

Abstract
The decrease in hemoglobin A (HbA, α2β2) synthesis in the erythroid cells of patients with β-thalassemia is due to a selective defect in β-chain synthesis. Since α-chains continue to be formed at a normal rate in these cells, this results in a marked relative excess of α-chain synthesis over β- and γ-chain synthesis. The α-chains uncombined with β- or β-like-chains (δ, γ) will be referred to as free α-chains. The experiments presented in this paper show that these free α-chains are capable of combining with β-chains to form HbA and are, therefore, structurally normal. Alternatively, in the absence of added β-chains, α-chains aggregates of various sizes are formed.
Peripheral blood from patients with β-thalassemia was incubated with radioactive amino acids and hemolysates were prepared. Column chromatography demonstrates that a majority of the free α-chains are not present in HbA. They are strongly bound to carboxymethylcellulose resin at pHs from 7.0 to 10.0, and do not elute with HbA. However, when chemically prepared hemoglobin H (Hbβ4) is added to the fresh hemolysates, the free α-chains are readily recovered in the HbA peak. This indicates that the free α-chains are able to combine normally with β-chains to form HbA. Freshly labeled hemolysates were also subjected to Sephadex G-100 chromatography. The free α-chains eluted as a broad peak migrating between myoglobin and hemoglobin, consistent with their forming α-chain aggregates of various mol wt between 16,000 and 64,000.
It is suggested that the chromatographic behavior of the free α-chains reported herein simply reflects the chemical properties of normal α-chains in the absence of adequate numbers of β- or γ-chains. The tendency of these free α-chains to aggregate may lead to their intracellular precipitation and the subsequent destruction of the cells containing them.
Full text
PDF
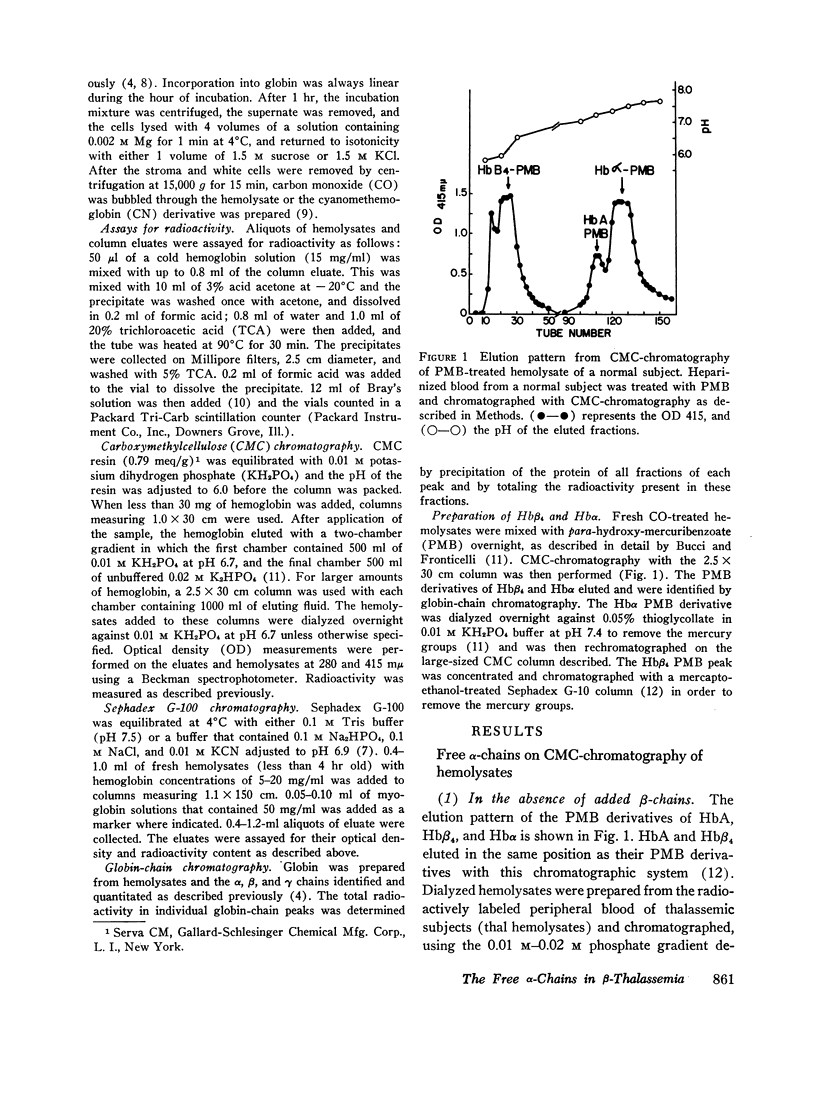
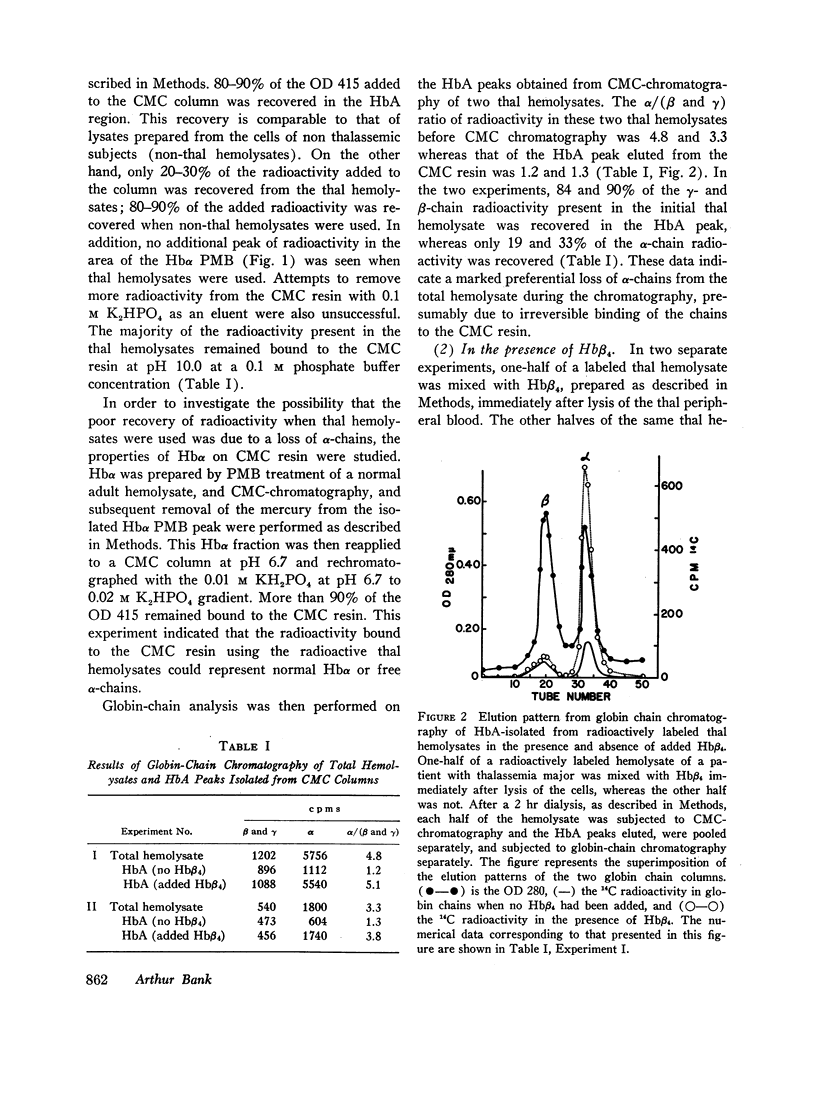
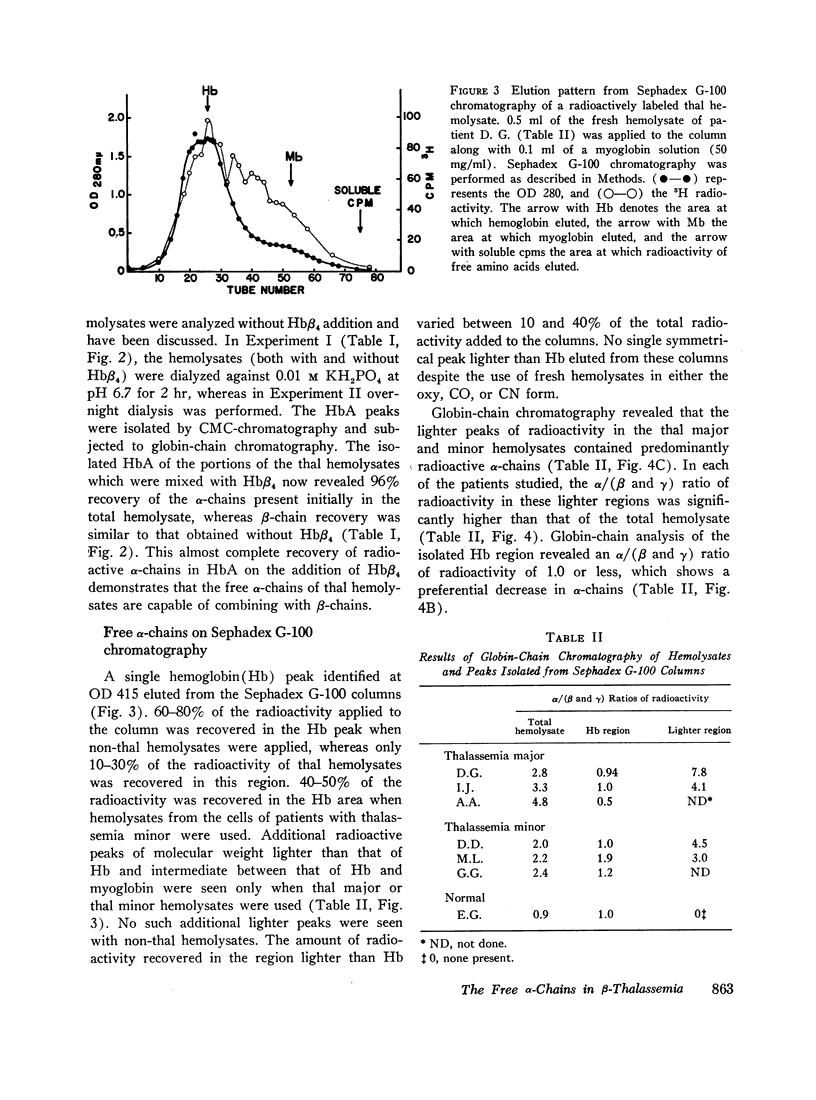
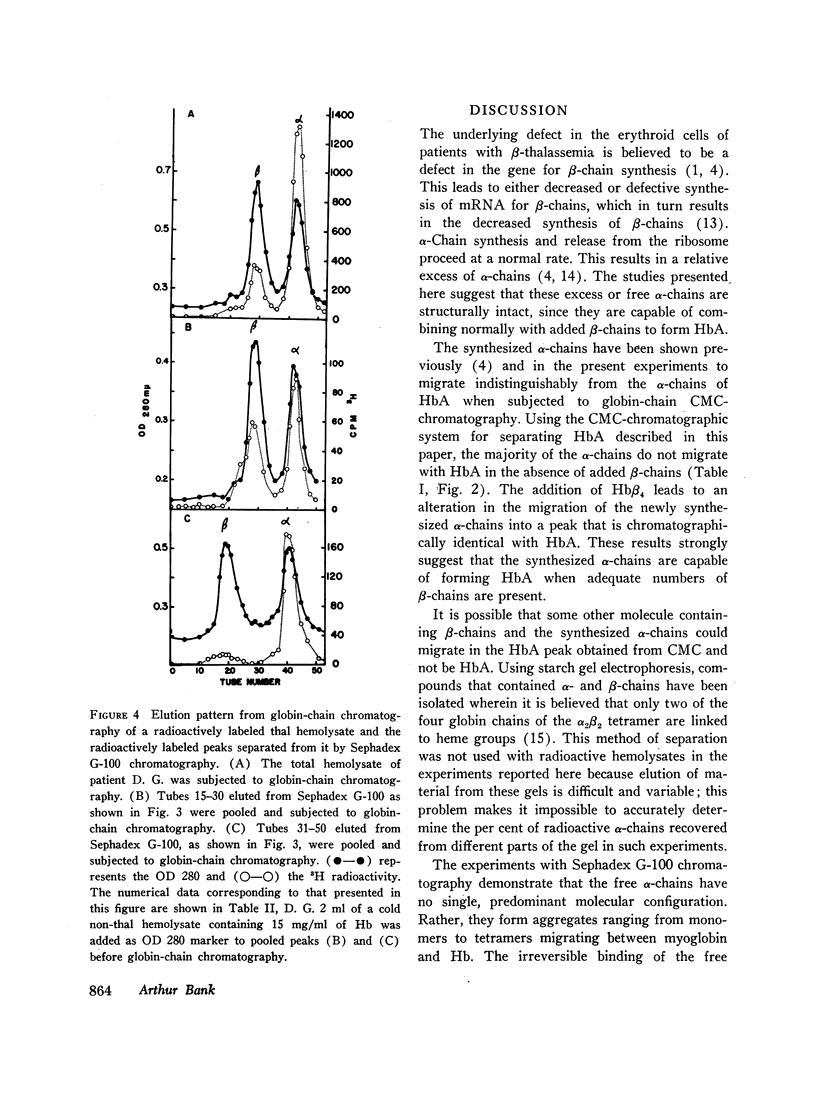


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Banerjee K., Lauffer M. A. Polymerization--depolymerization of tobacco mosaic virus protein. VI. Osmotic pressure studies of early stages of polymerization. Biochemistry. 1966 Jun;5(6):1957–1964. doi: 10.1021/bi00870a024. [DOI] [PubMed] [Google Scholar]
- Bank A., Braverman A. S., O'Donnell J. V., Marks P. A. Absolute rates of globin chain synthesis in thalassemia. Blood. 1968 Feb;31(2):226–233. [PubMed] [Google Scholar]
- Bank A., Marks P. A. Protein synthesis in a cell free human reticulocyte system: ribosome function in thalassemia. J Clin Invest. 1966 Mar;45(3):330–336. doi: 10.1172/JCI105347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargellesi A., Pontremoli S., Conconi F. Absence of beta-globin synthesis and excess of alpha-globin synthesis in homozygous beta-thalassemia. Eur J Biochem. 1967 Mar;1(1):73–79. doi: 10.1007/978-3-662-25813-2_13. [DOI] [PubMed] [Google Scholar]
- Colombo B., Baglioni C. Regulation of haemoglobin synthesis at the polysome level. J Mol Biol. 1966 Mar;16(1):51–66. doi: 10.1016/s0022-2836(66)80262-0. [DOI] [PubMed] [Google Scholar]
- FESSAS P. Inclusions of hemoglobin erythroblasts and erythrocytes of thalassemia. Blood. 1963 Jan;21:21–32. [PubMed] [Google Scholar]
- Fessas P., Loukopoulos D., Kaltsoya A. Peptide analysis of the inclusions of erythroid cells in beta-thalassemia. Biochim Biophys Acta. 1966 Aug 24;124(2):430–432. doi: 10.1016/0304-4165(66)90216-9. [DOI] [PubMed] [Google Scholar]
- Heywood D., Karon M., Weissman S. Asymmetrical incorporation of amino acids in the alpha and beta chains of hemoglobin synthesized by thalassemic reticulocytes. J Lab Clin Med. 1965 Sep;66(3):476–482. [PubMed] [Google Scholar]
- Huehns E. R. Further studies on the isolation and properties of alpha-chain sub-units of haemoglobin. Biochem J. 1966 Dec;101(3):843–851. doi: 10.1042/bj1010843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huehns E. R., Modell C. B. Haemoglobin synthesis in thalassaemia. Trans R Soc Trop Med Hyg. 1967;61(2):157–163. doi: 10.1016/0035-9203(67)90150-2. [DOI] [PubMed] [Google Scholar]
- Itano H. A. Genetic regulation of peptide synthesis in hemoglobins. J Cell Physiol. 1966 Jun;67(3 Suppl):65–76. doi: 10.1002/jcp.1040670408. [DOI] [PubMed] [Google Scholar]
- Marks P. A., Burka E. R., Schlessinger D. PROTEIN SYNTHESIS IN ERYTHROID CELLS, I. RETICULOCYTE RIBOSOMES ACTIVE IN STIMULATING AMINO ACID INCORPORATION. Proc Natl Acad Sci U S A. 1962 Dec;48(12):2163–2171. doi: 10.1073/pnas.48.12.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANNEY H. M., BRIEHL R. W., JACOBS A. S. OXYGEN EQUILIBRIA OF HEMOGLOBIN ALPHA-A AND OF HEMOGLOBIN RECONSTITUTED FROM HEMOGLOBINS ALPHA-A AND H. J Biol Chem. 1965 Jun;240:2442–2447. [PubMed] [Google Scholar]
- RIGAS D. A., KOLER R. D. Decreased erythrocyte survival in hemoglobin H disease as a result of the abnormal properties of hemoglobin H: the benefit of splenectomy. Blood. 1961 Jul;18:1–17. [PubMed] [Google Scholar]
- Weatherall D. J., Clegg J. B., Naughton M. A. Globin synthesis in thalassaemia: an in vitro study. Nature. 1965 Dec 11;208(5015):1061–1065. doi: 10.1038/2081061a0. [DOI] [PubMed] [Google Scholar]
- Winterhalter K. H. Sequence of linkage between the prosthetic groups and the polypeptide chains of haemoglobin. Nature. 1966 Aug 27;211(5052):932–934. doi: 10.1038/211932a0. [DOI] [PubMed] [Google Scholar]