

Summary
While numerous cell-intrinsic processes are known to play decisive roles in chemotherapeutic response, relatively little is known about the impact of the tumor microenvironment on therapeutic outcome. Here, we use a well-established mouse model of Burkitt's lymphoma to show that paracrine factors in the tumor microenvironment modulate lymphoma cell survival following the administration of genotoxic chemotherapy. Specifically, IL-6 and Timp-1 are released in the thymus in response to DNA damage, creating a “chemo-resistant niche” that promotes the survival of a minimal residual tumor burden and serves as a reservoir for eventual tumor relapse. Notably, IL-6 is released acutely from thymic endothelial cells in a p38-dependent manner following genotoxic stress, and this acute secretory response precedes the gradual induction of senescence in tumor-associated stromal cells. Thus, conventional chemotherapies can induce tumor regression while simultaneously eliciting stress responses that protect subsets of tumor cells in select anatomical locations from drug action.
Introduction
While significant progress has been made in the application of chemotherapy over the past 40 years, most chemotherapeutic regimens ultimately fail to cure cancer patients (Holen and Saltz, 2001). Even tumors that show dramatic initial responses to therapy frequently relapse as chemoresistant malignancies. This chemoresistance is thought to arise as a consequence of cell intrinsic genetic changes including upregulation of drug efflux pumps, activation of detoxifying enzymes or apoptotic defects (Bleau et al., 2009). However, recent data suggests that resistance to chemotherapy can also result from cell extrinsic factors such as cytokines and growth factors (Eckstein et al., 2009; Williams et al., 2007). Additionally, other studies have suggested that rare cancer stem cells are the source of eventual tumor relapse following therapy, as these cells are thought to be drug resistant due to increased genomic stability, decreased oxidative stress or the presence of multiple drug resistance transporters (Visvader and Lindeman, 2008).
Modern combinatorial chemotherapeutic regimes can reduce patient tumor burdens to undetectable levels, yet in many cases these tumors will relapse (Corradini et al., 1999). Thus, even when a patient is classified as being in complete remission, surviving cancer cells can persist in particular anatomical locations. This remnant population of cancer cells has been described as Minimal Residual Disease (MRD). MRD, is generally not macroscopic and may not be at the site of the primary tumor, making this phenomenon difficult to dissect experimentally (Ignatiadis et al., 2008). While MRD is a significant clinical problem, few models exist to study residual tumor burden following therapy. Thus, it remains unclear whether the cancer cells that compose the MRD burden are surviving following chemotherapy due to stochastic events, intrinsic drug resistance, or microenvironmental cues.
Efforts to experimentally recapitulate the response of human tumors in vivo to chemotherapy have generally relied upon xenografts of human tumors transplanted into immunodeficient mice (Sharpless and Depinho, 2006). These models have proven ineffective in predicting drug efficacy, likely due to a failure to reproduce the complexity of a tumor with its associated complement of stromal, immune and endothelial cells. This autochthonous tumor microenvironment includes a complex mixture of pro- and anti- neoplastic factors (Hideshima et al., 2007). Both malignant and untransformed cells within a tumor influence the balance of growth factors, chemokines and cytokines found in the tumor microenvironment. These factors play key roles in regulating tumor cell proliferation, and survival through the activation of diverse signaling pathways, including the Jak/Stat, NFκB, Smad, and PI3K pathways (Nguyen et al., 2009).
While numerous studies have addressed the role of tumor-proximal factors in tumor growth or metastasis, relatively few have addressed the role of the tumor microenvironment in chemotherapeutic outcome (Hanahan and Weinberg, 2000). Here we show that two cytokines, IL-6 and Timp-1, protect lymphoma cells from cell death induced by genotoxic chemotherapy, such that small molecule inhibition of cytokine-induced signaling potentiates chemotherapeutic efficacy. We further show that IL-6 release occurs as a result of p38 MAP Kinase activation in tumor-associated endothelial cells acutely following DNA damage. This acute cytokine release also occurs in treated human endothelial and hepatocellular carcinoma cells, suggesting that acute secretory responses may occur in numerous contexts. In the thymus, rapid cytokine release precedes the induction of senescence – a process recently shown to promote sustained cytokine release in cultured cells (Acosta et al., 2008; Coppe et al., 2008; Kuilman et al., 2008; Wajapeyee et al., 2008). Thus, genotoxic drugs can, paradoxically, elicit pro-survival signaling in select anatomical sites, providing a reservoir of minimal residual disease that subsequently fuels tumor relapse.
Results
The thymus represents a chemoprotective tumor microenvironment
To investigate the dynamics of lymphoma response and relapse following chemotherapy, we used a well-established preclinical model of human Burkitt's lymphoma – the Eμ-myc mouse (Adams et al., 1985). Tumors from these mice can be transplanted into immunocompetent syngeneic recipient mice, and the resulting tumors are pathologically indistinguishable from autochthonous tumors (Burgess et al., 2008). Six to 8 week old mice were tail vein injected with GFP-tagged Eμ-myc p19Arf-/- B lymphoma cells. At tumor onset all mice displayed a characteristic disseminated pattern of disease with lymphoma cells in the peripheral lymph nodes, spleen and mediastinum. Mice were treated with the maximum tolerated dose of the front-line chemotherapeutic doxorubicin at the time of lymphoma manifestation. Three days after administration of doxorubicin, all mice displayed tumor regression and peripheral tumor clearance, measured by lymph node palpation. These mice were sacrificed at four days post treatment and sites of minimal residual disease were identified by GFP imaging. Interestingly, the majority of surviving lymphoma cells were in the mediastinal cavity (Figure 1A), a central component of the thoracic cavity that encapsulates the heart, esophagus, trachea and a large amount of lymphatic tissue including the mediastinal lymph nodes and the thymus.
Figure 1. The thymus represents a chemoprotective niche that harbors surviving lymphoma cells following doxorubicin treatment.
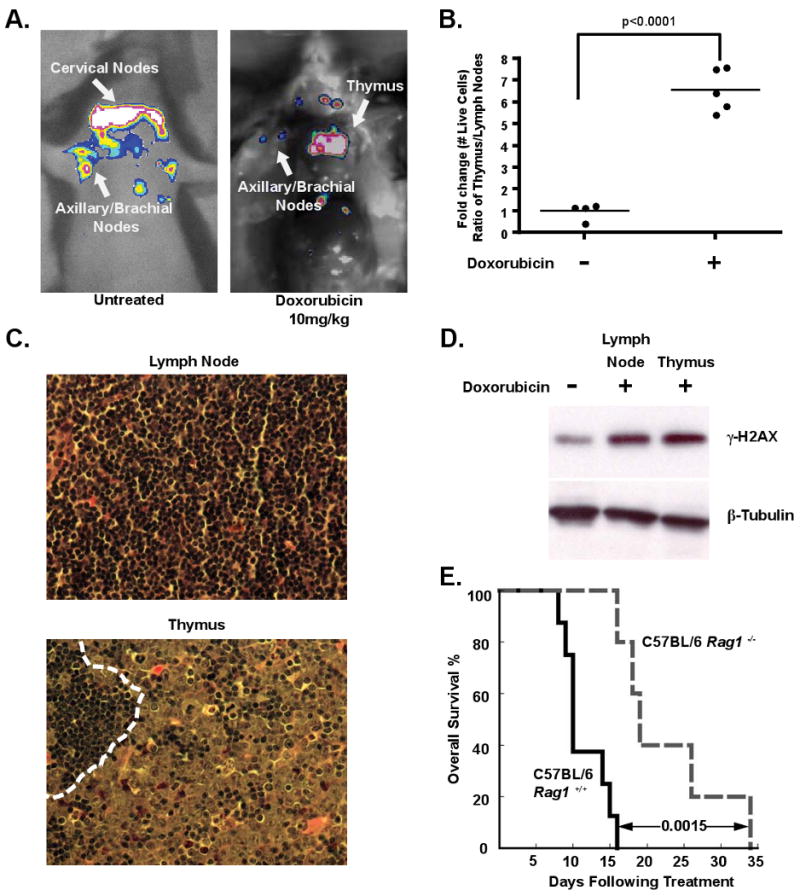
(A) Lymphoma-bearing mice were imaged for whole body fluorescence prior to treatment and 4 days following a single dose of 10mg/kg doxorubicin. Representative mice are shown. (B) Ratios of live GFP-tagged Eμ-myc p19Arf-/- B lymphoma cells in the thymus versus peripheral lymph nodes were quantified by flow cytometry, before (n=4 mice) and 48 hours after (n=5 mice) doxorubicin treatment. Average ratios are indicated with a line. (C) Hematoxylin and eosin (H&E) sections of lymph node and thymus from a tumor-bearing bearing mouse 48 hours after doxorubicin treatment. The dotted line in the thymus demarcates a small region of infiltrating lymphocytes neighboring a larger region of surviving lymphoma cells. Representative fields are shown at 40× magnification. (D) A western blot showing γ-H2AX levels in FACS sorted GFP-positive lymphoma cells from the thymus and peripheral lymph nodes following doxorubicin treatment. β-Tubulin serves as a loading control. The untreated sample is a lysate from cultured lymphoma cells. (E) A Kaplan-Meier curve showing the overall survival of tumor-bearing C57BL/6 (n=8) or C57BL/6 Rag1-/- (n=5) mice following doxorubicin treatment. The p value was calculated using a logrank test. See also Figure S1.
To analyze the effect of drug treatment on specific tumor niches, we harvested all primary lymphoid organs, including peripheral lymph nodes, thymus, spleen and bone marrow, following doxorubicin treatment. All tissues sampled showed extensive lymphoma cell apoptosis and restoration of normal organ architecture. Peripheral lymph nodes, spleen and bone marrow exhibited nearly complete tumor clearance with rare surviving lymphoma cells (Figure 1C and Figure S1A). In contrast, many surviving B lymphoma cells could be seen in the thymus. To quantify this phenotype, cells were harvested from peripheral lymph nodes and the thymus following treatment, and the number of surviving GFP positive lymphoma cells was assessed by flow cytometry. The number of viable lymphoma cells in the thymus relative to the lymph nodes increased 6.5 fold following doxorubicin treatment (Figure 1B). Thus, the thymus represents a chemoprotective niche that protects lymphoma cells from doxorubicin-induced cell death.
To rule out the possibility that the selective survival of tumor cells in the thymus was due to the specific exclusion of doxorubicin from the mediastinum, we sorted live GFP-positive tumor cells from the lymph nodes and thymus 12 hours after doxorubicin treatment and blotted for γ-H2AX, a marker of DNA damage (Morrison and Shen, 2005). Western blot analysis showed that cells in both anatomical locations undergo the same amount of DNA damage (Figure 1D). Additionally, flow cytometry of mediastinal lymphoma cells failed to identify any sub-population of lymphoma cells with decreased γ-H2AX fluorescence (Figure S1B). These data suggest that the thymus offers no physical barrier to drug delivery.
Minimal residual tumor burden in the thymus fuels tumor relapse following chemotherapy
Given the persistence of tumor cells in the thymus following chemotherapy, we next sought to determine whether tumor cells in the thymus contributed to lymphoma relapse. To this end, we examined therapeutic response in genetically and surgically athymic mice. We injected control or Rag1 deficient mice, which have severely atrophic thymuses, with lymphoma cells and then treated tumor-bearing recipient animals with doxorubicin. Overall survival and tumor free survival were significantly extended in tumor-bearing Rag1 deficient mice, relative to control animals, suggesting that the presence of a functional thymus promotes relapse and disease progression (Figure 1E and Figure S1C). Similarly, surgically thymectomized tumor-bearing mice also showed extended tumor-free and overall survival following therapy relative to control animals (Figure S1D and data not shown). Notably, overall survival in untreated tumor-bearing Rag1 deficient or thymectomized mice was indistinguishable from that in control animals (Figure S1E). Thus, the thymus harbors minimal residual disease that contributes to tumor relapse following therapy in this model.
Cultured thymuses secrete pro-survival factors in vitro
Preferential lymphoma cell survival in the thymus following doxorubicin treatment suggests that specific anatomical microenvironments may contain pro-survival factors absent in other lymphoid organs. There is precedence for this phenomenon in multiple myeloma, where the bone marrow microenvironment promotes myeloma cell survival (Hideshima et al., 2007). To address this possibility, we derived conditioned media from the thymus (TM, for Thymic Media), bone marrow (BMM) and lymph nodes (LNM) of mice treated with doxorubicin. Cultured lymphoma cells were then treated with doxorubicin, along with TM, LNM or BMM. Addition of TM provided a significant survival advantage, with 10 fold more cells surviving 48 hours following treatment (Figure 2A). This effect was specifically pro-survival, as opposed to pro-proliferative, as these same conditioned medias had little effect on lymphoma cell growth (Figure 2B). In contrast, conditioned media derived from peripheral lymph nodes had only a minimal effect on lymphoma cell survival (Figure 2A). Thus, soluble pro-survival factor(s) present in the thymic microenvironment protect tumor cells from genotoxic chemotherapy.
Figure 2. Thymic conditioned media contains soluble chemoprotective factors.
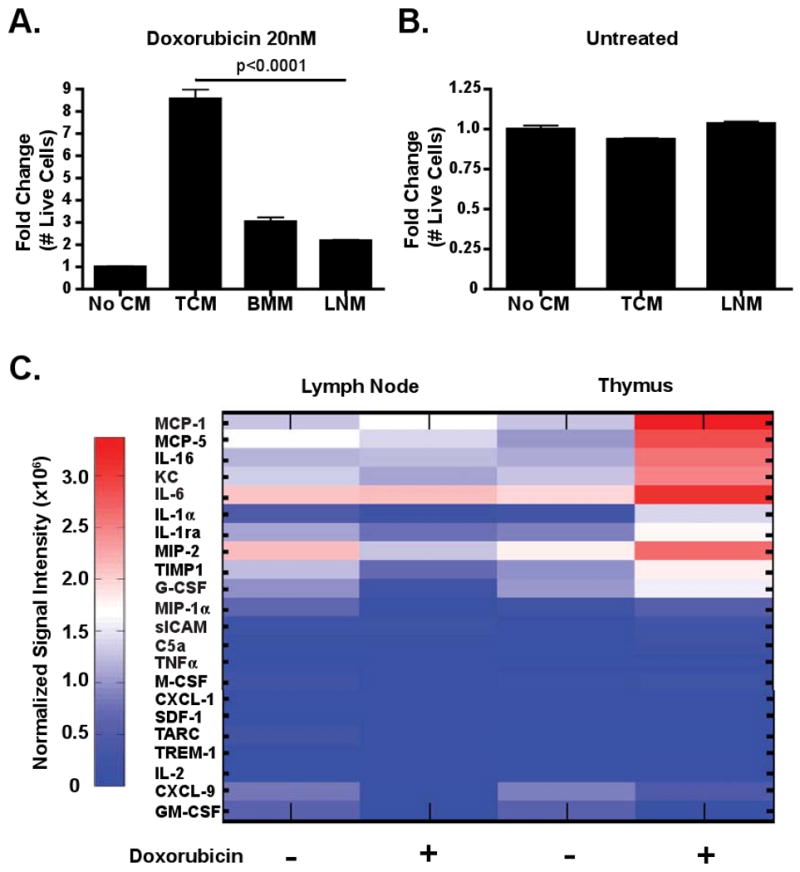
(A) A graph showing lymphoma cell survival in the presence of doxorubicin alone or in the presence of conditioned media. The data are represented as mean +/- standard error of the mean (SEM) (n=3). (B) A graph showing the growth of lymphoma cells cultured in the absence or presence of conditioned media. The data are represented as mean +/- SEM (n=3). (C) Cytokine array analysis of conditioned media from untreated and doxorubicin treated lymph nodes and thymus. The data is represented graphically as normalized signal intensity. Conditioned media was pooled from 3 or 4 mice for each array.
Cytokine levels vary between tumor-bearing anatomical locations
To identify the factor(s) contributing to lymphoma cell survival in the thymus, we performed cytokine arrays analyzing the abundance of 40 cytokines and chemokines in conditioned media from doxorubicin treated and untreated tumors (Figure 2C and data not shown). Analysis of cytokine expression showed significant differences between the thymic and lymph node tumor microenvironments. Multiple factors related to cell migration and cell cycle control were acutely upregulated in thymic lymphomas, but not in peripheral lymphomas or cultured lymphoma cells, following doxorubicin treatment. These included the cytokines G-CSF, IL-1α, IL-1ra, IL-6, IL-16 and the chemokines and growth factors KC, MCP-1, MCP-5, MIP-2 and Timp-1 (Figure 2C).
Each of the upregulated factors was tested in vitro for the ability to promote doxorubicin resistance. Of the 10 recombinant proteins examined, only two produced a significant effect on lymphoma cell survival following doxorubicin treatment in vitro. Recombinant Interleukin-6 (IL-6), as a single agent, was able to promote a 2.8-fold increase in the number of surviving lymphoma cells 72 hours following doxorubicin treatment (Figure 3A and Figure S2A). Similarly, addition of Tissue inhibitor of metalloproteases 1 (Timp-1) resulted in a 3-fold increase in surviving lymphoma cells following doxorubicin treatment (Figure 3B). These factors had a combinatorial effect (Figure 3B), as addition of both recombinant IL-6 and Timp-1 resulted in a 4.5-fold increase lymphoma cell number following treatment (Figure 3B). Importantly, neither factor alone or in combination affected lymphoma cell growth, suggesting that this increase in cell number was not due to enhanced cell proliferation (Figure S2B). Additionally, recombinant IL-6 had no effect on lymphoma cell motility in this setting (Figure S2C).
Figure 3. IL-6 and Timp-1 are chemoprotective in vitro and in vivo.
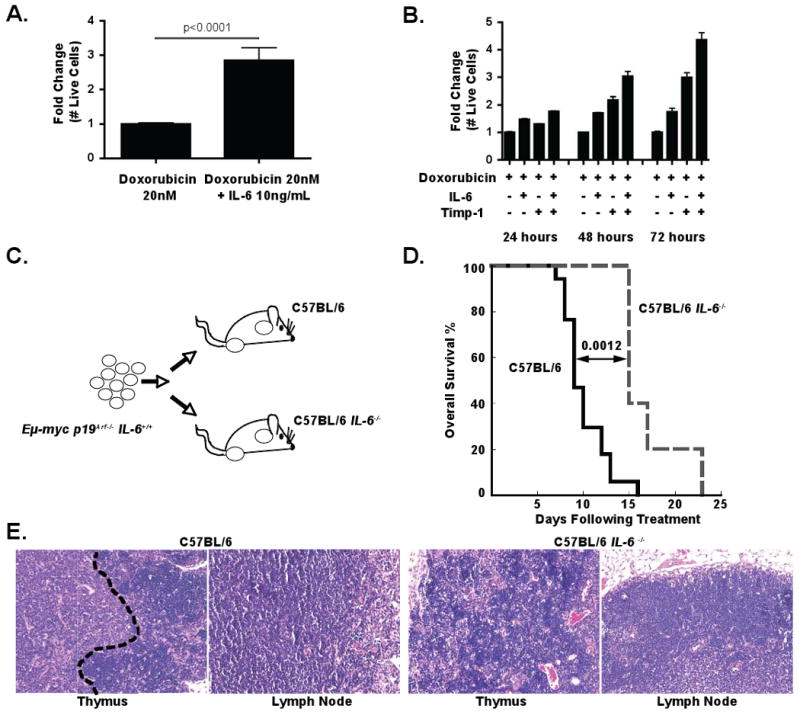
(A) A graph showing the fold change in lymphoma cell number 72 hours after treatment with doxorubicin as a single agent or doxorubicin plus recombinant IL-6. The data are represented as mean +/- SEM (n=4 independent experiments). (B) A graph showing the relative survival of cultured lymphoma cells at 24 hour intervals following treatment with doxorubicin alone, doxorubicin plus recombinant IL-6 or Timp-1, or doxorubicin plus both IL-6 and Timp-1. The data are represented as mean +/- SEM (n=3 independent experiments). (C) A schematic diagram of the lymphoma transplant experiment, showing injection of IL-6+/+ lymphoma cells into both IL-6+/+ and IL-6-/- recipients. (D) A Kaplan-Meier curve showing post-treatment survival of IL-6+/+ (n=17) or IL-6-/- (n=5) mice bearing IL-6+/+ lymphomas. All mice were treated with a single dose of 10mg/kg doxorubicin. The p value was calculated using a logrank test. (E) H&E stained sections of lymphomas 72 hours following doxorubicin treatment. The black dotted line shown in the thymus from the IL-6+/+ recipient mouse demarcates a zone of surviving lymphoma cells that is absent in the other sections. Representative fields are shown at 20× magnification. See also Figure S2.
While these data suggest that both Timp-1 and IL-6 promote chemoresistance in the thymus, we decided to focus our efforts on the contribution of IL-6 to therapeutic response. To determine whether IL-6 was acting to promote cell survival in an autocrine fashion following release from lymphoma cells or a paracrine fashion following release from surrounding thymic cells, we performed lymphoma transplant experiments in the presence or absence of IL-6. Specifically, IL-6+/+ lymphomas were transplanted into IL-6+/+ or IL-6-/- mice (Figure 3C and Figure S2D). Tumor-bearing recipient mice were then treated with the maximum tolerated dose of doxorubicin and monitored for tumor free survival and overall survival. Notably, while IL-6-/- and IL-6+/+ recipient mice developed pathologically indistinguishable tumors, IL-6-/- recipients displayed significantly longer tumor free survival and overall survival following treatment than their IL-6+/+ counterparts (Figure 3D and Figure S2F-G). Additionally, histological analysis confirmed the lack of surviving lymphoma cells in the thymus of IL-6-/- mice following treatment (Figure 3E). Thus, IL-6 release from the tumor microenvironment, rather than from the tumor itself, promotes tumor cell survival.
To further interrogate the source of thymic IL-6, IL-6 levels were examined in thymic lymphomas from doxorubicin-treated IL-6+/+ and IL-6-/- recipient mice, as well as doxorubicin treated lymphoma cells in vitro (Figure 3C). No IL-6 was detected in either IL-6+/+ lymphoma cells in vitro or in thymic or peripheral tumors from IL-6-/- lymphoma-bearing mice (data not shown). This result strongly suggests that the IL-6 present in the tumor microenvironment following genotoxic stress is secreted in a paracrine manner from resident thymic cells.
Genotoxic chemotherapy induces the release of pro-survival cytokines in vivo
Recent work has shown that DNA damage can induce a secretory phenotype in cultured cells (Rodier et al., 2009). To determine whether IL-6 is similarly induced as a consequence of genotoxic chemotherapy in vivo, we treated mice lacking tumors with the maximally tolerated dose of doxorubicin. 18 hours later we assayed IL-6 levels by ELISA in untreated and doxorubicin treated mice. As suggested by the cytokine array data, IL-6 was present at a constitutively higher level in the thymus versus the lymph nodes of mice (Figure 4A). Additionally, doxorubicin treatment significantly increased the amount of IL-6 in the thymus but not in peripheral lymph nodes or the spleen (Figure 4A and Figure S3A). Thus, genotoxic chemotherapy induces a stress response in the thymus that includes the release of IL-6, a pro-survival cytokine. Notably, IL-6 induction in the thymus occurred within 18 hours of treatment, much more acutely than has been reported for secretory phenotypes in cultured cells (Rodier et al., 2009).
Figure 4. Doxorubicin induces the release of IL-6, and inhibition of this cytokine signaling sensitizes tumor cells to chemotherapy.
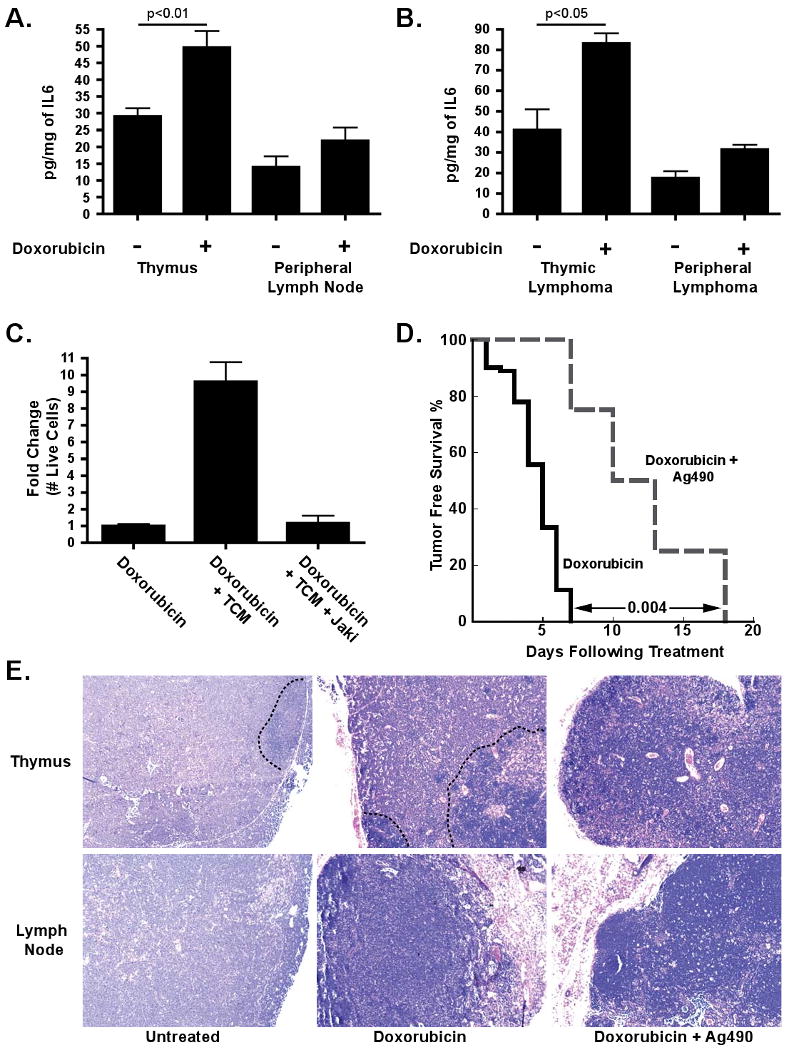
(A) Quantification of IL-6 levels in conditioned media from the thymus or lymph nodes of untreated mice (n≥10) or mice treated for 18 hours with 10mg/kg doxorubicin (n≥7). Values were normalized by tissue weight. The data are represented as mean +/- SEM. (B) Quantification of IL-6 levels in conditioned media derived from tumor-bearing thymuses or lymph nodes of doxorubicin treated (n=3) or untreated (n=3) mice. The data are represented as mean +/- SEM. (C) A bar graph showing the fold change in number of live cells following treatment with doxorubicin alone or in combination with conditioned media plus or minus a Jak2 inhibitor. The data are represented as mean +/- SEM (n=3). (D) A Kaplan-Meier curve showing tumor free survival of lymphoma-bearing mice treated with doxorubicin (n=9) or doxorubicin plus two doses of 50mg/kg AG-490 m-CF3 (n=4). The p value was calculated using a logrank test. (E) H&E sections of lymphomas 72 hours after treatment with doxorubicin or doxorubicin plus AG-490 m-CF3. Black dotted lines distinguish surviving lymphoma cells, which are largely absent in the presence of AG-490 m-CF3, from infiltrating immune cells. Representative fields are shown at 20× magnification. See also Figure S3.
To confirm that this acute DNA damage-induced secretory response also occurs in thymuses with substantial lymphoma infiltration, we examined IL-6 levels in tumor-bearing mice. Again, IL-6 was constitutively present at a higher level in the thymic tumor microenvironment versus the peripheral tumor microenvironment, and treatment with doxorubicin resulted in a rapid and significant increase in IL-6 levels in thymic lymphomas but not in peripheral lymphomas (Figure 4B). As chemotherapy rapidly induces apoptosis in lymphoid malignancies, these data are consistent with the idea that acute cytokine release following DNA-damage may directly impact therapeutic response.
Jak2 signaling is required for lymphoma cell survival following doxorubicin treatment in vivo and in vitro
Both IL-6 and Timp-1 have been shown to signal through Jak2 and Stat3 (Heinrich et al., 1998; Lambert et al., 2003), suggesting that doxorubicin efficacy could be potentiated if Jak signaling were chemically inhibited. We tested this hypothesis by treating lymphoma cells with doxorubicin and TM or doxorubicin and TM plus a Jak2/Jak3 inhibitor. Addition of the Jak inhibitor completely ablated the protective effect of TM (Figure 4C). Importantly, the Jak inhibitor had a minimal effect on lymphoma cell growth in the presence of TM (data not shown). Thus, Jak2/Jak3 signaling promotes the chemoprotective effect of TM in vitro.
To determine whether this effect could be recapitulated in vivo, we treated lymphoma-bearing mice with either doxorubicin, Ag490 (a Jak2/3 inhibitor previously used in murine in vivo studies) (Gu et al., 2005), or with a combination of both doxorubicin and Ag490. Mice treated with doxorubicin and Ag490 showed significantly longer tumor-free survival and overall survival than mice treated with doxorubicin alone (Figure 4D and Figure S3B and C). Histological analysis of the thymus in mice treated with both drugs showed few surviving lymphoma cells, in sharp contrast with those treated with doxorubicin alone (Figure 4E). Importantly, this was not due to a simple additive effect of doxorubicin and Ag490-induced cytotoxicity, as mice treated with Ag490 alone exhibited no tumor free survival or extended overall survival when compared to mice treated with a vehicle control (Figure S3D and data not shown). Thus, Jak2/Jak3 inhibition can eliminate pro-survival signaling in the thymic niche and potentiate doxorubicin cytotoxicity.
IL-6 is released from thymic endothelial cells
To identify the cell type(s) responsible for IL-6 release from the thymic stroma, we disassociated thymuses from untreated mice and sorted known resident cells by characteristic surface markers. Sorted cells were then plated in normal growth media, and IL-6 levels were assessed after 48 hours. Notably, the vast majority of IL-6 secreted following thymic disassociation was released from thymic endothelial cells, while B, T, dendritic and thymic epithelial cells failed to produce any IL-6 levels above background (Figure 5A). Resident macrophages produced trace levels of IL-6 (Figure 5A and Figure S4). However, they did so at a level that was more than ten-fold less than endothelial cells. Similar results were seen for Timp-1, which was released almost exclusively from thymic endothelial cells (Figure 5B).
Figure 5. Endothelial cells secrete IL-6 and Timp-1 in response to DNA damage in a p38 MAP kinase dependent manner.
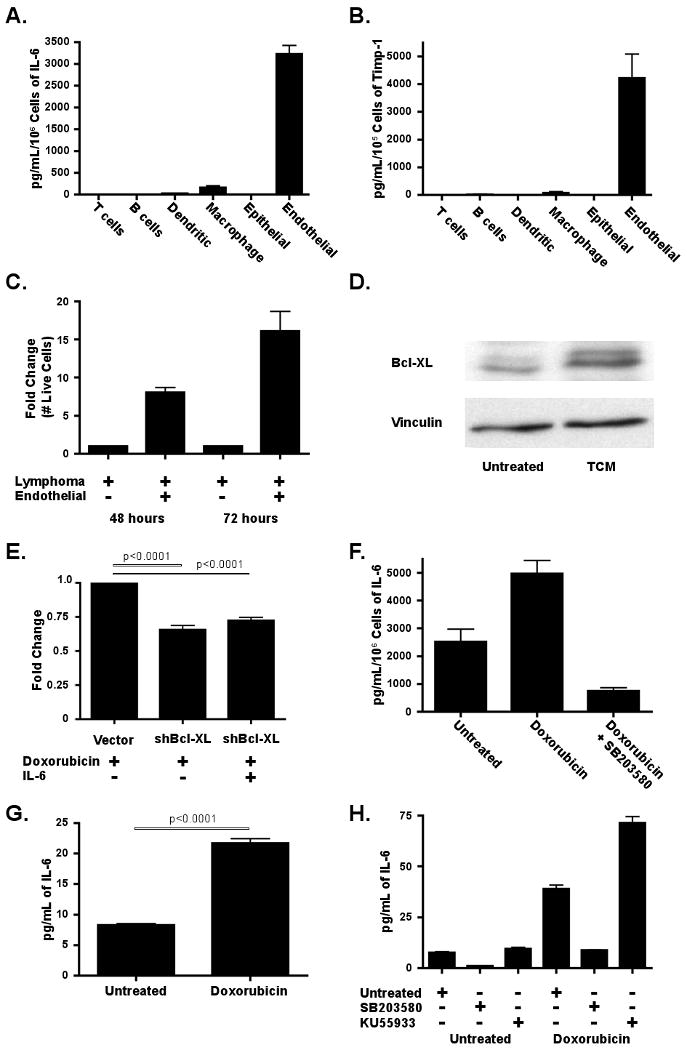
(A) IL-6 and (B) Timp-1 levels were quantified by ELISA in conditioned media derived from sorted thymic cell populations The data are represented as mean +/- SEM (n≥3 independent experiments). Values were normalized to the number of cells sorted. (C) A graph showing lymphoma cell survival in response to 20nM doxorubicin, with or without endothelial cell co-culture. Fold change in cell number was assessed at 48 and 72 hours post treatment. The data are represented as mean +/- SEM (n=6 independent experiments) (D) A western blot for Bcl-XL levels in lymphoma cells in the presence or absence of TCM for 24 hours. The blot is representative of three independent experiments. (E) A graph showing the results of a GFP competition assay in cells partially transduced with a Bcl-XL shRNA or a control vector. Fold change in GFP percentage was assessed 48 hours following treatment with 20nM doxorubicin. The data are represented as mean +/- SEM (n=3). (F) A bar graph showing the amount of IL-6 in conditioned media from endothelial cells sorted from the thymus of untreated mice (n=5), mice treated with doxorubicin (n=8) or mice treated with doxorubicin plus SB203580 (n=4). Values were normalized to cell number. The data are represented as mean +/- SEM. (G) A graph showing the amount of IL-6 present in conditioned media from untreated and treated human vascular endothelial cells (HUVECs). The data are represented as mean +/- SEM (n=3). (H) A graph showing the amount of IL-6 present in conditioned media from HUVECs 48 hours after treatment with doxorubicin alone or doxorubicin plus either SB203580 or KU55933. The data are represented as mean +/- SEM (n=3). See also Figure S4.
Importantly, in mice treated with doxorubicin, IL-6 levels were significantly induced in thymic endothelial cells (Figure 5F). While IL-6 levels were also elevated in treated macrophages (Figure S4A), this increase was not significant and represented less than one tenth of the amount released from treated endothelial cells - even when adjusted for total cell number. Additionally, no significant increase in infiltrating macrophages or dendritic cells was seen acutely following treatment (data not shown). Consistent with the central role of endothelial cells in this secretory response, pretreatment of mice with an inhibitor of VEGFR1/2 – receptors necessary for endothelial cell proliferation - partially inhibited doxorubicin-induced IL-6 release (Figure S4B). These data suggest that resident endothelial cells are largely responsible for the accumulation of pro-survival factors following chemotherapy in this model. To directly assess the relevance of endothelial cells to tumor cell survival, we co-cultured purified endothelial cells and lymphoma cells in the presence of doxorubicin (Figure 5C). The presence of endothelial cells dramatically increased lymphoma cell survival following treatment, with a 15-fold increase in lymphoma cell number in co-cultured populations relative to lymphoma cell-only populations 72 hours post treatment.
Several studies have indicated that cytokines, including IL-6, may exert a pro-survival benefit in target cells through induction of anti-apoptotic Bcl2 family members, including Bcl2, Bcl-XL and Mcl-1 (Jourdan et al., 2000). Thus, we examined the protein levels of Bcl2 family members in lymphoma cells treated with thymic conditioned media. While Bcl2 and Mcl-1 levels were unaffected (data not shown), Bcl-XL was consistently induced 2-4 fold (Figure 5D). To further examine whether Bcl-XL contributes to cell survival in this context, we treated cells expressing a Bcl-XL shRNA with doxorubicin alone or doxorubicin plus IL-6 (Figure 5E). Suppression of Bcl-XL blocked the ability of IL-6 to promote doxorubicin resistance, suggesting that IL-6 mediated induction of Bcl-XL may be necessary for its role in cell survival. This does not, however, preclude that other factors may contribute to cell survival following exposure to IL-6, as cytokines are known to activate numerous pro-survival pathways.
IL-6 is release from endothelial cells is dependent upon p38 MAP Kinase activity
The p38 MAP Kinase (p38) is known to be a key regulator of the expression of inflammatory cytokines, including IL-6 (Medzhitov and Horng, 2009). To determine whether p38 is required for DNA damage-induced IL-6 release, treated and untreated thymic endothelial cells were purified and probed by immunofluorescence for the presence of activated p38. Notably, treated endothelial cells showed significantly higher phospho-p38 levels than their untreated counterparts (Figure S4C). To examine the functional relevance of this p38 activation, we plated thymic endothelial cells from mice treated with doxorubicin in the presence or absence of a p38 inhibitor (Figure 5F). Strikingly, the addition of a p38 inhibitor not only prevented IL-6 induction, but actually reduced the level of secreted IL-6 to below the level in untreated cells. To investigate whether this DNA damage-induced IL-6 release is a conserved characteristic of endothelial cells, we performed similar experiments in human vascular endothelial cells (HUVECs). Cultured HUVECs were treated with doxorubicin and conditioned media was collected 24 hours after treatment. Here, doxorubicin elicited a threefold increase in the amount of secreted IL-6 (Figure 5G). This process was also dependent upon p38 activity, as concurrent treatment of HUVECs with doxorubicin and a p38 inhibitor blocked IL-6 induction (Figure 5F and Figure 5H).
Cell-based studies have implicated the ATM checkpoint kinase in senescence-associated secretory phenotypes (SASP) (Rodier et al., 2009). To examine the relevance of ATM to endothelial IL-6 release, we treated HUVECs with doxorubicin and an ATM inhibitor (Figure 5H). Surprisingly, as opposed to blocking cytokine secretion, ATM inhibition significantly increased the level of endothelial IL-6 release. These data suggest that the biology of acute cytokine release may be distinct from SASP.
Cytotoxic chemotherapy induces senescence in thymic stromal cells in vivo
Recent studies have shown that an autocrine IL-6 signaling loop is induced upon oncogene activation and that this autocrine loop reinforces oncogene induced senescence (OIS) (Coppe et al., 2008; Kuilman et al., 2008). This observation led us to investigate whether IL-6 secretion in the thymus is accompanied by drug-induced senescence. To determine whether doxorubicin induces senescence in vivo, we harvested the thymus and lymph nodes from mice 6 days following treatment with doxorubicin. Tissues were frozen, sectioned and stained for β-Galactosidase activity – a marker of cellular senescence (Dimri et al., 1995). Tissues from untreated mice showed no senescent cells (Figure 6A). In sharp contrast, β-Galactosidase-positive cells were abundant in the thymus, but not the lymph nodes, of doxorubicin-treated mice (Figure S5A). Notably, this “senescent” state was transient, as β-Galactosidase positive cells were no longer present at twelve days following treatment (Figure S5B). While the mechanism underlying the transient presence of senescent cells in this context is unclear, these data are consistent with the recognition and removal of senescent cells by the innate immune system (Krizhanovsky et al., 2008; Xue et al., 2007). Thus, doxorubicin can elicit the acute release of pro-survival cytokines from non-tumor cells in the thymus, coincident with a more gradual induction of senescence.
Figure 6. Genotoxic damage promotes cellular senescence in thymic stromal cells and subsequent IL-6 mediated thymic rebound.
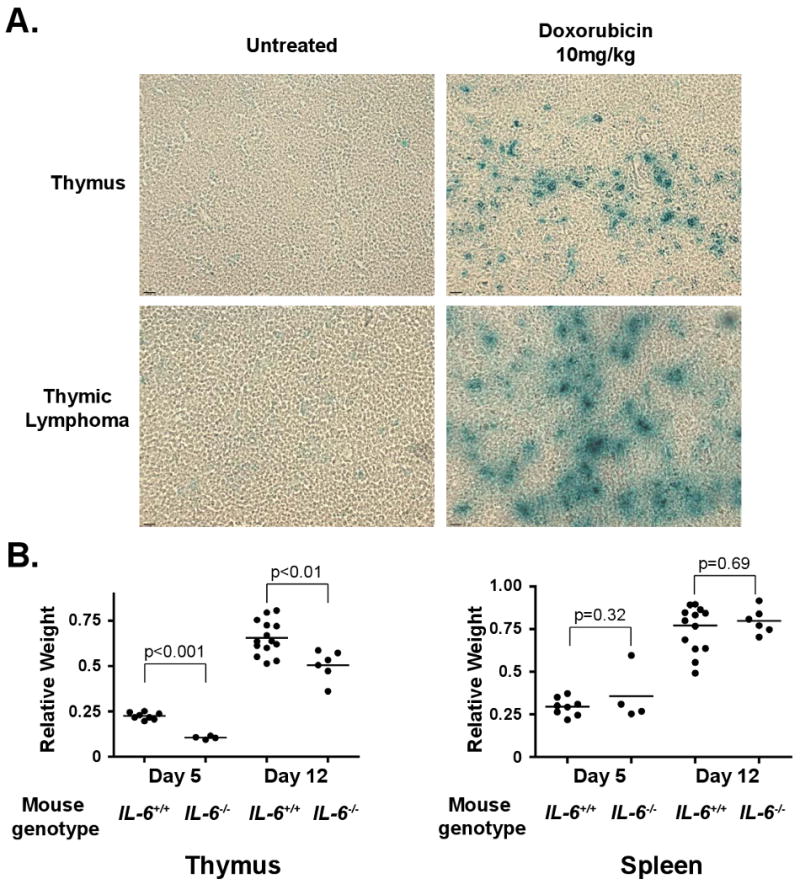
(A) β-galactosidase staining of normal and tumor-bearing thymuses and lymph nodes in the presence or absence of doxorubicin-induced DNA damage. Representative fields are shown at 20× magnification. (B) A graph showing relative thymic and splenic weight following genotoxic damage in the presence (n=14) and absence (n=6) of IL-6. Organ weights are shown as the ratio of individual irradiated thymus or spleen weights relative to the average un-irradiated thymic or spleen weight for each genotype. The data are represented as mean +/- SEM. See also Figure S5.
To confirm that a similar “senescent” state also occurs in the thymic tumor microenvironment, lymphoma-bearing mice were treated with doxorubicin. Six days following treatment, mice were sacrificed and tumors were harvested. Again, thymic tumor sites showed the presence of disseminated senescent cells, while the lymph nodes lacked any β-galactosidase positivity (Figure 6A and Figure S5A). At least a component of this treatment-induced senescent population was comprised of endothelial cells, as purified CD31+/CD34+ cells showed senescent phenotypes – including β–galactosidase activity (Figure S5C). Notably, tumor-bearing thymuses and lymph nodes showed similar numbers of macrophages and dendritic cells, suggesting that the β-galactosidase positive cells in the thymus were resident stromal cells as opposed to infiltrating immune cells (Figure S5D).
IL-6 modulates a general response to DNA damage in the thymus
The presence of a pro-survival secretory response in the thymus following chemotherapy led us to investigate whether IL-6 is involved more generally in stress-induced thymic homeostasis. Whole body irradiation, such as that occurring prior to bone marrow transplantation, induces thymocyte cell death, peripheral leucopenia and thymic involution. This acute wave of thymocyte death is followed by an acute regrowth of the thymus, termed “thymic rebound” (Delrez et al., 1978). To assess whether IL-6 induced by DNA damage is similarly cytoprotective in this setting, we irradiated wild type and IL-6-/- mice. 5 or 12 days later, all mice were sacrificed and the spleen and thymus were harvested and weighed. Thymic regrowth in IL-6-/- mice was significantly reduced when compared to wild type control mice (Figure 6B), while no difference was seen in the spleen. Therefore, IL-6 secretion in the thymus may be critical for thymic growth and repopulation following diverse genotoxic stresses.
Genotoxic damage promotes acute IL-6 release and chemoprotection in human liver cancer cells
Previous descriptions of secretory phenotypes have reported a gradual induction of cytokine release following the onset of stress-induced cellular senescence. This suggests that the release of pro-survival cytokines may not occur rapidly enough to impact chemotherapeutic response. The finding that doxorubicin can induce an acute secretory response led us to investigate whether IL-6 induction might be relevant to therapeutic response in contexts other than the thymic microenvironment. Recent reports have implicated IL-6 as a major contributor to the pathogenesis of hepatocellular carcinoma (HCC) (Naugler et al., 2007; Wong et al., 2009). In humans, activating mutations in gp130, the obligate signal transducing subunit of the IL-6 receptor, have been recently identified (Rebouissou et al., 2009). Additionally recent expression profiling identified the presence of an IL-6 induced transcriptional signature in the tumor stroma that is associated with poor prognosis in hepatocellular carcinoma (Hoshida et al., 2008).
We treated an HCC cell line, Focus cells, with doxorubicin - a front-line therapy for HCC - and measured the levels of secreted IL-6 after 24 hours. Consistent with our endothelial cell data, IL-6 secretion was increased over 3-fold acutely following treatment (Figure 7A). Notably, treated cells lacked any markers of senescence at this time point, indicating that senescence is not required for acute cytokine release. In contrast with endothelial cells, IL-6 secretion could be partially inhibited by either a p38 or an ATM inhibitor (Figure 7B). Thus, pathway requirements for acute secretory phenotypes may be somewhat variable between cell types.
Figure 7. DNA damage acutely induces IL-6 in human hepatocellular carcinoma, promoting both cellular survival and senescence.
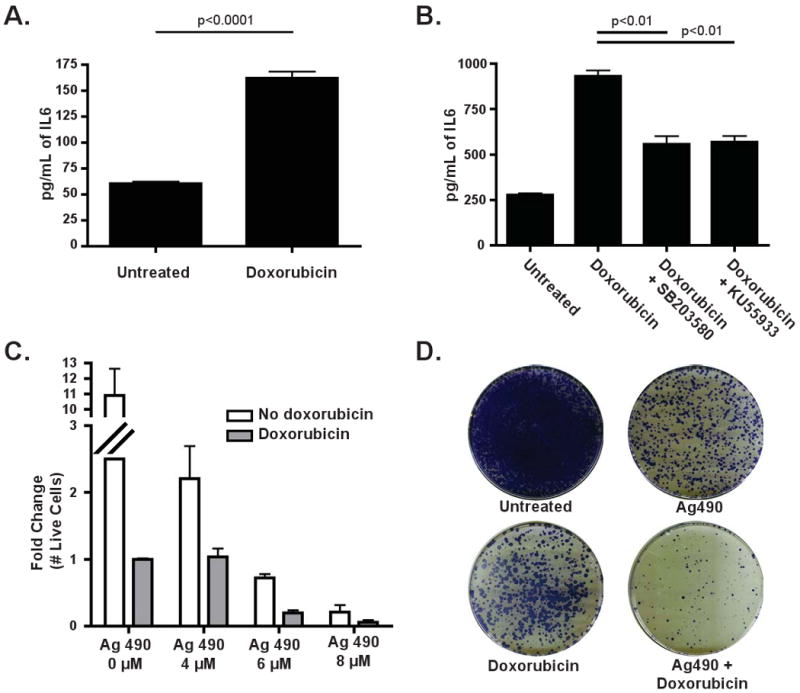
(A) IL-6 levels were quantified in conditioned media derived from Focus cells treated with 200nM doxorubicin for 24 hours. The data are represented as mean +/- SEM (n=3). (B) A graph showing the amount of IL-6 present in Focus cells following treatment with either SB203580 or KU55933, in the presence or absence of doxorubicin. The data are represented as mean +/- SEM (n=3). (C) A graph showing the results of an acute cell survival assay in which Focus cells were treated with doxorubicin and increasing doses of Ag490, as indicated, for 4 days. The data are represented as mean +/- SEM (n=3 independent experiments) (D) A colony formation assay showing Focus cells that were treated with doxorubicin, Ag490 or both for 24 hours before replating. Results are representative of 3 independent experiments.
We then investigated whether inhibition of IL-6 signaling could enhance doxorubicin-induced cell death in HCC cells. We treated Focus cells with doxorubicin alone, Ag490 alone or doxorubicin in combination with Ag490. Treatment with both doxorubicin and Ag490 resulted in more apoptosis and fewer surviving cells in acute survival assays than either single agent alone (Figure 7C). The combination treatment of doxorubicin and Ag490 was also more effective than single agent therapy when measured in a colony formation assay (Figure 7D). These data suggest that acute drug-induced IL-6 release is chemoprotective in HCC and may contribute to the intrinsic chemoresistance of these tumors.
Discussion
The persistence of minimal residual disease following anti-cancer therapy is strongly correlated with decreased survival in patients (Xenidis et al., 2009). However, the mechanisms by which cells survive in select contexts following chemotherapy are unclear. In a mouse model of Burkitt's lymphoma, we show that a specific anatomical location, the thymus, confers a potent cytoprotective benefit to lymphoma cells treated with genotoxic chemotherapy. This surviving cell population is functionally relevant to disease progression, as ablation of the thymus prolongs both tumor free survival and overall survival following treatment with chemotherapy. While the importance of the thymic microenvironment to tumor cell survival in human malignancy remains unclear, we expect that factors contributing to drug resistance at this site may also underlie MRD persistence at analogous locations in human cancers.
The establishment of the thymic pro-survival microenvironment occurs, paradoxically, as a response to genotoxic chemotherapy. Specifically, pro-survival chemokines and cytokines are acutely released following DNA damage. While the complete signaling network leading from a DNA damage response to cytokine release remains unclear, it involves the activation of stress responsive kinases – most notably the MAP kinase p38. Thus, cells exposed to genotoxic damage in vivo can engage well-described cell cycle arrest and apoptotic programs, as well as a physiological stress response pathway leading to survival signaling. Importantly, the resulting secretory response occurs not in the tumor cells themselves, but in proximal endothelial cells. Drug treated endothelial cells release IL-6 and Timp-1, which promote the induction of Bcl-XL in proximal lymphoma cells. As a result, pro-apoptotic signaling induced by the direct action of chemotherapy on tumor cells is countered by anti-apoptotic signaling emanating from the treated vascular compartment in the tumor microenvironment.
Recent literature has shown that the induction of oncogene-induced cellular senescence elicits a secretory response (Coppe et al., 2008; Kuilman et al., 2008). Here we find that IL-6 is induced acutely following DNA damage, prior to the onset of senescence. This difference between a pro-survival response that occurs within one day of treatment and a SASP that is detectable only after 3-4 days is critical. Chemotherapy-induced cell death generally occurs with 48 hours of treatment. Thus, a SASP simply cannot effectively alter treatment response, as it occurs well after tumor cell death decisions are made. However, our data does not preclude a role for senescence in overall tumor survival following therapy. Given that significant levels of senescence occur in the thymus days after doxorubicin treatment, it is possible that elevated IL-6 levels are maintained through the establishment of a SASP. Thus, acute cytokine release and subsequent senescence-related secretory phenotypes may represent a general strategy to promote paracrine cell survival in response to genotoxic stress in select microenvironments.
Secretory and inflammatory processes have been shown to be critical for tissue repair and regeneration (Grivennikov et al., 2009; Krizhanovsky et al., 2008). Thus, chemotherapy in the thymic setting may activate physiological mechanisms of tissue homeostasis. Consistent with this idea, the thymus is known to engage pro-survival and growth mechanisms in response to other cellular stresses. Following radiotherapy and subsequent thymic atrophy (Muller-Hermelink et al., 1987), the thymus re-grows and replenishes the peripheral T-cell population, leading to significant thymic hyperplasia – even in adults with limited remaining thymic tissue (Sfikakis et al., 2005). Factors that govern this process have not been previously identified. Here we show that IL-6 modulates thymic recovery in response to DNA damage. This suggests that lymphomas, and perhaps other malignancies, can co-opt organ-specific pro-survival mechanisms.
Interestingly, serum IL-6 levels are elevated in many types of cancer (Trikha et al., 2003), and high IL-6 levels are strongly correlated with poor overall survival and accelerated disease progression in a variety of cancers, including lymphomas (Seymour et al., 1995). Furthermore, IL-6 levels are greatly increased in metastatic disease versus non-metastatic disease (Salgado et al., 2003). Consequently, stromal or tumor up-regulation of IL-6 may contribute to the intrinsic chemoresistance commonly found in both primary and metastatic malignancies. Additionally, tumor-directed inflammatory responses that result in IL-6 release may similarly limit the efficacy of genotoxic agents. These data suggest that both intrinsic genetic alterations as well as chemoprotective microenvironments can play decisive roles in the cellular response to genotoxic insults. Thus, improved chemotherapeutic regimes may require a combination of cytotoxic agents, which target tumor cells, and targeted therapeutics that inhibit pro-survival signaling from the tumor-adjacent cells.
Experimental Procedures
Cell culture and chemicals
Eμ-Myc;p19Arf-/- mouse B cell lymphomas were cultured in B cell medium (45% DMEM/45% IMDM/10% FBS, supplemented with 2mM L-glutamine and 5μM β-mercaptoethanol). γ-irradiated NIH 3T3 cells were used as feeder cells. Focus cells were cultured in MEM with 10% FBS. HUVEC cells were cultured in Endothelial Cell Growth Medium 2 (Lonza). Doxorubicin, Jak Inhibitor 1 and Ag490 mCF3 were purchased from Calbiochem. SB203580 and KU55933 were purchased from Tocris Bioscience. Gefitinib was purchased from LC labs. For in vivo studies, Ag490 m-CF3 was dissolved in DMSO and then diluted 3:2 in DMEM plus 10% FBS.
Conditioned media
Conditioned media was made from mouse tissues 18 hours after doxorubicin treatment. Conditioned media for viability assays and cytokine arrays was derived from organs from 3-4 pooled mice, while media for ELISAs was generated from individual mice. All tissues were dissociated manually in B cell media. Thymic, bone marrow and lymph node conditioned media were conditioned for 6 hours at 37°C. To isolate single cell types from the thymus, tissue was manual dissociated and washed two times in serum free DMEM, followed by incubation for 1 hour at 37°C with Liberase™ (Roche, 1.3 Wunsch units/mL) and Dnase I (0.15 mg/mL). To aid in dissociation, samples were manually pipetted at 15-minute intervals. Single cell populations were sorted using FITC conjugated antibodies to the following cell surface markers: CD45, CD19, CD11b, CD11c, MHC II, CD31/CD34 for T cells, B cells, macrophages, dendritic cells, epithelial and endothelial cells, respectively. Cells were plated and allowed to condition media for 48 hours at 37°C and 5% CO2. All conditioned medias were cleared of tissue and cells by centrifugation. All values shown for viability assays, ELISAs and cytokine arrays are normalized to the weight of the dissected tissue or the number of sorted cells. For the viability assays, conditioned medias were diluted one to three. IL-6 ELISA kits were purchased from eBioscience. The Timp-1 ELISA kit and mouse cytokine arrays were purchased from R&D Biosystems.
In vitro viability, competition and cell growth assays
For viability, competition and growth assays Eμ-Myc;p19Arf-/- lymphoma cells were split into replicate wells of ≈500,000 cells in 24-well plates or ≈125,000 cells in a 48-well plate. Every 24 hours, cultured cells were resuspended by pipeting and half of the culture was replaced with fresh medium. Viability and cell number were determined by propidium iodide exclusion. For the competition assay, lymphoma cells were partially infected with the indicated retroviruses. The fold change for the competition assay is calculated by dividing the percentage of GFP positive lymphoma cells in the treated population by the percentage in untreated populations. Murine Timp-1 was purchased from R&D Biosystems and used at 100ng/mL. All other cytokines were purchased from Peprotech and used at 10ng/mL. Jak Inhibitor 1 was used at a final concentration of 500nM, and Gefitinib was used at a final concentration of 3μM.
In vivo response to chemotherapy
All mice were purchased from Jackson Laboratory. For survival assays, 1 × 106Eμ-Myc;p19Arf-/- mouse lymphoma cells were injected by tail-vein injection into syngenic C57BL/6J, C57BL/6J IL-6-/- or C57BL/6J Rag1-/- mice. Lymphoma burden was monitored by palpation of the axillary and brachial lymph nodes. At the presentation of a substantial tumor burden (12-13 days after injection), mice were treated with doxorubicin and/or Ag490 m-CF3. Tumor free survival was monitored by palpation and in vivo GFP imaging using a NightOwl imaging system (Berthold).
Thymic rebound in response to radiation
Untreated 6 to 8 week old C57BL/6J or C57BL/6J IL-6-/- mice were sacrificed to establish basal spleen and thymic weight. 6 to 8 week old C57BL/6J or C57BL/6J IL-6-/- mice were irradiated with 4 or 5 Gray. 5 or 12 days later all mice were sacrificed and the spleen and thymus were weighed.
Statistical analysis
Statistical analysis was performed using GraphPad Prism4 software. Two-tailed Student's t-tests were used, as indicated. Error bars represent mean +/- standard error of the mean. For comparison of survival curves, a Kaplan-Meier test was used.
Supplementary Material
Acknowledgments
We thank Holly Criscione and Tyler Miller for their experimental assistance. We thank Corbin Meacham for assistance with the cell migration assay. We would also like to acknowledge Eliza Vasile in the Koch Institute Microscopy Core Facility and Glen Paradis in the Koch Institute Flow Cytometry Core Facility for advice and services. Roderick Bronson provided expert pathology analysis, and Justin Pritchard performed bioinformatic analysis of cytokine arrays. We are grateful to Corbin Meacham, David Feldser and Ross Dickins for critically reading the manuscript and the entire Hemann lab for helpful discussions. M.T.H. is a Rita Allen Fellow and the Latham Family Career Development Assistant Professor of Biology and is supported by NIH RO1 CA128803 and Ludwig Center for Molecular Oncology at MIT. L.A.G. is supported by the MIT Herman Eisen fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A. 2008;105:9053–9058. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini P, Ladetto M, Pileri A, Tarella C. Clinical relevance of minimal residual disease monitoring in non-Hodgkin's lymphomas: a critical reappraisal of molecular strategies. Leukemia. 1999;13:1691–1695. doi: 10.1038/sj.leu.2401559. [DOI] [PubMed] [Google Scholar]
- Delrez M, Ikeh V, Maisin JR, Mattelin G, Haot J, Betz EH. Influence of a mixture of chemical protectors on the lymphoid regeneration of bone marrow and thymus in irradiated mice. Experientia. 1978;34:1221–1222. doi: 10.1007/BF01922970. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein N, Servan K, Hildebrandt B, Politz A, von Jonquieres G, Wolf-Kummeth S, Napierski I, Hamacher A, Kassack MU, Budczies J, et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69:2996–3003. doi: 10.1158/0008-5472.CAN-08-3153. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Zhuang H, Safina B, Xiao XY, Bradford WW, Rich BE. Combinatorial approach to identification of tyrphostin inhibitors of cytokine signaling. Bioorg Med Chem. 2005;13:4269–4278. doi: 10.1016/j.bmc.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- Holen KD, Saltz LB. New therapies, new directions: advances in the systemic treatment of metastatic colorectal cancer. Lancet Oncol. 2001;2:290–297. doi: 10.1016/S1470-2045(00)00324-7. [DOI] [PubMed] [Google Scholar]
- Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis M, Georgoulias V, Mavroudis D. Micrometastatic disease in breast cancer: clinical implications. Eur J Cancer. 2008;44:2726–2736. doi: 10.1016/j.ejca.2008.09.033. [DOI] [PubMed] [Google Scholar]
- Jourdan M, De Vos J, Mechti N, Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ. 2000;7:1244–1252. doi: 10.1038/sj.cdd.4400758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Lambert E, Boudot C, Kadri Z, Soula-Rothhut M, Sowa ML, Mayeux P, Hornebeck W, Haye B, Petitfrere E. Tissue inhibitor of metalloproteinases-1 signalling pathway leading to erythroid cell survival. Biochem J. 2003;372:767–774. doi: 10.1042/BJ20030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Shen X. DNA repair in the context of chromatin. Cell Cycle. 2005;4:568–571. [PubMed] [Google Scholar]
- Muller-Hermelink HK, Sale GE, Borisch B, Storb R. Pathology of the thymus after allogeneic bone marrow transplantation in man. A histologic immunohistochemical study of 36 patients. Am J Pathol. 1987;129:242–256. [PMC free article] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C, Izard T, Balabaud C, Bioulac-Sage P, Zucman-Rossi J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457:200–204. doi: 10.1038/nature07475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado R, Junius S, Benoy I, Van Dam P, Vermeulen P, Van Marck E, Huget P, Dirix LY. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J Cancer. 2003;103:642–646. doi: 10.1002/ijc.10833. [DOI] [PubMed] [Google Scholar]
- Seymour JF, Talpaz M, Cabanillas F, Wetzler M, Kurzrock R. Serum interleukin-6 levels correlate with prognosis in diffuse large-cell lymphoma. J Clin Oncol. 1995;13:575–582. doi: 10.1200/JCO.1995.13.3.575. [DOI] [PubMed] [Google Scholar]
- Sfikakis PP, Gourgoulis GM, Moulopoulos LA, Kouvatseas G, Theofilopoulos AN, Dimopoulos MA. Age-related thymic activity in adults following chemotherapy-induced lymphopenia. Eur J Clin Invest. 2005;35:380–387. doi: 10.1111/j.1365-2362.2005.01499.x. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9:4653–4665. [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 2007;21:2283–2287. doi: 10.1101/gad.1588607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, Ng EK, Chan FK, Sung JJ, Chan HL. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int J Cancer. 2009;124:2766–2770. doi: 10.1002/ijc.24281. [DOI] [PubMed] [Google Scholar]
- Xenidis N, Ignatiadis M, Apostolaki S, Perraki M, Kalbakis K, Agelaki S, Stathopoulos EN, Chlouverakis G, Lianidou E, Kakolyris S, et al. Cytokeratin-19 mRNA-positive circulating tumor cells after adjuvant chemotherapy in patients with early breast cancer. J Clin Oncol. 2009;27:2177–2184. doi: 10.1200/JCO.2008.18.0497. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.