

Abstract
Mutations in the Cu/Zn superoxide dismutase (SOD1), transactive response (TAR)-DNA binding protein (TARDBP) and fused in sarcoma (FUS) genes account for approximately one third of familial amyotrophic lateral sclerosis (ALS) cases. Mutations in these genes have been found in 1 to 2% of apparently sporadic cases. We present the first case of an ALS patient carrying a de novo missense mutation of the FUS gene (c.1561C>T, p.R521C). This report highlights the importance of screening ALS patients, both familial and sporadic, for FUS mutations and also suggests that de novo mutations is a relevant mechanism underlying sporadic neurodegenerative disease.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is neurodegenerative disorder of the adult life, characterized by a progressive loss of cortical, bulbar and spinal motor neurons. Approximately 5–10% of patients have a family history of disease, whereas the remaining 90–95% of cases appear to occur sporadically in the community. To date, mutations in three genes have been described as common causes of familial ALS, namely SOD1 (OMIM 147450), TARDBP (OMIM 605078), and FUS (OMIM 137070) (Rosen et al, 1993; Sreedharan et al, 2008; Kwiatkowski et al, 2009; Vance et al, 2009). Mutations of these genes account for about approximately one third of familial ALS cases. In contrast, the genetics of sporadic ALS is poorly understood. Genome-wide association studies of ALS have failed to yield a single replicating locus (Valdmanis et al, 2009), although possible susceptibility loci have been proposed (Van Es et al, 2009). Mutations in SOD1, FUS, and TARDBP have each been found in 1 to 2% of apparently sporadic cases, though it remains unclear if these cases are truly sporadic, or instead represent examples of familial ALS where the family history is not immediately obvious due to decreased disease penetrance or misdiagnosis in preceding generations (Sreedharan et al, 2008; Chiò et al, 2008; Corrado et al, 2009; Lai et al, 2010). There are two documented cases of genetically proven truly sporadic ALS patient, one resulting from a de novo SOD1 mutation (Alexander et al 2002) and one heterozygous splice-site mutation in FUS intron 13 (IVS13-2A>G) (Dejesus-Hernandez et al, 2010).
Here, we present a case of an ALS patient carrying a de novo missense mutation of the FUS gene.
2. Methods
While performing mutational screening of FUS in a large series of sporadic and familial ALS cases, we detected a young onset ALS patient carrying the p.R521C missense mutation (c.1561C>T) in exon 15 of FUS (Chiò et al, 2008; Lai et al, 2010. Since both his parents were still alive and not affected by ALS, we searched for the mutation in the parents and in the patient’s brother.
2.1 Genetic analysis
To ensure against possible laboratory sample mix-up, fresh blood samples were drawn from the patient and his parents. Genomic DNA was extracted using a Biorobot MDX DSP (Qiagen Inc.). Exons 1 to 15 of FUS were sequenced as previously described (Vance et al, 2009; Lai et al, 2010, Chiò et al, 2009). PCR products were sequenced using the Big-Dye Terminator v3.1 sequencing kit (Applied Biosystem) and run on an ABIPrism 3100Avant genetic analyzer. Exon 15 was also sequenced in 368 control Italian individuals (Chiò et al, 2009; Lai et al, 2010). Quantitative fluorescence polymerase chain reaction (QF-PCR) was performed to assess paternity and maternity of the proband, with a multiplex analysis of short tandem repeats (STRs) located on five chromosomes (Devyser Resolution kit, Devyser). The electropherograms in all 5 chromosomes confirmed the paternity and the maternity of the proband.
2.2 Standard Protocol Approvals and Patient Consents
The study was approved by the Ethical committee of our institution. The patient and his family members signed a written informed consent.
3. Case history
The patient’s family pedigree is shown in Figure 1. The patient (III-5) was a 38 year-old man who developed muscle weakness and atrophy at the shoulder girdle at the age of 34 years, spreading rapidly to involve the flexor and extensor neck muscles. Deep tendon reflexes were normal, with the exception of ankle jerk reflexes, which were hyperactive with clonus. Babinski and Hoffman signs were not present. He was cognitively normal. Neurophysiological examination demonstrated chronic and active denervation of all four limbs, and of the axial muscles of neck and trunk. Head and spinal cord MRI and cerebrospinal fluid examination were normal. Creatine kinase serum levels were raised, and muscle biopsy of right quadriceps confirmed the presence of a neurogenic muscle denervation. He was diagnosed as probable laboratory-supported ALS. In the following months, he developed progressive weakness of his lower limbs and both hands. Two years after the onset of the first symptoms, he showed exertional dyspnea, and was treated with non-invasive ventilation. Subsequently, he opted for invasive ventilation via tracheotomy at age 37. Currently, he has flaccid tetraparesis, and communicates with an eye-tracking system. He has no mutation in SMN1, SOD1, TARDBP and androgen receptor (AR) genes. He carries the c.1552A>G (p.R521C) missense mutation of the FUS gene (previously reported in Lai et al, 2010).
Figure 1.
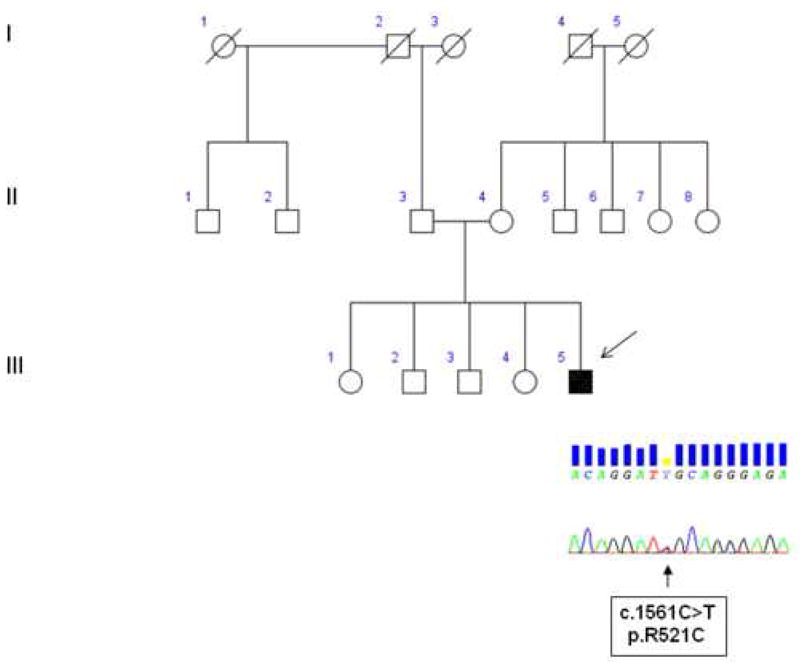
Family pedigree with chromatograms of part of exon 15 of FUS gene. Square indicates male; circle, female; slash, deceased; solid symbol, affected; and arrow, index patient.
His father (II-3) is 75 years of age and is healthy. His mother (II-4) is 70 years old and was diagnosed with definite remitting-relapsing multiple sclerosis since age 45. Neither the father nor the mother carry the FUS mutation identified in the proband, nor any other mutation of genes related to ALS. The proband’s sibs (III 1-4) are healthy and their ages range between 45 and 51.
4. Discussion
Here we report a proven case of de novoFUS mutation causing sporadic ALS. The parents and siblings did not carry the mutation, and highly informative polymorphic markers confirmed paternity and maternity. Furthermore, this particular mutation has previously been shown to segregate with disease in several ALS families, and has not been detected in a large number of neurologically normal controls (Kwiatkowski et al, 2009; Vance et al, 2009).
Our finding, together with our previous publication of a de novo mutation in H80A SOD1 (Alexander et al, 2002), and a recent publication of a heterozygous splice-site mutation in FUS intron 13 (IVS13-2A>G) (Dejesus-Hernandez et al, 2010) leading to C-terminal truncation of FUS, indicate that de novo mutations account for at least a portion of sporadic ALS cases. The rate of de novo mutations in the human genome is estimated to be ~2.5×10−8 mutations per nucleotide meaning that each individual carries ~175 mutations (Nachman et al, 2000). De novo mutations are a common cause of human disease. For example, 25% of familial adenomatous polyposis cases arise from de novo mutations in the APC gene (Bisgaard et al, 1994). The exact mechanism giving rise this type of mutations is not known, though certain spots in the genome are thought to be particularly susceptible to mutation (i.e. “mutational hotspots”), presumably because of the surrounding sequence. Indeed several lines of evidence now indicate that the c.1561C base pair represents such a hotspot for mutation: First, three different mutations of c.1561C base pair (i.e. C>A, C>G, C>T) have been described, each of which must have arisen by separate mutational events. Second, mutations of this particular codon account for more than 30% of all FUS-related ALS cases, making it by far the most common mutated codon in that gene (Kwiatkowski et al, 2009; Vance et al, 2009; Corrado et al, 2009; Lai et al, 2010; Belzil et al, 2009; Ticozzi et al, 2009; Groen et al, 2010; Drepper et al, 2009; Blair et al, 2009). Third, the c.1561C>T mutation must have arisen on at least two separate occasions (the current de novo mutation described in this paper and the occurrence of the same mutation in previously described familial ALS case).
In conclusion, we have identified a genetically proven case of a FUS mutation in a patient with true sporadic ALS, representing the first de novo mutation identified in this gene. Our data, in conjunction with other published studies, suggests that sequencing the FUS gene in both familial and sporadic ALS cases may be informative, particularly where the patient has a presentation characterized by a young age at onset, with rapid clinical progression and flail arm with dropped-head syndrome. This finding also indicates that de novo mutations is a relevant mechanism underlying sporadic neurodegenerative disease.
Acknowledgments
Adriano Chiò had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. We thank the patient and his family for having collaborated to this study.
Funding/Support: This work was funded by grants of Fondazione Vialli e Mauro for ALS Research Onlus, Federazione Italiana Giuoco Calcio (FICG), and Ministero della Salute (Ricerca Sanitaria Finalizzata 2007) (Dr Chiò); and Ministero della Salute (Ricerca Sanitaria Finalizzata 2007) (Dr Restagno). This work was supported in part by the Intramural Research Program of the NIH, and the National Institute on Aging (project Z01 AG000949-02). Funding organizations had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Footnotes
Financial Disclosure: None reported.
Data contained in the manuscript being submitted have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander MD, Traynor BJ, Miller N, Corr B, Frost E, McQuaid S, Brett FM, Green A, Hardiman O. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann Neurol. 2002;52:680–3. doi: 10.1002/ana.10369. [DOI] [PubMed] [Google Scholar]
- Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, Gauthier J, Hince P, Desjarlais A, Bouchard JP, Lacomblez L, Salachas F, Pradat PF, Camu W, Meininger V, Dupré N, Rouleau GA S2D team. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–9. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994;3:121–5. doi: 10.1002/humu.1380030206. [DOI] [PubMed] [Google Scholar]
- Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J, Blumbergs PC, Vucic S, Kiernan MC, Nicholson GA. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2009 Dec 3; doi: 10.1136/jnnp.2009.194399. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Chiò A, Traynor BJ, Lombardo F, Fimognari M, Calvo A, Ghiglione P, Mutani R, Restagno G. Prevalence of SOD1 mutations in the Italian ALS population. Neurology. 2008;70:533–7. doi: 10.1212/01.wnl.0000299187.90432.3f. [DOI] [PubMed] [Google Scholar]
- Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, Sorarù G, Carlomagno Y, Ghezzi S, Pensato V, Colombrita C, Gagliardi S, Cozzi L, Orsetti V, Mancuso M, Siciliano G, Mazzini L, Comi GP, Gellera C, Ceroni M, D’Alfonso S, Silani V. Mutations of FUS Gene in Sporadic Amyotrophic Lateral Sclerosis. J Med Genet. 2009 doi: 10.1136/jmg.2009.071027. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Dejesus-Hernandez M, Kocerha J, Finch N, Crook R, Baker M, Desaro P, Johnston A, Rutherford N, Wojtas A, Kennelly K, Wszolek ZK, Graff-Radford N, Boylan K, Rademakers R. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010 Mar 15; doi: 10.1002/humu.21241. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drepper C, Herrmann T, Wessig C, Beck M, Sendtner M. C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol Aging. 2009 Dec 15; doi: 10.1016/j.neurobiolaging.2009.11.017. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Groen EJ, van Es MA, van Vught PW, Spliet WG, van Engelen-Lee J, de Visser M, Wokke JH, Schelhaas HJ, Ophoff RA, Fumoto K, Pasterkamp RJ, Dooijes D, Cuppen E, Veldink JH, van den Berg LH. FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol. 2010;67:224–30. doi: 10.1001/archneurol.2009.329. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lai SL, Abramzon Y, Schymick JC, Stephan DA, Dunckley T, Dillman A, Cookson M, Calvo A, Battistini S, Giannini F, Caponnetto C, Mancardi GL, Spataro R, Monsurro MR, Tedeschi G, Marinou K, Sabatelli M, Conte A, Mandrioli J, Sola P, Salvi F, Bartolomei I, Lombardo F, Mora G, Restagno G, Chiò A, Traynor BJ the ITALSGEN Consortium. 2010 FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2010 Feb 4; doi: 10.1016/j.neurobiolaging.2009.12.020. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak–Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, Taroni F, Kwiatkowski TJ, Jr, McKenna-Yasek DM, Sapp PC, Brown RH, Jr, Landers JE. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73:1180–5. doi: 10.1212/WNL.0b013e3181bbff05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdmanis PN, Daoud H, Dion PA, Rouleau GA. Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep. 2009;9:198–205. doi: 10.1007/s11910-009-0030-9. [DOI] [PubMed] [Google Scholar]
- van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, Lemmens R, Schelhaas HJ, Groen EJ, Huisman MH, van der Kooi AJ, de Visser M, Dahlberg C, Estrada K, Rivadeneira F, Hofman A, Zwarts MJ, van Doormaal PT, Rujescu D, Strengman E, Giegling I, Muglia P, Tomik B, Slowik A, Uitterlinden AG, Hendrich C, Waibel S, Meyer T, Ludolph AC, Glass JD, Purcell S, Cichon S, Nöthen MM, Wichmann HE, Schreiber S, Vermeulen SH, Kiemeney LA, Wokke JH, Cronin S, McLaughlin RL, Hardiman O, Fumoto K, Pasterkamp RJ, Meininger V, Melki J, Leigh PN, Shaw CE, Landers JE, Al-Chalabi A, Brown RH, Jr, Robberecht W, Andersen PM, Ophoff RA, van den Berg LH. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009 Oct;41(10):1083–7. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]