuji, 2005). pFlagHIPK2 wild-type and K221A or K221R kinase-deficient HIPK2 were kindly provided by M. Lienhard Schmitz and Jun Ninomiya-Tsuji. ATF1 point mutations (63A, 99A, 164A and 198A) were constructed with the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Mouse PKA cDNA (Clontech) was subcloned into pCMV vector. Antibodies used in this study were: anti-HA (HA11, Covance), anti-Flag (M2, SIGMA), anti-HIPK2 (C15, Santa Cruz Biotechnology) and anti-ATF1 (C41-5.1, Santa Cruz Biotechnology).
DNA transfection and luciferase reporter assays
Transient DNA transfection into HepG2 cells was carried out by the calcium phosphate precipitation method as described previously (Tsuji et al., 1993b). Briefly, cells were plated at a density of 4×105 cells per 35 mm plate containing 2 ml of the culture medium (in duplicate per transfection) and a total of 0.2 ml of calcium phosphate solution containing 0.5–1 μg of each reporter plasmid DNA was added to the cells and incubated for 40–48 hours. The GAL4 reporter assays were performed as described previously (Iwasaki et al., 2006). To monitor and normalize the differences in transfection efficiency in each plate, 0.1 μg of pRL-CMV (Promega) or pRL-EF (elongation factor promoter) was simultaneously cotransfected. Preparation of cell extracts and luciferase assays were performed using Dual Luciferase Assay Reagents (Promega) and the luciferase activity was measured with Luminometer (Model 20E, Turner Designs). Luciferase expression in each transfected sample was normalized by Renilla luciferase activity.
Yeast two-hybrid assay
Yeast two-hybrid screening of a mouse B-cell pGAD cDNA library was performed using a human full-length ATF1 (pGBDATF1) as bait and the procedure was described previously (Iwasaki et al., 2007). Colonies of yeast PJ69-4A retransformed with pGADHIPK2 and pGBDATF1 (grown on tryptophan- and leucine-deficient synthetic dropout agar plates) were tested for growth on tryptophan-, leucine- and histidine-deficient agar media containing 2 mM 3-aminotriazole.
Immunoprecipitation and western blotting
HEK293 cells were transfected with 10 μg of pCMVFlagHIPK2 [wild-type (wt) or kinase-dead (kd) K221R] and 10 μg of pCMVHAATF1 plasmids by calcium phosphate transfection method. Whole-cell lysates were prepared at 40-48 hours after transfection and protein concentration of each cell lysate was measured with Bio-Rad protein assay reagent. Three hundred micrograms of the whole-cell lysates was immunoprecipitated with anti-HIPK2 antibody and then subjected to western blotting with anti-HA antibody. Expression levels of transfected Flag-HIPK2 and HAATF1 in the cell lysates (40 μg) were analyzed by western blotting with anti-HIPK2 and anti-HA antibodies, respectively. In the experiments of phosphatase treatment, 40 μg of HEK293 whole-cell lysates transfected with HAATF1 and Flag-HIPK2 (wt or kd K221R) were incubated with 800 units of λ-phosphatase (New England Biolabs) for 20 minutes at 37°C prior to western blotting with anti-HA antibody.
HIPK2 knockdown and real-time PCR
SH-SY5Y cells (1×107) were transfected with 100 pmol non-targeting siRNA (5′-UAGCGACUAAACACAUCAAUU-3′; D-001210-01; Dharmacon) or siHIPK2 (5′-GAGAAUCACUCCAAUCGAA-3′; J-003266-10; Dharmacon) using Gene Pulser X-Cell in 100 μl serum- and antibiotics-free media. After treatment with genotoxic chemicals, RNA was isolated with TRIzol reagent (Invitrogen) and real-time PCR was carried out to measure ferritin H, ferritin L, NQO1, GSTa, HO1, HIPK2 or GAPDH mRNA with SYBR Green PCR Master Mix (Applied Biosystems) in the presence of primer set for ferritin H (human, Qiagen QT00072681; mouse, 5′-GCCTCCTACGTCTATCTGTCTATGTC-3′ and 5′-TGGTGGAGAAAGTATTTGGC-3′), mouse ferritin L (5′-AATCAGGCCCTCTTGGATC-3′ and 5′-GATAGTGGCTTTCCAGGAAGTC-3′), NQO1 (human, Qiagen QT00050281; mouse, 5′-CATTCTGAAAGGCTGGTTTGA-3′ and 5′-CTAGCTTTGATCTGGTTGTCAG-3′), mouse GSTa (5′-GCCAAGTACCCTTGGTTGAA-3′ and 5′-AATCCTGACCACCTCAACA-3′), mouse HO1 (5′-CCCACCAAGTTCAAACAGCTC-3′ and 5′-AGGAAGGCGGTCTTAGCCTC-3′), HIPK2 (QT00051485, Qiagen) or GAPDH (QT00079247, Qiagen). The value of each mRNA expression was normalized by GAPDH mRNA expression. Approximately 70% HIPK2 knockdown in SH-SY5Y cells was achieved by our electroporation procedure (Sakamoto et al., 2010).
In vitro kinase assay
Recombinant HIPK2 (amino acids 165-564, Upstate/Millipore), PKA (human full-length catalytic subunit, Upstate/Millipore), and ATF1 (amino acids 39-271, Santa Cruz Biotechnology) proteins were used (Fig. 3A). His-tagged human ATF1 plasmids (pQE31ATF1 wt, 63A, and 198A) were constructed and recombinant full-length ATF1 proteins were purified as described (Sakamoto et al., 2010) with minor modifications. Indicated amounts of recombinant proteins were incubated at 30°C for 20–30 minutes in kinase buffer (10 mM HEPES, pH 7.4, 5 mM MgCl2, 1 mM DTT) in the presence of 5 uCi γ-32P-ATP and 100 μM unlabelled ATP. Samples were then separated on 10% SDS-PAGE and subjected to Coomassie Blue or Silver staining (SilverQuest, Invitrogen) and autoradiography.
Acknowledgments
We are grateful to Eric J. Huang at University of California San Francisco for providing us with the HIPK2 wild-type and knockout MEF cells. We thank M. Lienhard Schmitz at Justus-Liebig University, Germany for HIPK2 plasmids. We thank Jun Ninomiya-Tsuji at North Carolina State University for plasmids and discussion. This work was supported in part by National Institutes of Health research grant DK-60007 to Y.T. K.H. was supported by the National Institutes of Health training grant ES-007046 and the National Institutes of Health minority supplement grant DK-60007S. B.-W.H. was supported by North Carolina State University Research Assistant Fellowship. Deposited in PMC for release after 12 months.
References
- Aikawa Y., Nguyen L. A., Isono K., Takakura N., Tagata Y., Schmitz M. L., Koseki H., Kitabayashi I. (2006). Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 25, 3955-3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias J., Alberts A. S., Brindle P., Claret F. X., Smeal T., Karin M., Feramisco J., Montminy M. (1994). Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature 370, 226-229 [DOI] [PubMed] [Google Scholar]
- Arosio P., Levi S. (2002). Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 33, 457-463 [DOI] [PubMed] [Google Scholar]
- Atlas E., Stramwasser M., Mueller C. R. (2001). A CREB site in the BRCA1 proximal promoter acts as a constitutive transcriptional element. Oncogene 20, 7110-7114 [DOI] [PubMed] [Google Scholar]
- Belmonte N., Phillips B. W., Massiera F., Villageois P., Wdziekonski B., Saint-Marc P., Nichols J., Aubert J., Saeki K., Yuo A., et al. (2001). Activation of extracellular signal-regulated kinases and CREB/ATF-1 mediate the expression of CCAAT/enhancer binding proteins beta and -delta in preadipocytes. Mol. Endocrinol. 15, 2037-2049 [DOI] [PubMed] [Google Scholar]
- Bosilevac J. M., Olsen R. J., Bridge J. A., Hinrichs S. H. (1999). Tumor cell viability in clear cell sarcoma requires DNA binding activity of the EWS/ATF1 fusion protein. J. Biol. Chem. 274, 34811-34818 [DOI] [PubMed] [Google Scholar]
- Boucher M. J., Simoneau M., Edlund H. (2009). The homeodomain-interacting protein kinase 2 regulates insulin promoter factor-1/pancreatic duodenal homeobox-1 transcriptional activity. Endocrinology 150, 87-97 [DOI] [PubMed] [Google Scholar]
- Calzado M. A., Renner F., Roscic A., Schmitz M. L. (2007). HIPK2: a versatile switchboard regulating the transcription machinery and cell death. Cell Cycle 6, 139-143 [DOI] [PubMed] [Google Scholar]
- Calzado M. A., de la Vega L., Moller A., Bowtell D. D., Schmitz M. L. (2009a). An inducible autoregulatory loop between HIPK2 and Siah2 at the apex of the hypoxic response. Nat. Cell Biol. 11, 85-91 [DOI] [PubMed] [Google Scholar]
- Calzado M. A., De la Vega L., Munoz E., Schmitz M. L. (2009b). From top to bottom: the two faces of HIPK2 for regulation of the hypoxic response. Cell Cycle 8, 1659-1664 [DOI] [PubMed] [Google Scholar]
- Choi C. Y., Kim Y. H., Kim Y. O., Park S. J., Kim E. A., Riemenschneider W., Gajewski K., Schulz R. A., Kim Y. (2005). Phosphorylation by the DHIPK2 protein kinase modulates the corepressor activity of Groucho. J. Biol. Chem. 280, 21427-21436 [DOI] [PubMed] [Google Scholar]
- Choi D. W., Seo Y. M., Kim E. A., Sung K. S., Ahn J. W., Park S. J., Lee S. R., Choi C. Y. (2008). Ubiquitination and degradation of homeodomain-interacting protein kinase 2 by WD40 repeat/SOCS box protein WSB-1. J. Biol. Chem. 283, 4682-4689 [DOI] [PubMed] [Google Scholar]
- Chrivia J. C., Kwok R. P. S., Lamb N., Hagiwara M., Montominy M. R., Goodman R. H. (1993). Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365, 855-859 [DOI] [PubMed] [Google Scholar]
- Dong Y., Asch H. L., Ying A., Asch B. B. (2002). Molecular mechanism of transcriptional repression of gelsolin in human breast cancer cells. Exp. Cell Res. 276, 328-336 [DOI] [PubMed] [Google Scholar]
- D'Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G., et al. (2002). Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 4, 11-19 [DOI] [PubMed] [Google Scholar]
- Ebert B. L., Bunn H. F. (1998). Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell. Biol. 18, 4089-4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze M. W., Muckenthaler M. U., Andrews N. C. (2004). Balancing acts: molecular control of mammalian iron metabolism. Cell 117, 285-297 [DOI] [PubMed] [Google Scholar]
- Hofmann T. G., Mincheva A., Lichter P., Droge W., Schmitz M. L. (2000). Human homeodomain-interacting protein kinase-2 (HIPK2) is a member of the DYRK family of protein kinases and maps to chromosome 7q32-q34. Biochimie 82, 1123-1127 [DOI] [PubMed] [Google Scholar]
- Hofmann T. G., Moller A., Sirma H., Zentgraf H., Taya Y., Droge W., Will H., Schmitz M. L. (2002). Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4, 1-10 [DOI] [PubMed] [Google Scholar]
- Hsueh Y. P., Lai M. Z. (1995). Overexpression of activation transcriptional factor 1 in lymphomas and in activated lymphocytes. J. Immunol. 154, 5675-5683 [PubMed] [Google Scholar]
- Iwasaki K., Mackenzie E. L., Hailemariam K., Sakamoto K., Tsuji Y. (2006). Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol. Cell. Biol. 26, 2845-2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki K., Hailemariam K., Tsuji Y. (2007). PIAS3 interacts with ATF1 and regulates the human ferritin H gene through an antioxidant-responsive element. J. Biol. Chem. 282, 22335-22343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean D., Tellez C., Huang S., Davis D. W., Bruns C. J., McConkey D. J., Hinrichs S. H., Bar-Eli M. (2000). Inhibition of tumor growth and metastasis of human melanoma by intracellular anti-ATF-1 single chain Fv fragment. Oncogene 19, 2721-2730 [DOI] [PubMed] [Google Scholar]
- Kakhlon O., Gruenbaum Y., Cabantchik Z. I. (2001). Repression of the heavy ferritin chain increases the labile iron pool of human K562 cells. Biochem. J. 356, 311-316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur D., Yantiri F., Rajagopalan S., Kumar J., Mo J. Q., Boonplueang R., Viswanath V., Jacobs R., Yang L., Beal M. F., et al. (2003). Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron 37, 899-909 [DOI] [PubMed] [Google Scholar]
- Kim Y. H., Choi C. Y., Lee S. J., Conti M. A., Kim Y. (1998). Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J. Biol. Chem. 273, 25875-25879 [DOI] [PubMed] [Google Scholar]
- Kingsley-Kallesen M. L., Kelly D., Rizzino A. (1999). Transcriptional regulation of the transforming growth factor-beta2 promoter by cAMP-responsive element-binding protein (CREB) and activating transcription factor-1 (ATF-1) is modulated by protein kinases and the coactivators p300 and CREB-binding protein. J. Biol. Chem. 274, 34020-34028 [DOI] [PubMed] [Google Scholar]
- Kvietikova I., Wenger R. H., Marti H. H., Gassmann M. (1995). The transcription factors ATF-1 and CREB-1 bind constitutively to the hypoxia-inducible factor-1 (HIF-1) DNA recognition site. Nucleic Acids Res. 23, 4542-4550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok R. P. S., Lundblad J. R., Chrivia J. C., Richards J. P., Bachinger H. P., Brennan R. G., Roberts S. G. E., Green M. R., Goodman R. H. (1994). Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 370, 223-226 [DOI] [PubMed] [Google Scholar]
- Lee M. G., Pedersen P. L. (2003). Glucose metabolism in cancer: importance of transcription factor-DNA interactions within a short segment of the proximal region of the type II hexokinase promoter. J. Biol. Chem. 278, 41047-41058 [DOI] [PubMed] [Google Scholar]
- Lu Z., Sack M. N. (2008). ATF-1 is a hypoxia-responsive transcriptional activator of skeletal muscle mitochondrial-uncoupling protein 3. J. Biol. Chem. 283, 23410-23418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie E. L., Tsuji Y. (2008). Elevated intracellular calcium increases ferritin H expression through an NFAT-independent post-transcriptional mechanism involving mRNA stabilization. Biochem. J. 411, 107-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie E. L., Iwasaki K., Tsuji Y. (2008a). Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid. Redox. Signal. 10, 997-1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie E. L., Ray P. D., Tsuji Y. (2008b). Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radic. Biol. Med. 44, 1762-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B., Montminy M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599-609 [DOI] [PubMed] [Google Scholar]
- Motohashi H., Yamamoto M. (2004). Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549-557 [DOI] [PubMed] [Google Scholar]
- Nardinocchi L., Puca R., Sacchi A., D'Orazi G. (2009). Inhibition of HIF-1alpha activity by homeodomain-interacting protein kinase-2 correlates with sensitization of chemoresistant cells to undergo apoptosis. Mol. Cancer 8, 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama Y., Sowa Y., Fujita T., Mizuno T., Nomura H., Nikaido T., Endo T., Sakai T. (1996). ATF site of human RB gene promoter is a responsive element of myogenic differentiation. FEBS Lett. 397, 219-224 [DOI] [PubMed] [Google Scholar]
- Papanikolaou G., Pantopoulos K. (2005). Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 202, 199-211 [DOI] [PubMed] [Google Scholar]
- Pham C. G., Bubici C., Zazzeroni F., Papa S., Jones J., Alvarez K., Jayawardena S., De Smaele E., Cong R., Beaumont C., et al. (2004). Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119, 529-542 [DOI] [PubMed] [Google Scholar]
- Rehfuss R. P., Walton K. M., Loriaux M. M., Goodman R. H. (1991). The cAMP-regulated enhancer-binding protein ATF-1 activates transcription in response to cAMP-dependent protein kinase A. J. Biol. Chem. 266, 18431-18434 [PubMed] [Google Scholar]
- Rinaldo C., Prodosmo A., Siepi F., Soddu S. (2007). HIPK2: a multitalented partner for transcription factors in DNA damage response and development. Biochem. Cell Biol. 85, 411-418 [DOI] [PubMed] [Google Scholar]
- Rolli M., Kotlyarov A., Sakamoto K. M., Gaestel M., Neininger A. (1999). Stress-induced stimulation of early growth response gene-1 by p38/stress-activated protein kinase 2 is mediated by a cAMP-responsive promoter element in a MAPKAP kinase 2-independent manner. J. Biol. Chem. 274, 19559-19564 [DOI] [PubMed] [Google Scholar]
- Rouault T. A. (2006). The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406-414 [DOI] [PubMed] [Google Scholar]
- Sakamoto K., Iwasaki K., Sugiyama H., Tsuji Y. (2009). Role of the tumor suppressor PTEN in antioxidant responsive element-mediated transcription and associated histone modifications. Mol. Biol. Cell 20, 1606-1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K., Huang B. W., Iwasaki K., Hailemariam K., Ninomiya-Tsuji J., Tsuji Y. (2010). Regulation of genotoxic stress response by homeodomain-interacting protein kinase 2 through phosphorylation of cyclic AMP response element-binding protein at serine 271. Mol. Biol. Cell 21, 2966-2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K., Wang S., Costa M. (1997). Induction of activating transcription factor 1 by nickel and its role as a negative regulator of thrombospondin I gene expression. Cancer Res. 57, 5060-5066 [PubMed] [Google Scholar]
- Shaywitz A. J., Greenberg M. E. (1999). CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821-861 [DOI] [PubMed] [Google Scholar]
- Sombroek D., Hofmann T. G. (2009). How cells switch HIPK2 on and off. Cell Death Differ. 16, 187-194 [DOI] [PubMed] [Google Scholar]
- Theil E. C. (2003). Ferritin: at the crossroads of iron and oxygen metabolism. J. Nutr. 133, 1549S-1553S [DOI] [PubMed] [Google Scholar]
- Tsuji Y. (2005). JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 24, 7567-7578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y., Kwak E., Saika T., Torti S. V., Torti F. M. (1993a). Preferential repression of the H subunit of ferritin by adenovirus E1A in NIH-3T3 mouse fibroblasts. J. Biol. Chem. 268, 7270-7275 [PubMed] [Google Scholar]
- Tsuji Y., Ninomiya-Tsuji J., Torti S. V., Torti F. M. (1993b). Augmentation by IL-1a of tumor necrosis factor-a cytotoxicity in cells transfected with adenovirus E1A. J. Immunol. 150, 1897-1907 [PubMed] [Google Scholar]
- Tsuji Y., Akebi N., Lam T. K., Nakabeppu Y., Torti S. V., Torti F. M. (1995). FER-1, an enhancer of the ferritin H gene and a target of E1A-mediated transcriptional repression. Mol. Cell. Biol. 15, 5152-5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y., Ayaki H., Whitman S. P., Morrow C. S., Torti S. V., Torti F. M. (2000). Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell. Biol. 20, 5818-5827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Hofmann T. G., Runkel L., Haaf T., Schaller H., Debatin K., Hug H. (2001). Isolation and characterization of cDNAs for the protein kinase HIPK2. Biochim. Biophys. Acta 1518, 168-172 [DOI] [PubMed] [Google Scholar]
- Wasserman W. W., Fahl W. E. (1997). Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 94, 5361-5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G., Ku S., Ma G. K., Saito S., Tang A. A., Zhang J., Mao J. H., Appella E., Balmain A., Huang E. J. (2007). HIPK2 represses beta-catenin-mediated transcription, epidermal stem cell expansion, and skin tumorigenesis. Proc. Natl. Acad. Sci. USA 104, 13040-13045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin G. R., Soloaga A., Foster J. M., Murray-Tait V., Cohen P., Arthur J. S. (2002). MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol. 22, 2871-2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins A. K., Wei G., Doxakis E., Wong C., Tang A. A., Zang K., Luo E. J., Neve R. L., Reichardt L. F., Huang E. J. (2004). Interaction of Brn3a and HIPK2 mediates transcriptional repression of sensory neuron survival. J. Cell Biol. 167, 257-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M., Sombroek D., Dauth I., Moehlenbrink J., Scheuermann K., Crone J., Hofmann T. G. (2008). Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat. Cell. Biol. 10, 812-824 [DOI] [PubMed] [Google Scholar]
- Wu K. J., Polack A., Dalla-Favera R. (1999). Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 283, 676-679 [DOI] [PubMed] [Google Scholar]
- Zaman K., Ryu H., Hall D., O'Donovan K., Lin K. I., Miller M. P., Marquis J. C., Baraban J. M., Semenza G. L., Ratan R. R. (1999). Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J. Neurosci. 19, 9821-9830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Pho V., Bonasera S. J., Holtzman J., Tang A. T., Hellmuth J., Tang S., Janak P. H., Tecott L. H., Huang E. J. (2007). Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat. Neurosci. 10, 77-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. W., Klemm D. J., Vinson C., Lane M. D. (2004). Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J. Biol. Chem. 279, 4471-4478 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Yoshimatsu Y., Hildebrand J., Frisch S. M., Goodman R. H. (2003). Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 115, 177-186 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Nottke A., Goodman R. H. (2005). Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proc. Natl. Acad. Sci. USA 102, 2802-2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucman J., Delattre O., Desmaze C., Epstein A. L., Stenman G., Speleman F., Fletchers C. D., Aurias A., Thomas G. (1993). EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat. Genet. 4, 341-345 [DOI] [PubMed] [Google Scholar]