

Abstract
Low-density lipoprotein receptor-related protein-1 (LRP1), a member of the LDL receptor family, has major roles in the cellular transport of cholesterol, endocytosis of forty structurally diverse ligands, transcytosis of ligands across the blood-brain barrier, and transmembrane and nuclear signaling. Recent evidence indicates that LRP1 regulates brain and systemic clearance of Alzheimer's disease (AD) amyloid β-peptide (Aβ). According to the two hit vascular hypothesis for AD, vascular damage precedes cerebrovascular and brain Aβ accumulation (hit 1) which then further amplifies neurovascular dysfunction (hit 2) preceding neurodegeneration. In this study, we discuss the roles of LRP1 during the hit 1 and hit 2 stage of AD pathogenesis and describe a three-level serial LRP1-dependent homeostatic control of Aβ clearance including (i) cell-surface LRP1 at the BBB and cerebrovascular cells mediating brain-to-blood Aβ clearance (ii) circulating LRP1 providing a key endogenous peripheral ‘sink’ activity for plasma Aβ which prevents free Aβ access to the brain, and (iii) LRP1 in the liver mediating systemic Aβ clearance. Pitfalls in experimental Aβ brain clearance measurements with the concurrent use of peptides/proteins such as receptor-associated protein and aprotinin are also discussed. We suggest that LRP1 has a critical role in AD pathogenesis and is an important therapeutic target in AD.
Keywords: Aβ clearance, Alzheimer's disease, blood-brain barrier, LRP1, sLRP1
Introduction
The low-density lipoprotein receptor-related protein-1 (LRP1) is a multifunctional scavenger and signaling receptor that belongs to the low-density lipoprotein receptor family (Herz 2001; Herz and Strickland 2001). LRP1 has a major role in the transport and metabolism of cholesterol associated with apolipoprotein E (apoE)-containing lipoproteins (Herz 2001; Herz and Strickland 2001; Herz et al. 2009). The extracellular heavy α-chain of LRP1 (515 kDa) is noncovalently coupled to the 85 kDa transmembrane and cytoplasmic light β-chain domain (Fig. 1a). The α-chain contains four ligand-binding domains (clusters I-IV), consisting of 2, 8, 10 and 11 cysteine-rich complement-type repeats, respectively (Obermoeller-McCormick et al. 2001; Meijer et al. 2007). The LRP1 ligand-binding domains II and IV are the major LRP1 binding regions interacting with a diverse array of approximately forty structurally diverse ligands (Fig. 1b) including: apoE, α2-macroglobulin (α2M), tissue plasminogen activator (tPA), proteinase-inhibitors, blood coagulation factors (e.g., factor VIII), receptor-associated protein (RAP), Alzheimer's disease (AD) amyloid β-peptide (Aβ), prion protein and aprotinin (Hussain et al. 1999; Neels et al. 1999; Herz 2001; Herz and Strickland 2001; Croy et al. 2003; Deane et al. 2004a; Meijer et al. 2007; Demeule et al. 2008; Lillis et al. 2008; Parkyn et al. 2008; Herz et al. 2009).
Fig. 1.
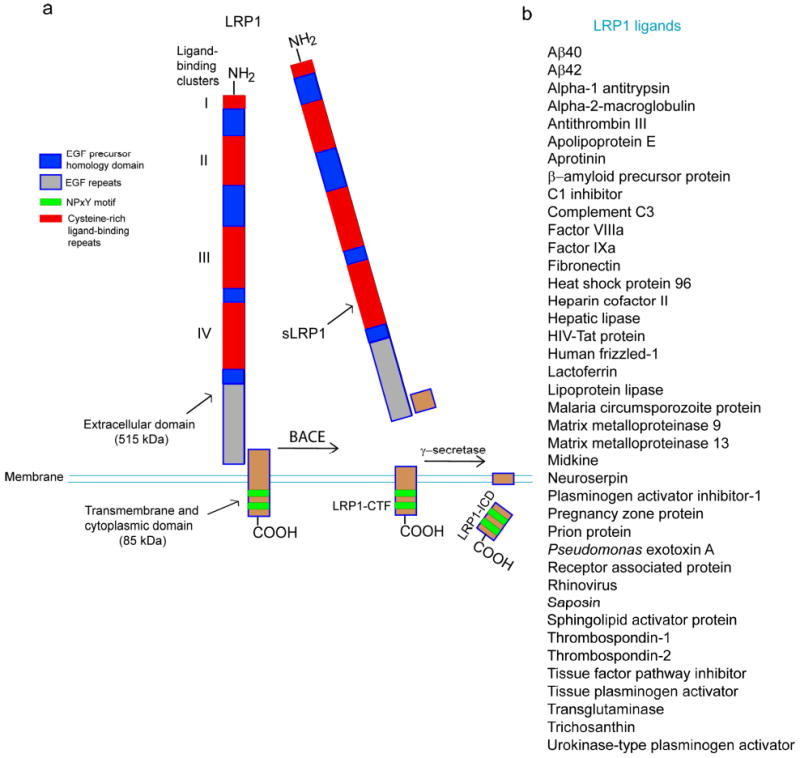
LRP1 schematic structure and ligands. (a) The extracellular heavy α-chain (515 kDa) of LRP1 containing four ligand binding domains (clusters I-IV) is non-covalently coupled to the transmembrane and cytoplasmic light β-chain (85 kDa). β-secretase (BACE) cleaves the N-terminal extracellular domain of LRP1 releasing soluble LRP1 (sLRP1) which circulates in plasma. γ-secretase cleaves the intracellular domain of LRP1 (LRP1-ICD) at the plasma membrane that is translocated from the plasma membrane to the nucleus. EGF, epidermal growth factor; LRP1-CTF, LRP1 C-terminal fragment; Green regions in LRP1-CTF denote two NPXY motifs, the distal NPXY motif overlaps with an YXXL internalization motif. (b) Structurally diverse ligands which bind to clusters II and IV within the extracellular domain of LRP1.
The cytoplasmic tail of LRP1 contains two NPXY motifs, one YXXL motif and two di-leucine motifs (Li et al. 2001) (Fig. 1a). It has been suggested that the YXXL motif and distal di-leucine repeats may be associated with the rapid endocytotic rate of LRP1 (i.e., < 0.5 s) (Li et al. 2001; Deane et al. 2004a, 2008). The cytoplasmic tail is phosphorylated on serine and/or tyrosine residues (Bu et al. 1998; van der Geer 2002) and can interact with different adaptor proteins associated with cell signaling, such as disabled-1, FE65 and postsynaptic density protein 95 (Trommsdorff et al. 1998; Gotthardt et al. 2000; Herz et al. 2009). Thus, LRP1 has a dual role as a rapid cargo endocytotic cellular transporter and a transmembrane cell signaling receptor.
LRP1 is expressed in the CNS in different cell types within the neurovascular unit including vascular cells such as brain endothelial cells, vascular smooth muscle cells and pericytes, and is also expressed in neurons and astrocytes (Herz and Bock 2002; Polavarapu et al. 2007). Although LRP1 has been regarded mainly as a receptor which internalizes its ligands and directs them to the lysosomes for proteolytic degradation, recent studies have demonstrated that LRP1 can also transport several ligands transcellularly across the blood-brain barrier (BBB) including Aβ (Shibata et al. 2000; Deane et al. 2004a), RAP (Pan et al. 2004), tissue plasminogen activator (Benchenane et al. 2005), lipid free and lipidated apoE2 and apoE3, and apoE2 and apoE3 complexes with Aβ (Deane et al. 2008) and a family of Kunitz domain-derived peptides (Demeule et al. 2008). These findings suggest that LRP1 can control transport exchanges of several ligands between the brain and the blood.
LRP1 and Alzheimer's disease
Some genetic studies have suggested that LRP1 is linked to AD and cerebral amyloid angiopathy (CAA) (Kang et al. 1997; Lambert et al. 1998; Wavrant-DeVrieze et al. 1999; Christoforidis et al. 2005; Ballatore et al. 2007). This, however, has not been confirmed by others (Bertram et al. 2000; Chalmers et al. 2010). Moreover, two recent genome-wide association studies have reported that phosphatidylinositol binding clathrin assembly protein (PICALM, also known as CALM, clathrin assembly lymphoid-myeloid leukemia gene) and apoJ (also known as clusterin) are the only two AD susceptibility genes (Harold et al. 2009; Lambert et al. 2009) in addition to the apoE4 gene. The exact roles of PICALM and apoJ in AD pathogenesis are unclear at present (Bertram and Tanzi 2010). It has been shown that apoJ, a ligand for the lipoprotein receptor related protein-2 (LRP2; also known as megalin) controls Aβ transcytosis across the BBB through megalin-dependent rapid Aβ42 efflux from brain to blood (Bell et al. 2007). However, apoJ can also mediate re-entry of circulating Aβ into the brain (Zlokovic et al. 1996). On the other hand, PICALM regulates clathrin-dependent receptor-mediated endocytosis of several ligands (Tebar et al. 1999; Bushlin et al. 2008). Whether PICALM is implicated in LRP1-mediated and/or megalin-mediated transcytosis of Aβ across the BBB is presently not known.
Earlier studies have demonstrated that LRP1 and many of its ligands are deposited in senile plaques (Rebeck et al. 1995; Arelin et al. 2002). In addition to regulating Aβ clearance from brain (Shibata et al. 2000; Deane et al. 2004a; see below), it has been shown that the LRP1 cytoplasmic C-terminal domain interacts with APP's (Aβ-precursor protein) cytoplasmic domain via FE65, an LRP1 adaptor protein, which in turn influences APP processing and Aβ generation (Pietrzik et al. 2004; Waldron et al. 2008). It has also been demonstrated that LRP1 in neurons mediates Aβ cellular uptake and possibly retention in the brain via LRP1 ligands α2M and apoE (Narita et al. 1997; Qiu et al. 1999; DeMattos et al. 2004; Zerbinatti et al. 2004; Zerbinatti and Bu 2005; Deane et al. 2008). The exact implications of these findings for the development of Aβ pathology and cognitive decline remain, however, unclear.
Recent findings have revealed the roles of γ-secretase (the APP processing enzyme) and LRP1 in the inhibition of the inflammatory response suggesting that both proteins may serve as potential therapeutic targets for the modulation of inflammation (Zurhove et al. 2008). Given the importance of neuroinflammation in the pathogenesis of late-stage sporadic AD (McGeer and Rogers 1992; McGeer and McGeer 2004), it is tempting to speculate that the downregulation of LRP1 expression in the brain vascular cells observed in AD (Deane et al. 2004a; Bell et al. 2009) may also contribute to the development of neuroinflammation through a loss of LRP1-dependent inhibition of the interferon-γ promoter and lipopolysaccharide (LPS)-inducible inflammatory genes (Zurhove et al. 2008).
Two hit vascular hypothesis for Alzheimer's disease
The amyloid hypothesis (Hardy and Allsop 1991; Selkoe 1991; Hardy and Higgins 1992; Hardy and Selkoe 2002) has been the basis for most work on the pathogenesis of AD over the past twenty years. More recently, however, both the direct Aβ neuronal toxicity and the clinical efficacy of the therapeutic approaches aimed at reducing Aβ levels in the brain of AD patients have been challenged by several investigators, as recently reviewed (for more details see, Hardy 2009). In this regard, it is of note that we proposed the two hit vascular hypothesis for AD a few years ago as an alternative mechanism for disease pathogenesis (Zlokovic 2005). According to the vascular hypothesis, an initial vascular damage to the brain mediated by hypoxia, perfusion stress and/or disruption of the BBB precedes Aβ accumulation (hit 1). Aβ then accumulates in the brain in response to this initial vascular damage in addition to re-entry of circulating Aβ into the brain and/or faulty Aβ clearance from the brain (hit 2). The two hit vascular hypothesis states that the role of Aβ in the disease process is to critically amplify the neurovascular damage and/or dysfunction which over time can lead to a chronic neurodegenerative process, cognitive decline and neuroinflammation. According to the vascular theory of AD, a primary damage to neurons by Aβ may contribute to but is not an absolute requirement for the development of a neurodegenerative process.
Hit 1
During the hit 1 disease stage, certain molecular mechanisms in the cerebrovascular system, such as the receptors for advanced glycation end products (RAGE) and LRP1, have been identified as important mediators of vascular damage. During normal brain aging or pathological brain aging accelerated by coexisting co-morbidities such as diabetes, hypertension, atherosclerosis, obesity, an acute ischemic insult, chronic hypoperfusion, brain trauma, etc., AGE proteins deposit in the basement membrane of the BBB and induce RAGE expression in the endothelium (Yan et al. 2010). RAGE-AGE interactions result in oxidant stress and expression of the cell adhesion proteins and proinflammatory cytokines (Yan et al. 2010). It is of note that Aβ also acts as a ligand for RAGE. It has been shown that RAGE mediates entry of circulating Aβ into the brain by a receptor-dependent transcytosis in those brain regions which express RAGE at the BBB during both normal and pathological aging (Mackic et al. 1998a; Deane et al. 2003). Alternatively, circulating Aβ can enter the brain across a disrupted BBB (Ujiie et al. 2003) which, again, is likely mediated by the proinflammatory cytokines in response to Aβ-RAGE interaction (Deane et al. 2003).
Pathological reductions in LRP1 expression at the BBB, on the other hand, may also initiate or contribute to the development of a proinflammatory brain endothelial cell phenotype associated with overexpression of interferon-γ and LPS-inducible inflammatory genes that are normally suppressed by the intracellular domain of LRP1 (LRP1-ICD; Fig 1a) that is translocated to the nucleus following γ-secretase-mediated cleavage at the plasma membrane (Zurhove et al. 2008). An increased expression of RAGE and downregulation of LRP1 at the BBB can suppress resting cerebral blood flow (CBF) and contribute to dysregulation of CBF responses to brain activation, respectively, as reported (Deane et al. 2003; Sagare et al. 2007a). RAGE expression has been found to be increased in brain endothelial cells and vascular smooth muscle cells (VSMC) in animal models of aging and AD as well as in human AD patients (Yan et al. 1996; Deane et al. 2003; Donahue et al. 2006; Miller et al. 2008), whereas LRP1 expression is decreased both at the BBB and in cerebral arterial VSMC (Shibata et al. 2000; Bading et al. 2002; Deane et al. 2004a; Donahue et al. 2006; Herring et al. 2008; Bell et al. 2009).
A moderate chronic hypoxia and/or chronic brain hypoperfusion can suppress the expression of the vascular-restricted mesenchyme homeobox gene 2 in brain endothelium in animal models and AD (Wu et al. 2005). Low expression of MEOX-2 mediates an aberrant brain angiogenesis characterized by premature capillary pruning and cell death ultimately leading to reductions in both microvascular length and resting CBF as well as downregulation of brain endothelial LRP1 (Wu et al. 2005). It has also been shown that an upregulation of two transcription factors that work in tandem, serum response factor and myocardin, in response to hypoxia and/or hypoperfusion leads to suppression of LRP1 in the VSMC in small cerebral arteries in animal models and in AD (Chow et al. 2007; Bell et al. 2009). Thus, hypoxia and perfusion stress can alter the phenotype of brain vascular cells amplifying the chronic neurovascular dysfunction that precedes neurodegenerative changes (Zlokovic 2005; Iadecola et al. 2009; de la Torre 2010). Brain ischemic events, ministrokes, brain hemorrhages, atherosclerosis, hypertension, diabetes, obesity, myocardial infarction, brain trauma and/or other vascular risk factors can set the stage for the development of chronic brain hypoxia, perfusion stress and/or BBB breakdown which may initiate neuronal degeneration independently of or synergistically with Aβ (for review see Zlokovic 2008).
Hit 2
To understand AD pathogenesis during the hit 2 phase, it is important to briefly review homeostasis of brain Aβ. Aβ concentration in the interstitial fluid in the brain is regulated by its rate of production from APP (Selkoe 2001a; Cirrito et al. 2003), influx of circulating Aβ into certain brain regions which depends on whether RAGE is expressed in the brain endothelium or not (Deane et al. 2003), rapid clearance across the BBB via LRP1 (Shibata et al. 2000; Deane et al. 2004a) which can be modulated by apoE isoforms (Zlokovic 1996; Martel et al. 1997; Tanzi et al. 2004; Moir and Tanzi 2005; Deane et al. 2008) and apoJ (Zlokovic et al. 1996; Calero et al. 2000; Bell et al. 2007), and the enzymatic degradation of Aβ (Selkoe 2001b). Although relatively little is known about the physiological functions of APP and Aβ, several studies have reported transport exchanges between peripheral and central Aβ pools from blood to brain (Zlokovic et al. 1993; Ghilardi et al. 1996; Martel et al. 1996; Poduslo et al. 1997; Mackic et al. 1998b, 2002; Banks et al. 2003; Deane et al. 2003; Ujiie et al. 2003) and from brain to blood (Shibata et al. 2000; Banks et al. 2003; Deane et al. 2004a, 2008; Ito et al. 2006, 2010; Bell et al. 2007).
In contrast to the fenestrated capillaries in peripheral organs which allow free exchange of many molecules between the blood and the ISF (Mann et al. 1985), the BBB is normally impermeable to small polar molecules, peptides and proteins (Zloković et al. 1985). Thus, transport of Aβ from blood to brain across the BBB requires the presence of a specialized transport system such as RAGE mediating a continuous re-entry of Aβ into the brain during the hit 2 stage that is associated with oxidant stress and pro-inflammatory response (Deane et al. 2003). It is noteworthy that RAGE-Aβ interactions at the BBB downregulates LRP1 (Deane et al. 2004b), thus making it unfavorable for Aβ clearance from the brain (see below). This in turn leads to a greater accumulation of different Aβ species in the brain (Kayed et al. 2003) including neurotoxic Aβ oligomers (Walsh et al. 2005; Lesné et al. 2006).
LRP1: a three-step homeostatic control of Aβ clearance
A continuous removal of Aβ from brain, blood and the entire organism is essential for preventing its accumulation in the brain (Zlokovic et al. 2000; Zlokovic 2008). As illustrated in Fig. 2, LRP1 plays a key role in the three-step serial clearance mechanism mediating Aβ elimination from the brain.
Fig. 2.
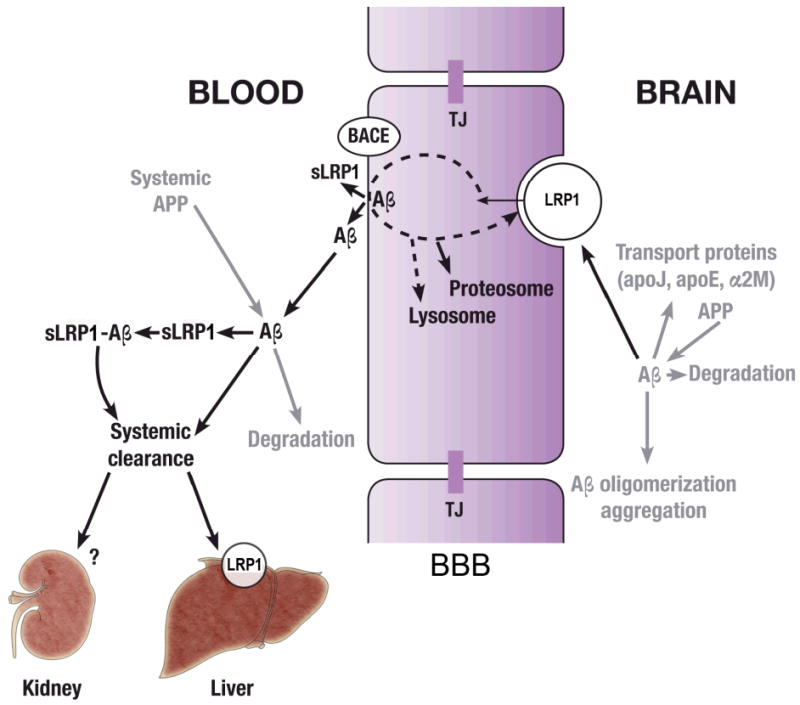
Schematic diagram illustrating the key role of LRP1 in the three-step serial clearance mechanism mediating Alzheimer's amyloid β-peptide (Aβ) elimination from the brain and the blood. Step 1: The cell surface LRP1 at the abluminal side of the blood-brain barrier (BBB) binds Aβ from the brain interstitial fluid initiating its transcytosis across the BBB to blood. Apolipoprotein J (apoJ), apoE2, apoE3 and apoE4 and α2-macroglobulin (α2M) influence differentially LRP1-mediated Aβ clearance at the BBB. Aβ is generated from Aβ precursor protein (APP) by all types of cells within the neurovascular unit. Degradation indicates enzymatic clearance of Aβ by different enzymes such as for example neprilysin and insulin degrading enzyme. TJ, the tight junctions between brain endothelial cells form an anatomical barrier for transport exchanges of solutes between blood and brain and vice versa, brain and blood. Step 2: β-secretase (BACE) in many organs including the BBB cleaves the N-terminal extracellular domain of LRP1 releasing soluble LRP1 (sLRP1) in the circulation. Circulating sLRP1 provides a key endogenous peripheral ‘sink’ activity for Aβ. sLRP1 normally binds 70-90% of plasma Aβ40 and Aβ42 preventing free Aβ access to the brain. Systemic APP indicates Aβ generation by different peripheral organs and its secretion back into the circulation. Step 3: The cell surface LRP1 in the liver mediates systemic clearance of sLRP1-Aβ complexes and free Aβ ultimately eliminating Aβ from the body. In addition, kidney removes sLRP1-Aβ complexes and Aβ. Whether LRP1 in the kidney can also mediate sLRP1-Aβ and Aβ systemic clearance as in the liver in presently unknown.
Level 1 – LRP1 at the blood-brain barrier and in brain vascular cells
LRP1 is the main cell surface receptor mediating brain Aβ clearance at the BBB. It has been demonstrated that binding of Aβ to LRP1 at the abluminal side of the BBB in vivo initiates a rapid Aβ clearance across the BBB into the blood (Shibata et al. 2000; Deane et al. 2004a, 2008; Cirrito et al. 2005; Ito et al. 2006; Bell et al. 2007; Sagare et al. 2007a). Human Aβ injected into different brain regions in mice was found intact in murine plasma confirming its elimination from the brain (Shiiki et al. 2004; Bell et al. 2007). It has been shown that clearance of human 125I-labeled Aβ40 and Aβ42 injected into the caudate nucleus in mice is significantly inhibited (i.e., >70%) by RAP which blocks binding of ligands to both LRP1 and megalin, as well as by an LRP1 specific antibody, but not by antibodies against LRP2 (megalin), low density lipoprotein receptor (LDLR), very low density lipoprotein receptor (VLDLR) or apoE2R, thereby suggesting that LRP1 mediates clearance of Aβ (Shibata et al. 2000; Deane et al. 2004a). A significant reduction of human 125I-Aβ40 and 125I-Aβ42 clearance from the brain has been reported in transgenic RAP null mice exhibiting a greater than 70% decrease in the LRP1 levels in brain microvessels, but not in transgenic mice lacking LDLR or VLDLR, demonstrating again that cerebrovascular LRP1 mediates Aβ clearance across the BBB (Shibata et al. 2000; Deane et al. 2004a). It has been shown that LRP1 is involved in the vascular clearance of human 125I-Aβ40 from the rat secondary somatosensory cortex (S2 region) (Ito et al. 2006).
Using human specific ELISA to determine the levels of unlabeled human Aβ40 and Aβ42 in brain and plasma after their respective microinjections into the mouse caudate nucleus, it has been independently confirmed that both RAP and an anti-LRP1 antibody inhibit Aβ40 clearance from the brain and the appearance of intact Aβ in plasma, indicating LRP1 involvement (Bell et al. 2007). Furthermore, it has been shown that inhibition of LRP1 expression in brain microvessels of CD-1 mice by a cocktail of the LRP1 antisense RNAs results in impaired Aβ clearance from the brain associated with an increase in endogenous brain Aβ levels and impaired cognition (Jaeger et al. 2009). Studies in mouse models with genetically manipulated LRP1 expression at the BBB, such as RAP-null mice expressing substantially reduced LRP1 levels in brain microvessels and Tie-2-LRP1-cluster-IV transgenic mice expressing the LRP1 cluster IV minigene in cerebral microvasculature, have demonstrated increased and reduced mouse endogenous Aβ40 and Aβ42 brain levels, suggesting a major role of LRP1 in regulating brain clearance of Aβ under physiological conditions (Deane et al. 2004a; LaRue et al. 2007; Sagare et al. 2007b). Recently, it has been reported that the levels of LRP1 in brain microvessels can be increased by fluvastatin resulting in an enhanced Aβ clearance from the brain (Shinohara et al. 2010).
It is of note, in addition to LRP1-mediated clearance of Aβ from the brain, there is also clearance of different Aβ isoforms from the CSF to the blood (Ghersi-Egea et al. 1996; Monro et al. 2002; Silverberg et al. 2003). Although the preponderance of intraparenchymally-injected biologically active test-molecules in the rodent brain is likely reabsorbed across brain microvessels, the bulk flow clearance from brain interstitial fluid to CSF (Szentistvanyi et al. 1984) and along perivascular spaces (Weller et al. 2008) has been estimated to contribute up to 15-20%. In the case of Aβ, it has been determined that the CSF bulk flow in the normal mouse brain can mediate ∼ 15% of total Aβ clearance (Shibata et al. 2000). It is conceivable that under pathological conditions associated with amyloid accumulation and diminished overall Aβ clearance from the CNS, the CSF bulk flow component can eventually become > 15%. However, this relative increase in the CSF clearance contribution would likely reflect a diminished LRP1-mediated Aβ clearance across the BBB, rather than an increase in the clearance capacity for Aβ through the choroid plexus and via the CSF pathway.
It is of note that LRP1 is also expressed in the choroid plexus epithelium of healthy young rats and its expression is sustained during aging (Johanson et al. 2006). Moreover, exposure to lead (Pb) has been shown to decrease expression of LRP1 in the choroid plexus epithelium that has been associated with Aβ accumulation in the choroid plexus (Behl et al. 2009, 2010). The exact role of LRP1 in the choroid plexus epithelium in mediating the CSF-to-blood Aβ clearance and for brain Aβ homeostasis during normal and pathological aging is an important topic deserving further research.
Studies using isolated murine cerebral microvessels have demonstrated LRP1-dependent clearance of Aβ40 and Aβ42 at the abluminal side of the BBB (Deane et al. 2004a). It has also been shown that RAP blocks an apoE-dependent uptake of Aβ peptides by astrocytes indicating that LRP1 and/or another member of the LDLR receptor family are likely involved in the astrocyte-mediated clearance of Aβ (Koistinaho et al. 2004). Studies using in vitro BBB models with a conditional immortalized cell line derived from brain capillary endothelial cells of transgenic rats expressing temperature-sensitive large T antigen (Yamada et al. 2008) and with the polarized Madin-Darby canine kidney cells expressing LRP1 mini-receptors (Nazer et al. 2008), have also importantly demonstrated the role of LRP1 in Aβ endothelial cellular uptake and endocytosis, respectively, resulting in Aβ clearance. Moreover, our preliminary observations using a human BBB in vitro model with primary brain endothelial cells and pericyte-conditioned media to direct LRP1 distribution mainly to the basolateral side of an endothelial monolayer have revealed LRP1-mediated transcytosis of Aβ40 and Aβ42 in the basolateral-to-apical direction corresponding to the abluminal and luminal sides of the BBB in vivo, respectively (E. A. Winkler, Y. Sallstrom, D. Zhu, R. Deane and B. V. Zlokovic, unpublished data).
LRP1 that is expressed at the abluminal side of the BBB was shown to mediate Aβ transport from brain to blood, but others have also reported that LRP1 can be utilized for delivery of therapeutics to the brain, as for example angiopeps (Demeule et al. 2008), implying that LRP1 might also be expressed at the luminal side of the BBB. The exact distribution of LRP1 between the luminal side of the BBB, the cytoplasmic endothelial pool and the abluminal side of the BBB is presently unknown. Our work in progress using high resolution confocal microscopy analysis indicates, however, that LRP1 is mainly confined to the abluminal side of the BBB, but a smaller portion of LRP1 is also expressed at the luminal side of the BBB (E. A. Winkler, Y. Sallstrom, D. Zhu, R. Deane and B. V. Zlokovic, unpublished data). It is possible that luminal LRP1 may participate in transport of angiopeps from blood-to-brain and that LRP1 in the cerebral vascular smooth muscle cells (Bell et al. 2009) can be utilized as well for delivery of therapeutics to the brain and cerebral arteries.
Reduced levels of LRP1 in brain microvessels correlating with endogenous Aβ deposition have been shown in a chronic hydrocephalus model in rats (Klinge et al. 2006). Moreover, reduced levels of LRP1 in brain microvessels associated with Aβ cerebrovascular and brain accumulation have been reported in AD patients (Shibata et al. 2000; Donahue et al. 2006). Several studies have indicated that LRP1 expression in the brain capillary endothelium is reduced during normal aging in rodents, non-human primates and humans, as well as in AD models and AD patients (Kang et al. 2000; Shibata et al. 2000; Bading et al. 2002; Deane et al. 2004a; Donahue et al. 2006; Bell and Zlokovic 2009). Similar reductions in LRP1 expression have been reported in cerebral vascular smooth muscle cells in small pial and intracerebral arteries regulating blood flow to the brain which was shown to be associated with Aβ accumulation within the wall of these brain arteries (Bell et al. 2009). Therefore, it is likely that LRP1 downregulation in the brain endothelium and vascular cells in patients with mild cognitive impairment during the hit 1 stage and in AD patients during the hit 2 stage would lead to faulty vascular Aβ clearance promoting cerebrovascular and focal parenchymal Aβ accumulations contributing to AD pathogenesis.
Pitfalls in Aβ clearance measurements in animal models
Reproducible and accurate measurements of Aβ clearance from the brain are challenging because of the hydrophobic nature of the full length peptide, possible conformational and structural changes of Aβ and heterogeneity of truncated Aβ fragments. Studies with 125I-labeled Aβ have generated critical data for the field. However, work with 125I-Aβ preparations also requires special precautions due to rapid radiolysis of the labeled peptide. We have recommended that the radiolabeled Aβ should be used either immediately after labeling within 24 h or alternatively can be stored in ethanol over a short period of time (i.e., 3-4 days), and re-purified before use on the day of the experiment by HPLC to eliminate free iodine and possible Aβ degradation products, and separate mono-iodinated from di-iodinated Aβ species and oxidized from reduced Aβ (LaRue et al. 2004). It is of note, Aβ radioiodination by a mild lactoperoxidase method typically provides less damage to the peptide than a more robust chloramine-T method (LaRue et al. 2004). In brain clearance studies, the integrity of 125I-Aβ in the brain should be additionally confirmed by different analytical methods such as sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and HPLC (Deane et al. 2004a, 2008).
It has also been raised that iodination can potentially introduce a conformational change in the peptide. Therefore, more recently some important control measurements have been performed by different groups (as discussed above) to complement the radiotracer studies. This includes, but is not limited to the use of unlabeled Aβ and measurements of its concentrations in brain and blood by ELISA, as well as measurements of endogenous Aβ levels in transgenic models with manipulated LRP1 expression, or LRP1 silencing and/or by pharmacologically manipulating LRP1 expression in brain microvessels.
In a recent paper, Ito et al. (2010) have confirmed 125I-Aβ40 clearance from the mouse S2 region using an established brain clearance technique (Kakee et al. 1996), but failed to demonstrate inhibition of Aβ clearance by RAP. Based on this single negative result with RAP, Ito et al. have suggested that the members of the LDLR receptor family including LRP1 do not participate in Aβ40 clearance, challenging findings from several different groups that have demonstrated a major role of LRP1 in Aβ clearance, as discussed above. However, Ito et al. have not performed any control experiments to determine 125I-Aβ integrity immediately prior to its use on the day of the experiment and/or at the end of the experiment in brain extracts. They also did not use unlabeled Aβ as a control for radiolabeled Aβ. Moreover, they have not employed any complementary approach to determine the role of LRP1 in Aβ clearance, such as blockade of LRP1 by an LRP1-specific antibody, inhibition of LRP1 expression by LRP1 silencing by siRNA and/or antisense RNAs strategies, or use of transgenic models with genetically manipulated LRP1 expression.
By analyzing the experimental design in Ito et al.'s (2010) study, one can find out that Aβ40 was radiolabeled with chloramine-T and after labeling it was lyophilized and mixed with different excipients including aprotinin, as per the PerkinElmer Technical Data Certificate of Analysis, NEX361 sheet and Dr. Terasaki's (senior author in Ito et al. 2010) personal communication to us. Aprotinin is a protease inhibitor, but it unfortunately also binds to LRP1 and prevents an LRP1-dependent uptake of RAP (Demeule et al. 2008), which therefore could potentially interfere with Aβ clearance measurements in vivo as performed by Ito et al. (2010). According to table 1 [from Ito et al. 2010], one can calculate the final concentration of aprotinin in the injectate in their experiments which was about 400 KIU (Kallikrein Inhibitor Unit)/mL of aprotinin or about 8.6 μM.
To test whether co-administered aprotinin interferes with the RAP-mediated blockade of Aβ clearance, we have determined clearance of unlabeled Aβ40 from the mouse caudate nucleus in the presence or absence of RAP and with or without aprotinin by using human specific Aβ40 ELISA over a 30 min period of time. In all experiments, 14C-inulin (an inert polar molecule) was administered into the brain simultaneously with Aβ as a reference standard (Shibata et al. 2000). As we have reported previously by using unlabeled Aβ (Bell et al. 2007), RAP (5 μM) decreased human Aβ40 clearance from the brain by ∼50% (Fig. 3a; for details of calculations please see Bell et al. 2007). However, in the presence of aprotinin (8.6 μM), RAP did not have any significant effect on Aβ40 clearance (Fig. 3a) corroborating data by Ito et al. (2010). The appearance of an intact human Aβ40 in plasma was abolished by RAP in the absence of aprotinin (Fig. 3b). However, the presence of aprotinin completely inhibited RAP-mediated blockade of Aβ brain-to-blood transfer (Fig. 3b).
Fig. 3.
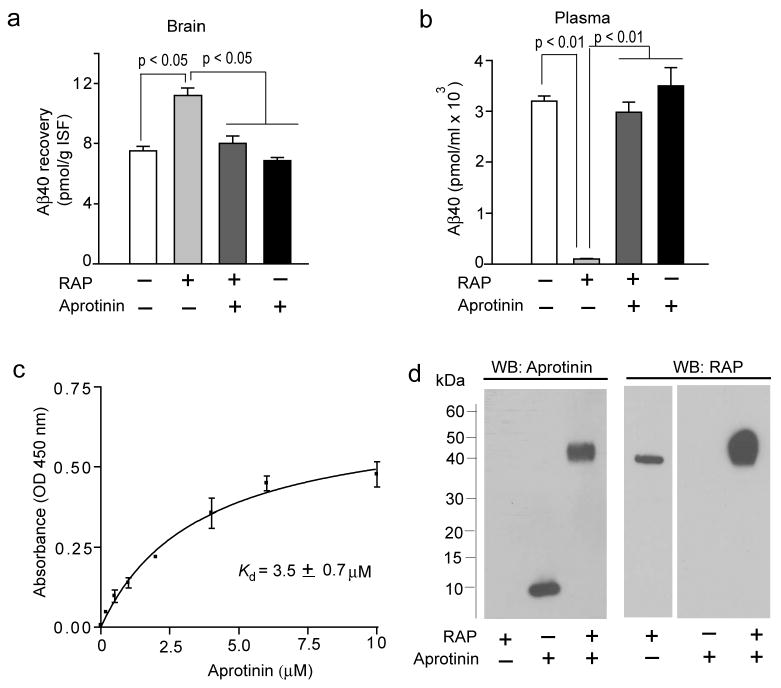
Levels of human Aβ40 in the brain (a) and plasma (b) 30 min after microinjection of human Aβ40 (40 nM) and 14C-inulin (0.023 μCi) into the mouse caudate nucleus in the presence and absence of RAP (5 μM) with and without aprotinin (8.6 μM). Human unlabeled Aβ40 peptide levels in the brain and plasma were determined by using human-specific ELISA, as described (Bell et. al. 2007). The brain sample used for analysis was approximately 15 mg of tissue adjacent to the site of microinjection in the caudate nucleus as described (Bell et al. 2007). In (a) and (b), values are mean ± SEM from 3 to 5 independent experiments. (c) Binding of aprotinin to immobilized human RAP, by ELISA. Briefly, 5 μg/mL repurified human recombinant RAP (Oxford Biomedical Research, Oxford, MI, USA) was coated on microtiter plate and wells were blocked with 1% BSA. Varying concentrations of aprotinin (Sigma, St Louis, MO, USA) were added to the wells and incubated for 1 h at 25°C. Bound aprotinin was detected by mouse anti-aprotinin antibody (Abcam, Cambridge, MA, USA), followed by goat anti-mouse HRP conjugate (Bio-Rad Laboratories, Hercules, CA, USA). The reaction was developed using tetramethyl benzidine substrate (TMB; KPL), stopped with 1M HCl and quantified at 450 nm. Values are mean ± s.e.m. from 3 independent experiments. (d) Formation of RAP and aprotinin complexes detected by the Western blot analysis for aprotonin and RAP. Aprotinin (2 μM; Sigma) was incubated with human recombinant RAP (2 μM; Oxford Biomedical Research) for 1 h at 37°C in PBS and the complex formation was confirmed by cross-linking with Bis(sulfosuccinimidyl)suberate (BS3; Pierce, Rockford, IL, USA) followed by 4-12% SDS-PAGE separation of proteins under non-reducing conditions and Western blot analysis for aprotinin using mouse anti-aprotinin (Abcam) antibody and for RAP using mouse anti-RAP (Oxford Biomedical Research). Representative WB analysis from 3 independent experiments were shown.
Consistent with a previous report (Ito et al. 2010), Aβ recovery in brain and its appearance in plasma after intracerebral administration were not affected by aprotinin alone (Fig. 3a and b), suggesting that aportinin alone does not influence LRP1-mediated Aβ clearance from brain and that at least in vivo Aβ and aprotinin likely bind to different exosites on LRP1. Overall, this data shows that only in the presence of RAP aprotinin substantially affects the measurement of Aβ clearance from the mouse brain.
As aprotinin prevented RAP-mediated inhibition of Aβ40 clearance from the brain, we next explored whether aprotinin interacts directly with RAP. Here, we have demonstrated by ELISA that aprotinin binds to immobilized RAP (Fig. 3c). By incubating aprotinin and RAP for 1 h at 37°C we have additionally confirmed by immunoblotting analysis after cross-linking with bis[sulfosuccinimidyl]suberate (BS3) that aprotinin interacts directly with RAP (Fig. 3d). Thus, binding of RAP to aprotinin would prevent RAP from blocking 125I-Aβ40 binding to LRP1 at the abluminal side of the BBB in vivo which in turn may confound measurements of Aβ clearance and data interpretation.
Level 2 – Soluble LRP1 in plasma
We have demonstrated that circulating plasma sLRP1 provides a key endogenous peripheral ‘sink’ activity for Aβ by promoting a continuous removal of Aβ from brain (Sagare et al. 2007a). In neurologically healthy humans and mice, sLRP1 normally binds > 70% of circulating Aβ preventing free Aβ access to the brain (Sagare et al. 2007a) (Fig. 1a). In AD patients and AD transgenic mice, however, Aβ binding to sLRP1 is compromised because of increased levels of oxidized sLRP1 which does not bind Aβ (Sagare et al. 2007a) resulting in elevated levels of free Aβ40 and Aβ42 that can re-enter the brain via RAGE-mediated transport (Deane et al. 2003; Ujiie et al. 2003; Donahue et al. 2006; Sagare et al. 2007a).
In the human hippocampus, it was shown that RAGE expression in brain endothelium is increased with advanced AD compared to early stage AD and/or individuals with mild cognitive impairment (MCI) (Miller et al. 2008) which may further contribute to Aβ accumulation into the brain via enhanced Aβ influx from blood to brain. Moreover it has been recently shown that a diminished sLRP1-Aβ ‘sink’ activity precedes an increase in the tau/Aβ42 CSF ratio and a drop in global cognitive decline in individuals with MCI converting into AD, and is therefore a useful early biomarker for AD-type dementia (Sagare et al. 2009). It has also been shown that recombinant LRP1 fragments can effectively replace oxidized sLRP1 and sequester free Aβ in plasma in AD patients and AD transgenic mice ultimately reducing Aβ-related pathology in the brain (Sagare et al. 2007a). It is of note, a recent Phase II clinical trial in patients with mild AD with Baxter's intravenous immunoglobulin preparation Gammagard Liquid containing both sLRP1 and anti-RAGE immunoglobulins (Weber et al. 2009) has shown encouraging results (Relkin et al. 2009). It has been suggested that both sLRP1 and anti-RAGE may contribute to the observed beneficial effects of GGL by improving peripheral sink for Aβ and preventing Aβ influx into the brain, respectively (Dodel et al. 2010).
Level 3 – LRP1 in liver
In aged Squirrel monkeys, Aβ systemic clearance is reduced and is associated with increased Aβ levels in the brain (Mackic et al. 1998b, 2002). Also, an increased entry of circulating Aβ42 into the brain and its deposition onto senile plaques in aged Rhesus monkeys (Mackic et al. 2002) or accumulation of circulating Aβ40 in the cerebral vessels in aged Squirrel monkeys with CAA (Ghilardi et al. 1996), have been demonstrated. An age-dependent reduction in the systemic Aβ clearance may diminish the ‘sink action’ for Aβ clearance from the brain which in turn could increase the RAGE-dependent free Aβ transport across the BBB into brain regions expressing RAGE.
It has also been reported that a rapid peripheral clearance of Aβ is mediated mainly by LRP1 in the liver (Tamaki et al. 2006). Using the perfused rat liver preparation, Tamaki et al. (2007) were able to convincingly show that RAP blocks 125I-Aβ uptake by the liver likely because in these experiments RAP was pre-infused into the liver before administration of 125I-labeled Aβ and aprotinin, and was not mixed with Aβ/aprotinin. In addition, the dilution of aprotinin in the liver uptake measurements (Tamaki et al. 2007) was at least by ∼ 40-fold greater than in the brain clearance measurements (Ito et al. 2010). These differences in the experimental design were likely to minimize the interaction between RAP and aprotinin in the Aβ liver uptake study.
It is of note that reduced hepatic LRP1 levels have been shown to be associated with decreased peripheral Aβ clearance in the aged rats (Tamaki et al. 2006, 2007). In addition, both sLRP1-Aβ complexes and free Aβ are eliminated via the kidneys (Sagare et al. 2007a), but whether LRP1 is involved in Aβ clearance by the kidneys as in the liver is presently unknown.
Conclusions and future therapeutic directions
In conclusion, we have reviewed recent evidence suggesting that LRP1 has a major role in regulating brain and systemic clearance of Alzheimer's Aβ. Specifically, we have discussed the role of LRP1 during hit 1 (i.e., before Aβ accumulation) and hit 2 (i.e., the Aβ accumulation stage) phases of AD pathogenesis. According to our vascular two hit hypothesis for AD an initial vascular damage such as hypoxia, perfusion stress and/or disruption of the BBB functional integrity drive the disease process and Aβ accumulation in the brain. The multiple regulatory roles of cell surface LRP1 at the BBB, the circulating soluble LRP1 and LRP1 in the peripheral organs such as liver suggest a three-level serial LRP1 homeostatic mechanism for Aβ clearance from the brain that could be a major, but still underexplored therapeutic target in AD.
It has been shown that pharmacological and/or gene therapy strategies to increase the levels of LRP1 in the cerebrovascular system and at the BBB may hold potential to alleviate initial vascular damage by suppressing brain endothelial cell activation during the hit 1 stage, and prevent and/or reduce Aβ accumulation during both the hit 1 and hit 2 stages of AD pathogenesis. The oxidized sLRP1 in plasma, which does not bind peripheral Aβ and therefore does not have an Aβ ‘sink’ activity, is also an important early biomarker in MCI individuals converting into AD as well as in AD patients. Thus, replacement therapy for the native oxidized circulating sLRP1 with recombinant LRP1 clusters may help maintain the peripheral Aβ ‘sink’ activity at an early stage of the disease in patients with MCI by reducing the influx of circulating Aβ into the brain which may slow down disease progression. Finally, approaches to increase LRP1 expression in peripheral organs, such as liver, through pharmacologic agents and/or gene therapy might help promote systemic Aβ elimination, which has been shown to improve peripheral Aβ-‘sink’ activity by reducing sLRP1-Aβ and free Aβ levels in plasma and promoting Aβ clearance from the brain.
As a note of caution, we would like to stress that the development of LRP1-based therapies for AD requires careful toxicity and safety monitoring of unwanted potential side effects given that LRP1 participates in multiple control systems in the body, ranging from the cellular transport and metabolism of cholesterol to anticoagulation and inflammation. However, our preliminary findings suggest that LRP1-based therapeutics such as the recombinant LRP1 clusters (Zlokovic et al. 2009) can be tailored by genetic engineering to be more specifically directed at Aβ with minimal effects on other systems in the body. These new LRP1 variants produced by site-directed LRP1 mutagenesis may have potential either as recombinant proteins adapted for systemic administration and/or as Aβ-selective LRP1 gene constructs adapted for tissue-specific expression in the brain, liver and/or other peripheral organs.
Acknowledgments
This study was supported by US National Institutes of Health (NIH) grants R37AG023084 and R37NS34467 to BVZ.
Abbreviations used
- AD
Alzheimer's disease
- apoE
apolipoprotein E
- APP
Aβ-precursor protein
- Aβ
amyloid β-peptide
- BBB
blood-brain barrier
- CBF
cerebral blood flow
- LDLR
low-density lipoprotein receptor
- LRP1
low-density lipoprotein receptor-related protein-1
- MCI
mild cognitive impairment
- PICALM
phosphatidylinositol binding clathrin assembly protein
- RAGE
receptor for advanced glycation end products
- RAP
receptor associated protein
- VSMC
vascular smooth muscle cells
References
- Arelin K, Kinoshita A, Whelan CM, Irizarry MC, Rebeck GW, Strickland DK, Hyman BT. LRP and senile plaques in Alzheimer's disease: colocalization with apolipoprotein E and with activated astrocytes. Brain Res Mol Brain Res. 2002;104:38–46. doi: 10.1016/s0169-328x(02)00203-6. [DOI] [PubMed] [Google Scholar]
- Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV. Brain clearance of Alzheimer's amyloid-beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target. 2002;10:359–368. doi: 10.1080/10611860290031831. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Banks WA, Robinson SM, Verma S, Morley JE. Efflux of human and mouse amyloid beta proteins 1-40 and 1-42 from brain: impairment in a mouse model of Alzheimer's disease. Neuroscience. 2003;121:487–492. doi: 10.1016/s0306-4522(03)00474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M, Zhang Y, Monnot AD, Jiang W, Zheng W. Increased beta-amyloid levels in the choroid plexus following lead exposure and the involvement of low-density lipoprotein receptor protein-1. Toxicol Appl Pharmacol. 2009;240:245–254. doi: 10.1016/j.taap.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M, Zhang Y, Shi Y, Cheng J, Du Y, Zheng W. Lead-induced accumulation of β-amyloid in the choroid plexus: Role of low density lipoprotein receptor protein-1 and protein kinase C. Neurotoxicology. 2010;31:524–532. doi: 10.1016/j.neuro.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Deane R, Chow N, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchenane K, Berezowski V, Ali C, et al. Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein-related protein-mediated transcytosis. Circulation. 2005;111:2241–2249. doi: 10.1161/01.CIR.0000163542.48611.A2. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Alzheimer disease: new light on an old CLU. Nat Rev Neurol. 2010;6:11–13. doi: 10.1038/nrneurol.2009.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Blacker D, Crystal A, et al. Candidate genes showing no evidence for association or linkage with Alzheimer's disease using family-based methodologies. Exp Gerontol. 2000;35:1353–1361. doi: 10.1016/s0531-5565(00)00193-5. [DOI] [PubMed] [Google Scholar]
- Bu G, Sun Y, Schwartz AL, Holtzman DM. Nerve growth factor induces rapid increases in functional cell surface low density lipoprotein receptor-related protein. J Biol Chem. 1998;273:13359–13365. doi: 10.1074/jbc.273.21.13359. [DOI] [PubMed] [Google Scholar]
- Bushlin I, Petralia RS, Wu F, Harel A, Mughal MR, Mattson MP, Yao PJ. Clathrin assembly protein AP180 and CALM differentially control axogenesis and dendrite outgrowth in embryonic hippocampal neurons. J Neurosci. 2008;28:10257–10271. doi: 10.1523/JNEUROSCI.2471-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero M, Rostagno A, Matsubara E, Zlokovic BV, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer's disease. Microsc Res Tech. 2000;50:305–315. doi: 10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Chalmers KA, Barker R, Passmore PA, Panza F, Seripa D, Vincenzo S, Love S, Prince JA, Kehoe PG. LRP-1 variation is not associated with risk of Alzheimer's disease. Int J Mol Epidemiol Genet. 2010;1:104–113. [PMC free article] [PubMed] [Google Scholar]
- Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, Van Nostrand W, Miano JM, Zlokovic BV. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proc Natl Acad Sci USA. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis M, Schober R, Krohn K. Genetic-morphologic association study: association between the low density lipoprotein-receptor related protein (LRP) and cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2005;31:11–19. doi: 10.1111/j.1365-2990.2004.00614.x. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy JE, Shin WD, Knauer MF, Knauer DJ, Komives EA. All three LDL receptors homology regions of the LDL receptor-related protein bind multiple ligands. Biochemistry. 2003;42:13049–13057. doi: 10.1021/bi034752s. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, et al. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2004a;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004b;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- Demeule M, Currie JC, Bertrand Y, Ché C, Nguyen T, Régina A, Gabathuler R, Castaigne JP, Béliveau R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J Neurochem. 2008;106:1534–1544. doi: 10.1111/j.1471-4159.2008.05492.x. [DOI] [PubMed] [Google Scholar]
- Dodel R, Neff F, Noelker C, Pul R, Du Y, Bacher M, Oertel W. Intravenous immunoglobulins as a treatment for Alzheimer's disease. Drugs. 2010;70:513–528. doi: 10.2165/11533070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Donahue JE, Flaherty SL, Johanson CE, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- van der Geer P. Phosphorylation of LRP1: regulation of transport and signal transduction. Trends Cardiovasc Med. 2002;12:160–165. doi: 10.1016/s1050-1738(02)00154-8. [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD. Fate of cerebrospinal fluid-borne amyloid β-peptide: Rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996;67:880–883. doi: 10.1046/j.1471-4159.1996.67020880.x. [DOI] [PubMed] [Google Scholar]
- Ghilardi JR, Catton M, Stimson ER, Rogers S, Walker LC, Maggio JE, Mantyh PW. Intra-arterial infusion of [125I]Aβ1-40 labels amyloid deposits in the aged primate brain in vivo. Neuroreport. 1996;7:2607–2611. doi: 10.1097/00001756-199611040-00040. [DOI] [PubMed] [Google Scholar]
- Gotthardt M, Trommsdorff M, Nevitt MF, Shelton J, Richardson JA, Stockinger W, Nimpf J, Herz J. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J Biol Chem. 2000;275:25616–25624. doi: 10.1074/jbc.M000955200. [DOI] [PubMed] [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease, the amyloid cascade hypothesis. Science. 1992;286:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring A, Yasin H, Ambrée O, Sachser N, Paulus W, Keyvani K. Environmental enrichment counteracts Alzheimer's neurovascular dysfunction in TgCRND8 mice. Brain Pathol. 2008;18:32–39. doi: 10.1111/j.1750-3639.2007.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J. The LDL receptor gene family: (un)expected signal transducers in the brain. Neuron. 2001;29:571–581. doi: 10.1016/s0896-6273(01)00234-3. [DOI] [PubMed] [Google Scholar]
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Bock HH. Lipoprotein receptors in the nervous system. Ann Rev Biochem. 2002;71:405–434. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]
- Herz J, Chen Y, Masiulis I, Zhou L. Expanding functions of lipoprotein receptors. J Lipid Res. 2009;50:S287–S292. doi: 10.1194/jlr.R800077-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain MM, Strickland DK, Bakillah A. The mammalian low-density lipoprotein receptor family. Ann Rev Nutr. 1999;19:141–172. doi: 10.1146/annurev.nutr.19.1.141. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Park L, Capone C. Threats to the mind. Aging, amyloid, and hypertension. Stroke. 2009;40:S40–S44. doi: 10.1161/STROKEAHA.108.533638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Ohtsuki S, Terasaki T. Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1-40) across the rat blood-brain barrier. Neurosci Res. 2006;56:246–252. doi: 10.1016/j.neures.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Ito S, Ueno T, Ohtsuki S, Terasaki T. Lack of brain-to-blood efflux transport activity of low-density lipoprotein receptor-related protein-1 (LRP-1) for amyloid-β peptide(1-40) in mouse: involvement of an LRP-1-independent pathway. J Neurochem. 2010;113:1356–1363. doi: 10.1111/j.1471-4159.2010.06708.x. [DOI] [PubMed] [Google Scholar]
- Jaeger LB, Dohgu S, Hwang MC, et al. Testing the neurovascular hypothesis of Alzheimer's disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-β protein, and impairs cognition. J Alzheimer's Dis. 2009;17:553–570. doi: 10.3233/JAD-2009-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson C, Flaherty S, Messier A, Duncan J, III, Silverberg G. Expression of the beta-amyloid transporter, LRP-1, in aging choroid plexus: implications for the CSF-brain system in NPH and Alzheimer's disease. Cerebrospinal Fluid Res. 2006;3:S29. [Google Scholar]
- Kakee A, Terasaki T, Sugiyama Y. Brain efflux index as a novel method of analyzing efflux transport at the blood-brain barrier. J Pharmacol Exp Ther. 1996;277:1550–1559. [PubMed] [Google Scholar]
- Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, Thomas RG, Thal LJ, Katzman R. Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer's disease. Neurology. 1997;49:56–61. doi: 10.1212/wnl.49.1.56. [DOI] [PubMed] [Google Scholar]
- Kang DE, Pietrzik CU, Baum L, et al. Modulation of amyloid beta-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Klinge PM, Samii A, Niescken S, Brinker T, Silverberg GD. Brain amyloid accumulates in aged rats with kaolin-induced hydrocephalus. Neuroreport. 2006;17:657–660. doi: 10.1097/00001756-200604240-00020. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10:716–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Wavrant-De Vrieze F, Amouyel P, Chartier-Harlin MC. Association at LRP gene locus with sporadic late-onset Alzheimer's disease. Lancet. 1998;351:1787–1788. doi: 10.1016/s0140-6736(05)78749-3. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- LaRue B, Hogg E, Sagare A, Jovanovic S, Maness L, Maurer C, Deane R, Zlokovic BV. Method for measurement of the blood-brain barrier permeability in the perfused mouse brain: application to amyloid-β peptide in wild type and Alzheimer's Tg2576 mice. J Neurosci Methods. 2004;138:233–242. doi: 10.1016/j.jneumeth.2004.04.026. [DOI] [PubMed] [Google Scholar]
- LaRue BA, Cheng T, Pinkert C, Deane R, Zlokovic BV. 2007 Neuroscience meeting planner. San Diego, CA: Society for Neuroscience; Online: 2007. A new mouse model expressing the amyloid beta clearance receptor at the blood-brain barrier. Program No. 156.1/T13. [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu W, Marzolo MP, Bu G. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J Biol Chem. 2001;276:18000–18006. doi: 10.1074/jbc.M101589200. [DOI] [PubMed] [Google Scholar]
- Lillis AP, van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackic JB, Stins M, McComb JG, et al. Human blood-brain barrier receptors for Alzheimer's amyloid-beta 1-40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998a;102:734–743. doi: 10.1172/JCI2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer's amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem. 1998b;70:210–215. doi: 10.1046/j.1471-4159.1998.70010210.x. [DOI] [PubMed] [Google Scholar]
- Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV. Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer's disease lesions. Vascul Pharmacol. 2002;38:303–313. doi: 10.1016/s1537-1891(02)00198-2. [DOI] [PubMed] [Google Scholar]
- Mann GE, Zlokovic BV, Yudilevich DL. Evidence for a lactate transport system in a sarcolemmal membrane of the perfused rabbit heart: kinetics of unidirectional influx, carrier specificity and effects of glucagon. Biochim Biophys Acta. 1985;819:241–248. doi: 10.1016/0005-2736(85)90179-8. [DOI] [PubMed] [Google Scholar]
- Martel CL, Mackic JB, McComb JG, Ghiso J, Zlokovic BV. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer's amyloid beta in guinea-pigs. Neurosci Lett. 1996;206:157–160. doi: 10.1016/s0304-3940(96)12462-9. [DOI] [PubMed] [Google Scholar]
- Martel CL, Mackic JB, Matsubara E, et al. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid beta. J Neurochem. 1997;69:1995–2004. doi: 10.1046/j.1471-4159.1997.69051995.x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer's disease. Neurology. 1992;42:447–449. doi: 10.1212/wnl.42.2.447. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- Meijer AB, Rohlena J, van der Zwaan C, van Zonneveld AJ, Boertjes RC, Lenting PJ, Mertens K. Functional duplication of ligand-binding domains within low-density lipoprotein receptor-related protein for interaction with receptor associated protein, alpha2-macroglobulin, factor IXa and factor VIII. Biochim Biophys Acta. 2007;1774:714–722. doi: 10.1016/j.bbapap.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Miller MC, Tavares R, Johanson CE, Hovanesian V, Donahue JE, Gonzalez L, Silverberg GD, Stopa EG. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer's disease. Brain Res. 2008;1230:273–280. doi: 10.1016/j.brainres.2008.06.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Tanzi RE. LRP-mediated clearance of Aβ is inhibited by KPI-containing isoforms of APP. Curr Alz Res. 2005;2:269–273. doi: 10.2174/1567205053585918. [DOI] [PubMed] [Google Scholar]
- Monro OR, Mackic JB, Yamada S, Segal MB, Ghiso J, Maurer C, Calero M, Frangione B, Zlokovic BV. Substitution at codon 22 reduces clearance of Alzheimer's amyloid-beta peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol Aging. 2002;23:405–412. doi: 10.1016/s0197-4580(01)00317-7. [DOI] [PubMed] [Google Scholar]
- Narita M, Holtzman DM, Schwartz AL, Bu G. Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem. 1997;69:1904–1911. doi: 10.1046/j.1471-4159.1997.69051904.x. [DOI] [PubMed] [Google Scholar]
- Nazer B, Hong S, Selkoe DJ. LRP promotes endocytosis and degradation, but not transcytosis, of the amyloid-beta peptide in a blood-brain barrier in vitro model. Neurobiol Dis. 2008;30:94–102. doi: 10.1016/j.nbd.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neels JG, van den Berg BMM, Lookene A, Olivecrona G, Pannekoek H, van Zonneveld AJ. The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J Biol Chem. 1999;274:31305–31311. doi: 10.1074/jbc.274.44.31305. [DOI] [PubMed] [Google Scholar]
- Obermoeller-McCormick LM, Li Y, Osaka H, FitzGerald DJ, Schwartz AL, Bu G. Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J Cell Sci. 2001;114:899–908. doi: 10.1242/jcs.114.5.899. [DOI] [PubMed] [Google Scholar]
- Pan W, Kastin AJ, Zankel TC, van Kerkhof P, Terasaki T, Bu G. Efficient transfer of receptor-associated protein (RAP) across the blood-brain barrier. J Cell Sci. 2004;117:5071–5078. doi: 10.1242/jcs.01381. [DOI] [PubMed] [Google Scholar]
- Parkyn CJ, Vermeulen EGM, Mootoosamy RC, et al. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci. 2008;121:773–783. doi: 10.1242/jcs.021816. [DOI] [PubMed] [Google Scholar]
- Pietrzik CU, Yoon IS, Jaeger S, Busse T, Weggen S, Koo EH. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci. 2004;24:4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Haggard JJ, Biere AL, Selkoe DJ. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid β protein 1-40 at the blood-brain barrier. Neurobiol Dis. 1997;4:27–34. doi: 10.1006/nbdi.1997.0132. [DOI] [PubMed] [Google Scholar]
- Polavarapu R, Gongora MC, Yi H, Ranganathan S, Lawrence DA, Strickland D, Yepes M. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270–3278. doi: 10.1182/blood-2006-08-043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Strickland DK, Hyman BT, Rebeck GW. Alpha2-macroglobulin enhances the clearance of endogenous soluble beta-amyloid peptide via low-density lipoprotein receptor-related protein in cortical neurons. J Neurochem. 1999;73:1393–1398. doi: 10.1046/j.1471-4159.1999.0731393.x. [DOI] [PubMed] [Google Scholar]
- Rebeck GW, Harr SD, Strickland DK, Hyman BT. Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein E receptor, the alpha 2-macroglobulin receptor/low-density-lipoprotein receptor-related protein. Ann Neurol. 1995;37:211–217. doi: 10.1002/ana.410370212. [DOI] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, et al. 18-month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Sagare A, Deane R, Bell RD, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007a;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare AP, Deane R, Zlokovic BV. 2007 Neuroscience meeting planner. Society for Neuroscience; San Diego, CA: 2007b. LRP1 and RAGE regulate normal amyloid β-peptide homeostasis in the CNS. Program No 689.19/L3. Online. [Google Scholar]
- Sagare AP, Deane R, Zetterberg H, Blennow K, Zlokovic BV. 2009 Neuroscience meeting planner. Chicago, IL: 2009. sLRP1: a potential biomarker of mild cognitive impairment and Alzheimer's disease?. Program No. 139.21/D31. Online. [Google Scholar]
- Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001a;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001b;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer's amyloid-β1-40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiiki T, Ohtsuki S, Kurihara A, Naganuma H, Nishimura K, Tachikawa M, Hosoya K, Terasaki T. Brain insulin impairs amyloid-beta (1-40) clearance from the brain. J Neurosci. 2004;24:9632–9637. doi: 10.1523/JNEUROSCI.2236-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Sato N, Kurinami H, Takeuchi D, Takeda S, Shimamura M, Yamashita T, Uchiyama Y, Rakugi H, Morishita R. Reduction of brain Aβ by fluvastatin, an HMG-CoA reductase inhibitor, through increase in degradation of APP-CTFs and Aβ clearance. J Biol Chem. 2010;285:22091–22102. doi: 10.1074/jbc.M110.102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg GD, Mayo M, Saul T, Rubenstein E, McGuire D. Alzheimer's disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2003;2:506–511. doi: 10.1016/s1474-4422(03)00487-3. [DOI] [PubMed] [Google Scholar]
- Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF. Drainage of interstitial fluid from different regions of rat brain. Am J Physiol. 1984;246:F835–F844. doi: 10.1152/ajprenal.1984.246.6.F835. [DOI] [PubMed] [Google Scholar]
- Tamaki C, Ohtsuki S, Iwatsubo T, Hashimoto T, Yamada K, Yabuki C, Terasaki T. Major involvement of low-density lipoprotein receptor-related protein 1 in the clearance of plasma free amyloid β-peptide by the liver. Pharm Res. 2006;23:1407–1416. doi: 10.1007/s11095-006-0208-7. [DOI] [PubMed] [Google Scholar]
- Tamaki C, Ohtsuki S, Terasaki T. Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (1-40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol Pharmacol. 2007;72:850–855. doi: 10.1124/mol.107.036913. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Aβ peptide: the many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell. 1999;10:2687–2702. doi: 10.1091/mbc.10.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC. Vascular risk factor detection and control may prevent Alzheimer's disease. Ageing Res Rev. 2010;9:218–225. doi: 10.1016/j.arr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer's disease model. Microcirculation. 2003;10:463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- Waldron E, Heilig C, Schweitzer A, Nadella N, Jaeger S, Martin AM, Weggen S, Brix K, Pietrzik CU. LRP1 modulates APP trafficking along early compartments of the secretory pathway. Neurobiol Dis. 2008;31:188–197. doi: 10.1016/j.nbd.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, et al. The role of cell-derived oligomers of Aβ in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- Wavrant-DeVrieze F, Lambert JC, Stas L, et al. Association between coding variability in the LRP gene and the risk of late-onset Alzheimer's disease. Hum Genet. 1999;104:432–434. doi: 10.1007/s004390050980. [DOI] [PubMed] [Google Scholar]
- Weber A, Engelmaier A, Teschner W, Ehrlich HJ, Schwarz HP. Intravenous immunoglobulin (IVIg) gammagard liquid contains anti-RAGE IgG and sLRP. Alzheimer Dement. 2009;5:P416. [Google Scholar]
- Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008;18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo H, Chow N, et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- Yamada K, Hashimoto T, Yabuki C, et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J Biol Chem. 2008;283:34554–34562. doi: 10.1074/jbc.M801487200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yan SF, Ravichandran R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Wozniak DF, Cirrito J, et al. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci USA. 2004;101:1075–1080. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Bu G. LRP and Alzheimer's disease. Rev Neurosci. 2005;16:123–135. doi: 10.1515/revneuro.2005.16.2.123. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Cerebrovascular transport of Alzheimer's amyloid beta and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996;59:1483–1497. doi: 10.1016/0024-3205(96)00310-4. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zloković BV, Segal MB, Begley DJ, Davson H, Rakić L. Permeability of the blood-cerebrospinal fluid and blood-brain barriers to thyrotropin-releasing hormone. Brain Res. 1985;358:191–199. doi: 10.1016/0006-8993(85)90963-1. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B. Blood-brain barrier transport of circulating Alzheimer's amyloid beta. Biochem Biophys Res Commun. 1993;197:1034–1040. doi: 10.1006/bbrc.1993.2582. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, Frangione B, Ghiso J. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA. 1996;93:4229–4234. doi: 10.1073/pnas.93.9.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B. Clearance of amyloid beta-peptide from brain: transport or metabolism? Nat Med. 2000;6:718. doi: 10.1038/77397. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Srivastava A, Bell RD, Sagare A, Larson A, Van Nostrand WE, Singh I, Deane R. 2009 Neuroscience meeting planner. Society for Neuroscience; Chicago, IL: 2009. LRP1-cluster IV with an improved Alzheimer's Aβ binding specific activity exerts an enhanced Aβ sink action in mice. Program No. 236.10/F10. Online. [Google Scholar]
- Zurhove K, Nakajima C, Herz J, Bock HH, May P. γ-Secretase limits the inflammatory response through the processing of LRP1. Sci Signal. 2008;1:ra15. doi: 10.1126/scisignal.1164263. [DOI] [PMC free article] [PubMed] [Google Scholar]