

Abstract
Status epilepticus (SE) in adulthood dramatically alters the hippocampus and produces spatial learning and memory deficits. Some factors, like environmental enrichment and exercise, may promote functional recovery from SE. Prenatal choline supplementation (SUP) also protects against spatial memory deficits observed shortly after SE in adulthood, and we have previously reported that SUP attenuates the neuropathological response to SE in the adult hippocampus just 16 days after SE. It is unknown whether SUP can ameliorate longer-term cognitive and neuropathological consequences of SE, whether repeatedly engaging the injured hippocampus in a cognitive task might facilitate recovery from SE, and whether our prophylactic prenatal dietary treatment would enable the injured hippocampus to more effectively benefit from cognitive rehabilitation. To address these issues, adult offspring from rat dams that received either a control (CON) or SUP diet on embryonic days 12–17 first received training on a place learning water maze task (WM) and were then administered saline or kainic acid (KA) to induce SE. Rats then either remained in their home cage, or received three additional WM sessions at 3, 6.5, and 10 weeks after SE to test spatial learning and memory retention. Eleven weeks after SE, the brains were analyzed for several hippocampal markers known to be altered by SE. SUP attenuated SE-induced spatial learning deficits and completely rescued spatial memory retention by 10 weeks post-SE. Repeated WM experience prevented SE-induced declines in glutamic acid decarboxylase (GAD) and dentate gyrus neurogenesis, and attenuated increased glial fibrilary acidic protein (GFAP) levels. Remarkably, SUP alone was similarly protective to an even greater extent, and SUP rats that were water maze trained after SE showed reduced hilar migration of newborn neurons. These findings suggest that prophylactic SUP is protective against the long-term cognitive and neuropathological effects of KA-induced SE, and that rehabilitative cognitive enrichment may be partially beneficial.
Keywords: seizure, kainic acid, neurogenesis, glutamic acid decarboxylase, glial fibrillary acidic protein
Introduction
Status epilepticus (SE), a period of prolonged seizures, is a serious neurological condition that produces a myriad of degenerative and regenerative changes in the hippocampus, which are thought to contribute to the development of temporal lobe epilepsy in rodent models and in humans. Hippocampal pathophysiology following SE includes considerable neuronal loss (e.g., Cavazos et al., 1994; Haas et al., 2001; Gorter et al., 2003), γ-aminobutyric acid (GABA) system alterations (Morimoto et al., 2004; Sperk et al., 2004; Brooks-Kayal et al., 2009), reactive gliosis (e.g., Jorgensen et al., 1993; Niquet et al., 1994a; Stringer, 1996; Kang et al., 2006), aberrant mossy fiber innervation of the dentate gyrus (Sutula et al., 1988; Ben-Ari and Represa, 1990), altered growth factor levels (Shetty et al., 2003; Morimoto et al., 2004), and perturbed dentate cell proliferation and neurogenesis (Bengzon et al., 1997; Parent et al., 1997; Scharfman et al., 2000; Hattiangady et al., 2004). These plastic changes following SE are accompanied by cognitive deficits on hippocampal-dependent tasks, which are present both before and after the emergence of spontaneous recurrent motor seizures (Stafstrom et al., 1993; Liu et al., 1994; Sarkisian et al., 1997; Hort et al., 1999; Mikati et al., 2001; Kemppainen and Pitkanen, 2004; McKay and Persinger, 2004; Detour et al., 2005; Lin et al., 2009). As has been suggested (Prince et al., 2009), to develop prophylactic or rehabilitative strategies that might be applied either before or after brain injuries that lead to epilepsy, it is critical to understand the pathophysiological and cognitive processes to target.
Both environmental enrichment and physical exercise are known to enhance hippocampal plasticity and cognitive function in the absence of injury (van Praag et al., 2000; Cotman et al., 2007), and may also be beneficial for the functional recovery from SE and temporal lobe epilepsy. Enrichment and/or exercise have been shown to reduce the frequency and severity of seizures, prevent neurodegeneration, and reverse deficits in synaptic plasticity (Dhanushkodi and Shetty, 2008; Hattiangady and Shetty, 2008b; Arida et al., 2009). Post-SE housing of immature rats in an enriched environment with access to a running wheel has been shown to ameliorate spatial learning and memory deficits in the water maze (Young et al., 1999; Faverjon et al., 2002; Rutten et al., 2002), although studies of the effects of experience on cognitive recovery after SE in adult animals are very limited. Recent evidence suggests that voluntary wheel running after SE in adult mice aids recovery of spatial learning and memory in the water maze (Sartori et al., 2009).
Similar to environmental enrichment and physical exercise, supplementation of pregnant rats during embryonic days (ED) 12–17 with the nutrient choline (about 4.5 times the amount of choline in standard rat chow) also enhances several features of hippocampal plasticity of adult offspring in the absence of injury and improves cognitive function. Choline is a precursor to the neurotransmitter acetylcholine that is critical for learning, memory, and attentional processes; it is one of the building blocks of all biological membranes; and it contributes to methyl donation for the epigenetic modification of histones and DNA (Blusztajn, 1998; Zeisel, 2004; Zeisel, 2006). Compared to control-fed offspring, prenatal choline supplemented adult rats have increased basal neurogenesis (Glenn et al., 2007), increased levels of various neurotrophic/growth factors (Sandstrom et al., 2002; Glenn et al., 2007; Glenn et al., 2008; Napoli et al., 2008; Wong-Goodrich et al., 2008a; Wong-Goodrich et al., 2008b), a reduced threshold to induce long-term potentiation (LTP, Pyapali et al., 1998; Jones et al., 1999), enhanced depolarization-induced mitogen-activated protein kinase (MAPK) and cAMP-response element binding protein (CREB) activation (Mellott et al., 2004), and improved memory capacity and precision (Meck and Williams, 1997b; Meck and Williams, 1997c; Meck and Williams, 1997a; Ricceri and Berger-Sweeney, 1998; Williams et al., 1998; Meck and Williams, 1999; Tees and Mohammadi, 1999; Meck et al., 2008). Remarkably, prophylactic prenatal dietary choline supplementation has also been shown to protect adult rats against seizure-induced spatial learning and memory retention deficits observed 1–2 weeks after SE (Yang et al., 2000; Holmes et al., 2002). And, we have recently reported that prenatal choline supplementation attenuates some features of the neuropathological response to SE in adult offspring. Prenatal choline supplemented rats have drastically reduced histopathology, aberrant dentate cell proliferation, reactive gliosis, and GABAergic dysfunction in the hippocampus 16 days after SE compared to offspring of standard control-fed rats (Wong-Goodrich et al., 2008b). It is unknown, however, whether prenatal choline supplementation is a prophylactic treatment that can ameliorate more long-term cognitive and neuropathological consequences of SE and/or whether the added plasticity conferred by prenatal choline supplementation might aid an injured hippocampus to engage in a cognitive task, which might facilitate the long-term recovery from SE.
In the present study, we utilized a kainic acid model of excitotoxic injury in adult rats to investigate whether prenatal choline supplementation and/or repeated cognitive testing in the water maze can alter the long-term effects of SE on spatial learning and memory retention and on hippocampal neuropathology. This study had several goals. Place learning in the water maze offers an enriching learning experience that both engages hippocampal networks (Jenkins et al., 2004; Kee et al., 2007) and has an exercise component. No study to date has examined whether repeated cognitive testing can rehabilitate spatial learning and memory and alter hippocampal recovery after SE. Second, while both prenatal choline supplementation and enrichment/exercise increase hippocampal plasticity and aid recovery from hippocampal injury, no study has directly compared the effects of these two manipulations on brain and cognitive function to determine if there are similar mechanisms underlying these effects. In particular, we focused on the effects of SE on a number of markers in the hippocampus that contribute to the neuropathological response to seizures, which we have shown to be attenuated by prenatal choline supplementation shortly after SE (Wong-Goodrich et al., 2008b), and that may be potential therapeutic targets for the treatment of SE and epilepsy. These measures included hippocampal histopathology, glutamic acid decarboxylase (GAD) expression, astrogliosis as measured by glial fibrillary acidic protein (GFAP) expression, dentate neurogenesis, and levels of brain derived neurotrophic factor (BDNF). Third, because prenatal choline supplementation enhances hippocampal plasticity in the intact adult hippocampus (e.g., Sandstrom et al., 2002; Mellott et al., 2004; Glenn et al., 2007; Wong-Goodrich et al., 2008b), we were interested in whether prenatal choline supplementation would better enable the adult injured hippocampus to take advantage of the rehabilitative effects of cognitive enrichment after SE.
Materials and Methods
Animals
Fifty-six timed-pregnant Sprague-Dawley rats (CD strain, Charles River, Kingston, NY) were obtained on day 9 of gestation (ED9). All dams were individually housed in clear polycarbonate cages (27.9 × 27.9 × 17.8 cm) that were individually ventilated, and the colony was maintained at 21°C on a 12-h light/dark cycle with lights on at 7 a.m. Dams were fed a control diet ad libitum (AIN76-A from Dyets, American Institute of Nutrition, ICN, Nutritional Biochemical, Cleveland, Ohio; 1.1 g/kg choline chloride substituted for choline bitartrate). Prenatal diet treatments were the same as those used in studies showing memory enhancing and memory protecting effects of prenatal choline supplementation (Meck et al., 1988; Meck et al., 1989; Yang et al., 2000; Holmes et al., 2002; Meck and Williams, 2003; Wong-Goodrich et al., 2008a; Wong-Goodrich et al., 2008b). On the evening of ED11 to the morning of ED18 (ED 12–17), pregnant dams were either given ad libitum access to a control diet (n = 29) or a choline supplemented diet (n = 17). Control dams were given the AIN76-A diet and water sweetened with 50 mM saccharine. Choline supplemented dams received the AIN76-A diet and water containing 25 mM choline chloride and sweetened with 50 mM saccharine. On the morning of ED18, all dams were returned to normal drinking water. There were no significant differences in the amount of water intake, food consumed, or body weights on ED11–18 between control and choline supplemented dams (ps > .05; data not shown). After birth, offspring from the control and choline supplemented dams were toe clipped for identification and then were selected randomly and cross-fostered to dams that consumed the control diet throughout pregnancy to yield 10 pups per litter (5 males and 5 females, half from different control dams and half from supplemented dams). There were no significant differences between control and prenatally choline supplemented litter size or pup birth weights (ps > .05). On postnatal day (P) 25, pups were weaned and pair-housed with a rat of the same sex and prenatal diet condition. All offspring were given ad libitum access to the control diet through the duration of the study. Male offspring were used as subjects. All animal procedures were in compliance with the Institutional Animal Care and Use Committee of Duke University.
Timeline of Experimental Procedures
The experimental design and timeline of procedures is presented in Figure 1. Adult male control (CON) and prenatally choline supplemented (SUP) offspring were first trained in the water maze at approximately 56 days of age. Twenty-four hours after the last day of water maze training, CON and SUP rats were injected with saline or of kainic acid (KA) to induce status epilepticus (SE) and were given 5 days to recover. All rats were then injected with the cell division marker, bromodeoxyuridine (BrdU), every other day for 10 days. A subgroup of saline-and KA-treated rats were then given three additional water maze testing sessions at 3, 6.5, and 10 weeks post-SE, while the remaining rats remained pair-housed in their home cage, yielding the following experimental groups: saline-treated home cage rats (CON, n = 6; SUP, n = 5), saline-treated water maze rats (CON, n = 10; SUP, n = 9), KA-treated home cage rats (CON, n = 4; SUP, n = 6), and KA-treated water maze rats (CON, n = 7; SUP, n = 8). All rats were sacrificed at approximately 11 weeks after saline or KA treatment. The overall design was a 2 (prenatal diet: CON vs. SUP) × 2 (seizure: saline vs. KA) × 2 (experience: home cage vs. water maze) between-subjects design.
Figure 1.
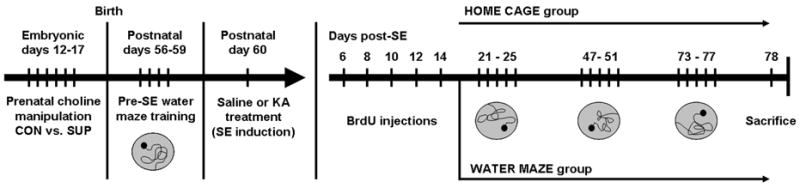
Timeline of experimental procedures. Adult rat offspring from dams that received either a choline control (CON) or supplemented (SUP) diet during embryonic days 12–17 were all trained in the water maze at postnatal day (P) 56 for 4 days. On P60, CON and SUP rats were given injections of saline or kainic acid (KA) to induce status epilepticus (SE). All rats were then given an injection of BrdU on days 6, 8, 10, 12, and 14 after SE. Saline- and KA-treated CON and SUP rats then either remained in their home cage, or were given three additional water maze testing sessions at 3, 6.5, and 10 weeks after SE, each session lasting 5 days. All rats were then sacrificed one day after the last water maze testing session at approximately 11 weeks after SE.
Induction of Status Epilepticus
Status epilepticus (SE) induction procedures were adapted from previous reports and were based on a model of chronic temporal lobe epilepsy that yields a low mortality rate and mimics several aspects of human temporal lobe epilepsy (Hellier et al., 1998; Hellier and Dudek, 1999; Hattiangady et al., 2004; Hellier and Dudek, 2005; Pitkanen et al., 2007; Sharma et al., 2007). KA was obtained from Tocris Bioscience (Ellisville, MO) and was dissolved in 0.9% sterile saline (Sigma, St. Louis, MO). A group of adult male CON and SUP offspring were injected with KA (2.5 mg/kg, i.p.) every hour and observations of seizure behavior were recorded according to Racine’s scale (Racine, 1972). A separate group of CON and SUP male offspring was similarly treated with hourly injections of 1 ml/kg saline. All KA-treated animals initially showed wet dog shakes and head-nodding, followed by Class III (unilateral forelimb clonus), Class IV (bilateral forelimb clonus with rearing), and Class V (bilateral forelimb clonus with rearing and falling over) motor seizures. KA treatment continued until rats displayed at least one Class IV/V seizure per hour for 3 consecutive hours, although most rats exhibited many more seizures during these 1-hour periods and all rats eventually entered a state of continuous Class III–V seizures for over 3 hours. If a rat displayed ≥ 10 Class IV/V seizures in an hour, administration of the next injection was delayed to the next hour. If a rat displayed bouncing seizures after reaching SE, it was given a low dose of diazepam (2.5 mg/kg) to reduce the risk of mortality. We have observed and others have demonstrated (Scharfman et al., 2000; Pierce et al., 2005) that administering a low dose of diazepam following SE does not compromise the development of spontaneous epileptic motor seizures. The total KA dosage administered was titrated for each rat according to each rat’s motor seizure activity. Because it has been found that seizures, as opposed to neuronal cell death per se, lead to increases in hippocampal cell proliferation and neurogenesis (Parent et al., 1997; Tooyama et al., 2002; Smith et al., 2005), our procedures were designed such that all KA-treated rats experienced similar duration and severity of seizure activity (see Results).
After KA treatment, rats were injected subcutaneously with 5 ml of lactated Ringer’s solution (Abbott Laboratories, Chicago, IL) to prevent dehydration and were monitored closely until all seizure activity subsided. Saline-treated rats were also given 5 ml of lactated Ringer’s solution to equate their post-treatment experience to that of KA-treated rats. For the next 5 days, all rats continued to receive daily subcutaneous injections of 1–5 ml of lactated Ringer’s solution and were provided with moistened chow and slices of fresh fruit to aid recovery. Mortality rates did not differ significantly between CON and SUP rats, χ2 (1, N = 57) = 0.12, p = 0.74. Rats remained pair-housed throughout the duration of the experiment. All KA-treated rats were checked at least 3 times per week in their home cage for fighting and for spontaneous motor seizures. During these brief observations, we did not see spontaneous motor seizures in any rat in its home cage. However, 2 CON and 2 SUP rats that were treated with KA did exhibit a spontaneous motor seizure during the last spatial learning component of water maze testing (see water maze procedures), but it did not prevent the animals from completing the water maze task. In this case, these rats were immediately removed from the maze, given a short rest period, followed by a “make-up” trial. Analysis of each data point collected indicated that these 4 animals performed similarly to other animals in their respective treatment group. It should be noted, however, that we did not systematically monitor the occurrence of spontaneous seizures during and after SE induction and that others have used electroencephalographic (EEG) monitoring to show that abnormal spiking activity and nonconvulsive EEG seizures can occur after excitotoxic injury before the onset of the first motor seizure (Bertram and Cornett, 1993; Jessberger et al., 2007a; Raedt et al., 2009). It is therefore possible that we underestimated the number of rats undergoing spontaneous seizure activity weeks following SE.
Bromodeoxyuridine Injections
Five days after being treated with KA or saline, all rats were administered a total of 5 daily injections of 5-bromo-2-deoxyuridine (BrdU; 100mg/kg/day, i.p.; Sigma, St. Louis, MO) one injection every other day for 10 days to label dividing cells. This injection regimen was based on our previous report and was designed to capture the period of time during which we have shown alterations in SE-induced dentate cell proliferation and hippocampal pathology as a result of prenatal choline supplementation (Wong-Goodrich et al., 2008b).
Water Maze Procedures
The maze consisted of a circular pool approximately 1.8 m in diameter and filled with room temperature water. A circular platform 10 cm in diameter was submerged 2 cm below the surface of the water and the water was clouded by non-toxic powdered tempera paint to ensure that the rats could not see the platform. The pool was located in a well-lit room (approximately 5.8 m × 2.6 m in dimension) with salient extramaze cues, such as a table with a computer, shelving that contained large objects, pictures of large black shapes adhered to a curtain and room walls, a large metal trash bin, and the experimenter who sat in a chair near the computer. All rats received 2 days of pretraining to adapt to the room, experimenter, and water, and to learn that there was an escape platform in the pool. Rats were first placed on a hidden platform in the middle of the maze for ~10 s, then placed in the water near the hidden platform and gently guided toward the platform. This was repeated 2–3 times per day. For all training trials, rats were placed in a random start location facing the wall of the pool and given 60 s to locate a hidden platform. If a rat did not find the platform by the end of 60 s, it was gently guided to the platform. Rats were allowed to sit on the platform for 15 s after climbing on to it. Following each trial, rats were removed from the water maze and dried with a towel. Performance on the task, including latency and pathlength to locate a hidden platform as well as swim speed, was measured and recorded using a computerized tracking system (HVS Image, Hampton, UK).
Prior to saline or KA treatment (pre-SE), all rats were given four consecutive days (6 training trials per day with an inter-trial interval of 5–8 min) of water maze training where rats were trained to navigate the maze to learn a single platform location. Each rat’s total latency to locate the hidden platform (summed across 6 trials) was recorded for each day. This measure has been used previously to assess water maze performance before and after excitotoxin-induced SE (Yang et al., 2000; Holmes et al., 2002). Following saline or KA treatment, a subgroup of CON and SUP rats were given three additional water maze testing sessions at 3, 6.5, and 10 weeks post-SE that included both a spatial learning and a spatial memory retention component. Each post-SE testing session was 5 days long and was separated by 21 days. On the first day of each session, a single spatial memory retention test was administered. Rats were required to locate the hidden platform that was placed in the same spatial location learned 3 weeks prior to the retention test. Latency and pathlength to locate the previously learned platform was recorded for each rat. In the first post-SE session (3 weeks post-SE), rats were tested for their ability to remember the platform location they learned during the pre-SE training period (prior to saline or KA treatment). During the remaining 4 days of each session, rats were administered 6 training trials per day where they were required to learn a new platform location. Total latency and pathlength (summed across the 6 training trials) was recorded for each rat across each day.
Tissue Harvesting, Reverse Transcriptase PCR, Western Blot Analysis, and ELISA
At approximately 11 weeks after saline or KA treatment (24 hours after the last day of water maze training), rats were given an overdose of a ketamine/xylazine cocktail, decapitated, and brains were rapidly removed and midsagitally sectioned. The entire hippocampus from one half-brain was immediately dissected out and divided into pieces of approximately equal length along the rostral-caudal extent of the hippocampus. Pieces for reverse transcriptase PCR assays (see Reverse transcriptase PCR section) were immediately homogenized in cold guanidine isothiocyanate solution, frozen on dry ice, and stored at −80°C for. The remaining pieces were immediately stored at −80° C until assayed for protein. The other half-brain from each rat was immediately post-fixed in 4% paraformaldehyde for 72 hours at 4° C and then cryoprotected in a 30% sucrose solution in 1M phosphate buffer (PB). These half-brains were then sectioned coronally at 60 μm on a freezing microtome through the rostral-caudal extent of the hippocampus and every fifth section was collected in 0.1% sodium azide in 1M PB to yield five series of sections. The first and second series were processed for BrdU and doublecortin (DCX) immunohistochemistry, respectively, for subsequent cell counting. Representative samples of four to five 60 μm sections that captured the rostral-caudal extent of the hippocampus were selected from the third and fourth series and were processed for NeuN immunohistochemistry and triple immunofluorescent labeling procedures, respectively (see immunohistochemistry procedures). The fifth series was mounted and stained with cresyl violet for quantification of characterization of hippocampal histopathology and quantification of total hippocampal volume.
Reverse transcriptase (RT)-PCR procedures for GAD65, GAD67, and GFAP were performed according to our initial study (Wong-Goodrich et al., 2008b). In brief, total RNA was extracted from tissues by phenol and chloroform method (Chomczynski and Sacchi, 1987; Sambrook, 1989) and precipitated. RNA was resuspended and its quantity was determined using Quant-iT™ RiboGreen® RNA assay kit (Molecular Probes) and the Victor3 multi-label plate reader (PerkinElmer Life Sciences). Hippocampal RNA was used for RT-PCR using Superscript One-Step RT-PCR with Platinum Taq (Invitrogen). PCR products were separated on a 10% TBE polyacrylamide gel and stained with ethidium bromide. PCR products were then visualized with the Kodak Image Station 440 (Rochester, NY) and product intensities were quantified using Kodak software. PCR product levels were normalized with β-actin values.
Western blot analysis procedures were also performed according to our initial report (Wong-Goodrich et al., 2008b). In brief, extracts were normalized for total protein and subjected to SDS-PAGE. After transfer of protein to an Immoblin P membrane (Millipore), the membrane was blocked with 5% nonfat dry milk in 1X Tris-buffered saline (TBS) containing 0.1% Tween 20 for 2 hours and then probed overnight with either anti-GFAP monoclonal antibody GA5 (1:1000) (Cell Signaling Technology), anti-GAD65/67 polyclonal antibody (1:1000) (Chemicon), or anti-β-actin monoclonal A5441 (1:5000; Sigma). The antibody/antigen complexes on the membranes were detected using a peroxidase-conjugated anti-mouse IgG for GFAP and β-actin (1:2000) or anti-rabbit IgG (1:5000) for GAD65/67 and visualized using the enhanced chemiluminescence method (Western Lightning, Perkin Elmer) and a Kodak Image Station 440. Digitized images of immunoblots were quantified using Kodak ID software. Protein levels were normalized with β-actin values.
BDNF ELISA assay procedures were performed according to our previous studies (Glenn et al., 2007; Wong-Goodrich et al., 2008b). The ChemiKine™ BDNF sandwich ELISA kit (Chemicon Int., Inc.) was used to assay the BDNF levels in hippocampal lysates, according to the manufacturer’s instructions. A biotinylated mouse anti-BDNF monoclonal antibody (1:1000) was used for 2 hours at room temperature. After rinsing, diluted streptavidin-HRP conjugate solution (1:1000) was added to wells and incubated for 1 hour at room temperature. The optical density of each well was measured using the Victor3 microplate reader (PerkinElmer Life Sciences). The intensity of color was measured at a wavelength of 450 nm. In order to correct for optical imperfections in the plate, readings at 540 nm were subtracted from readings at 450 nm. The standard curve was used to assess the validity of the protocol and to determine the relative concentrations of the growth factors. Values in all samples were normalized per gram of tissue assayed, and the average value for each sample was calculated separately before determining the group means.
BrdU, DCX, and NeuN Immunohistochemistry
Immunohistochemical procedures for BrdU-, DCX-, and NeuN-labeling were based on our and others’ previous reports (Kuhn et al., 1996; Hattiangady et al., 2004; Glenn et al., 2007; Wong-Goodrich et al., 2008b). Free-floating coronal sections were rinsed with tris-buffered saline (TBS: pH 7.3) followed by 30 min in 50% methanol and 30 min in 3% hydrogen peroxide in TBS at room temperature to reduce nonspecific staining, and then rinsed in TBS. For BrdU-labeling, sections were first treated for 2 hours in 50% Formamide/2x SSC (0.3 M NaCl, 0.03 M sodium citrate) at 65°C, rinsed in 2x SSC for 10 min, incubated in 2 N HCl for 30 min at 37°C, rinsed in 0.1 M boric acid (pH 8.5) for 15 min, and rinsed again in TBS. Sections were incubated in 0.1% Triton X-100 (TTX; Sigma) and 3% normal horse serum (Vector Laboratories, Burlingame, CA) in TBS for 30 min at room temperature, and then incubated with the primary antibody (monoclonal mouse anti-BrdU, 1:400, Roche; polyclonal goat anti-DCX, 1:200, Santa Cruz Biotechnology, Inc.; monoclonal mouse anti-NeuN, 1:500, Millipore) for 48 hours at 4 °C. Following this, sections were rinsed with TBS and incubated with the secondary antibody (biotinylated horse anti-mouse or biotinylated horse anti-goat, 1:200; Vector Laboratories) for 2 hours at room temperature. Sections were then rinsed in TBS, incubated in an avidin-biotinylated peroxidase complex (ABC, Vector Laboratories) for 1 hour at room temperature, rinsed again in TBS, and treated for peroxidase detection with diaminobenzidine (for BrdU and NeuN; Vector Laboratories, nickel intensified) or with vector grey substrate (for DCX; Vector Laboratories) for 4 min. Stained sections were mounted on gelatin-coated slides, dehydrated, and coverslipped. Prior to being coverslipped, BrdU-labeled sections were first counterstained with cresyl violet.
Dual Immunofluorescence for BrdU and NeuN
Double immunofluorescent labeling procedures were adapted from previous reports (Mirescu et al., 2006; Segi-Nishida et al., 2008). Four to five free-floating sections were rinsed with phosphate buffered saline (PBS) followed by 30 min in 50% methanol at room temperature to quench unperfused blood vessels and reduce nonspecific staining, and 30 min in 0.1% sodium tetrahydridoborate to reduce autofluorescence. After rinsing again in PBS, sections were treated for 10 min in 0.9% saline, followed by 30 min in 2N HCl at 37°C. Sections were rinsed in PBS, incubated in 0.3% Triton X-100 (TTX; Sigma) and 5% normal donkey serum (Jackson Immuno) in PBS for 30 min at room temperature, and then incubated with a primary antibody cocktail (polyclonal sheep anti-BrdU, 1:100, Abcam Inc.; monoclonal mouse anti-NeuN, 1:50, Millipore) for 48 hours at room temperature. Following this, sections were rinsed with PBS and incubated with the fluorescent secondary antibody Alexa Fluor 488 anti-mouse, 1:200 (Molecular Probes) for 24 hours at 4 °C. Sections were then rinsed in PBS, incubated in a biotinylated anti-sheep secondary antibody, 1:500 (Jackson Immuno) for 2 hours, rinsed in PBS, and then incubated in a streptavidin-conjugated Alexa Fluor 555, 1:500 (Molecular Probes) for 2 hours. Stained sections were then rinsed in PBS, mounted on gelatin-coated slides with Vectashield anti-fading mounting medium (Vector Labs), coverslipped, and stored in the dark at 4 °C.
Quantification of BrdU+, DCX+, and NeuN+ Cells Using Unbiased Stereology
BrdU-, DCX-, and NeuN-labeled cells in the dentate gyrus were counted using a modified optical fractionator method (West, 1993; West, 1999; Mouton, 2002). For counting of BrdU+ and DCX+ cells, we sampled every fifth section through the rostral-caudal extent of the dentate gyrus in two sampling regions: one region included the subgranular zone (SGZ), which was designated as an approximately 2-cell thick zone between the inner rim of the GCL, and the hilus, and the granule cell layer (GCL) that encompassed the suprapyramidal and infrapyramidal granule cell blades, and the other region included the hilus, which did not include the CA3c region. StereoInvestigator (Microbrightfield Inc., Williston, VT) was used to sample systematically throughout each SGZ-GCL region of all rats and count numbers of immunopositive cells, using an 80 x 80 μm counting frame and ~10–60 sites per section in 8 sections that captured the rostral-caudal extent of the dentate gyus. Optical fractionator estimates were multiplied by 2 to account for both hemispheres. For the hilus region, there was sporadic labeling of BrdU+ cells in all saline-treated rats, and sporadic labeling (or no labeling) of DCX+ cells in all treatment groups. In these cases, the hilus region was sampled exhaustively for immunopositive cells (as such, total counts were multiplied by 5 and then by 2 to account for both hemispheres). However, due to the high density of BrdU+ cells in the hilus of KA-treated rats, we systematically sampled using the optical fractionator throughout the hilus region in this case. These parameters ensured adequate sampling of BrdU+ and DCX+ immunolabeled cells throughout the SGZ-GCL and hilus. The same parameters were used for counting NeuN+ cells with the exception of using 5 representative sections that captured the rostral-caudal extent of the hippocampus and a smaller counting frame (15 x 15 μm) due to the very high density of NeuN+ neurons in the GCL. For analysis, we set an optical dissector height of 20 μm with a 2-μm guard zone to avoid over-sampling and counted stained cells in each frame using a 40x objective lens. Gundersen coefficient of error values were ≤ 0.10 for all optical fractionator estimates for each animal, with a range of 0.04 to 0.10. For each section examined, the area of the dentate gyrus was calculated by the StereoInvestigator software and was based on the boundaries of the contour tracings. Volume estimates were obtained by multiplying the section area estimates with the spacing between sampled areas. Spacing was derived by multiplying the measured, post-histology thickness of each sample by the number of sections examined, which was constant for all sections for all rats (~50% shrinkage). Estimates of total hippocampal volume and of granule cell density were generated for all KA-treated rats and a subgroup of saline-treated rats (n = 4 per treatment group). For total hippocampal volume, contours were made around the entire half hippocampus in each coronal section of a single one-in-five series (stained with cresyl violet) throughout the rostral-caudal extent of the hippocampus. Total hippocampal volume and GCL volume estimates were generated according to Cavalleri’s principle (Mouton, 2002) and were multiplied by 2 to account for both hemispheres. Granule cell density estimates were generated by dividing the optical fractionator estimate of the total number of NeuN+ neurons in the GCL by the volume estimate of the GCL.
Phenotypic Analysis of BrdU+ Cells Using Confocal Microscopy
Procedures used to phenotype and quantify BrdU+ cells co-labeled with NeuN were adapted from previous reports (Mirescu et al., 2006; Hattiangady and Shetty, 2008a; Segi-Nishida et al., 2008). For dual immunofluorescence analyses, 25–50 BrdU+ cells in the SGZ-GCL of saline- and KA-treated rats (n = 4 per group) and all BrdU+ cells that met our selection criteria (see below) in the hilus of KA-treated rat (n = 4 per group) were analyzed for co-labeling of NeuN using a Zeiss Axio Observer inverted confocal laser-scanning microscope equipped with LSM 510 software. Rat brains selected for confocal analysis (n = 4 per group) yielded treatment group means of BrdU+ cell counts that were comparable to overall treatment group mean BrdU+ cell counts. We did not analyze the hilus of saline-treated rats due to a very small number of observed hilar BrdU+ cells. Due to the increased density of BrdU immunofluorescence of varying intensities in the hilus of KA-treated rats, we selected BrdU+ cells that exhibited strong immunofluroescence (i.e., that could be detected with a pinhole ≤ 1 μm and a detector gain of ≤ 500) with a more pronounced nuclei for phenotypic analyses.
Putative BrdU+ endothelial cells that were dispersed throughout the entire hilar region and had a nuclear morphology and placement consistent with endothelial cells were excluded (Scott et al., 2000; Hellsten et al., 2004). These cells had very weak immunofluorescence, were smaller and had more elongated nuclei, and did not express NeuN. BrdU+ cells were individually examined for the coexpression of NeuN using z-sectioning at 1 μm intervals at a 40x objective. Percentages of BrdU+ cells that coexpressed NeuN were individually calculated for each rat analyzed (n = 4 per group) and then multiplied by the total number BrdU+ cells for each rat to yield an estimated number of new neurons.
Statistical Analyses
Data were analyzed using ANOVAs, post-hoc tests, and a priori comparisons to evaluate differences between specific group means where appropriate. A significance level of .05 was set for all statistical tests. Values are reported in the text as means ± standard error of the mean (SEM). Analyses of water maze performance are presented first for pre-SE spatial learning performance, followed by post-SE spatial learning performance, and then post-SE spatial memory retention performance. Our analyses of all brain measures were organized to adequately address three main points of focus: 1) the long-term effects of KA-induced SE, 2) how prenatal choline supplementation modulates these effects, and 3) how the experience of additional water maze training following SE modulates the effects of SE and/or interacts with prenatal choline availability. Thus, for brain measures, our 2 (prenatal diet) × 2 (seizure) × 2 (experience) design was decomposed into separate 2 (seizure) × 2 (experience) ANOVAs for CON vs. SUP rats such that the effects of SE and water maze experience were evaluated with respect to each within-diet’s (CON or SUP) own baseline (i.e., saline-treated home cage) condition. Results are first presented for CON rats, followed by SUP rats. Specific planned comparisons between CON and SUP rats are highlighted where appropriate. Note that subjects in each experimental condition were randomly selected from different litters (n of 1/litter). Thus, we have taken the necessary precautions to be sure that our findings are not contaminated by a lack of within litter variability.
Results
Induction of Status Epilepticus
All rats treated with KA reached SE and exhibited continuous Class III–V seizures for over 3 hours. Careful observation and analyses of behavioral activity of rats receiving KA revealed that the pattern of seizure activity (i.e., progression, number, and severity of seizures) was similar for both CON and SUP rats. There were no significant differences between CON and SUP rats in the number of Class III (CON = 0.82 ± 0.44; SUP = 0.86 ± 0.27), Class IV (CON = 5.64 ± 1.19; SUP = 4.14 ± 0.88), Class V (CON = 4.55 ± 1.61; SUP = 4.77 ± 1.02), or total number of motor seizures (CON = 11.00 ± 2.58; SUP = 9.43 ± 2.01) observed within the first 1-hour period of observable seizure activity (all ps > 0.05). After exhibiting their first motor seizure, the majority of both CON and SUP rats either entered a continuous Class III–V seizure state or exhibited ≥ 10 Class IV/V seizures in the second 1–hr period, and by the third hour all rats entered a continuous Class III–V seizure state. No differences in seizure severity or duration occurred beyond the 3-hr time window used to define SE. All rats gradually dropped out of SE within 2–3 hours of the last KA injection. During this time window, continuous seizures subsided, followed by the emergence of 1–3 discrete Class III/IV/V seizures, and all seizure activity abated by 5 hours after the last injection. There were no significant differences between CON and SUP rats in the total amount of KA needed to induce SE (CON = 13.77 ± 1.44 mg/kg; SUP = 13.13 ± 0.64 mg/kg) or in the latency to the first motor seizure (CON = 270.4 ± 29.5 min; SUP = 252.3 ± 12.1 min). The variability in dose and latency to the first motor seizure in our KA-treated rats is consistent with our previous report (Wong-Goodrich et al., 2008b) and other reports showing that the Sprague Dawley rats show a more variable convulsant response to KA than other rat strains (Golden et al., 1991; Golden et al., 1995). Thus, while the dose of KA needed to induce SE varied across rats, the SE produced was comparable between our CON and SUP rats. All KA-treated rats were then pseudo-randomly assigned to either a home cage or water maze post-SE condition to ensure there were no significant differences in any of the above seizure measures between CON and SUP rats in either home cage or water maze post-SE condition (all ps > 0.05; data not shown).
Water Maze Performance
Prior to saline or KA injections, all rats were trained in the water maze for four days. During pre-SE training, all CON and SUP rats learned the platform location, as revealed by a decreased latency to locate the hidden platform over four days of training (Fig. 2A). A two-way repeated measures ANOVA revealed a main effect of day (F(3,162) = 76.54, p < 0.001) with significant differences between all days except days 3 and 4 (ps < 0.001), but no effect of prenatal diet (F < 1). Analyses of pathlength data revealed the same pattern of findings (data not presented) and there were no significant group differences in overall swim speed (CON = 0.23 ± 0.01 m/s; SUP = 0.24 ± 0.01 m/s; p = 0.51), confirming that both diet groups did, in fact, learn the platform location with the same proficiency.
Figure 2.
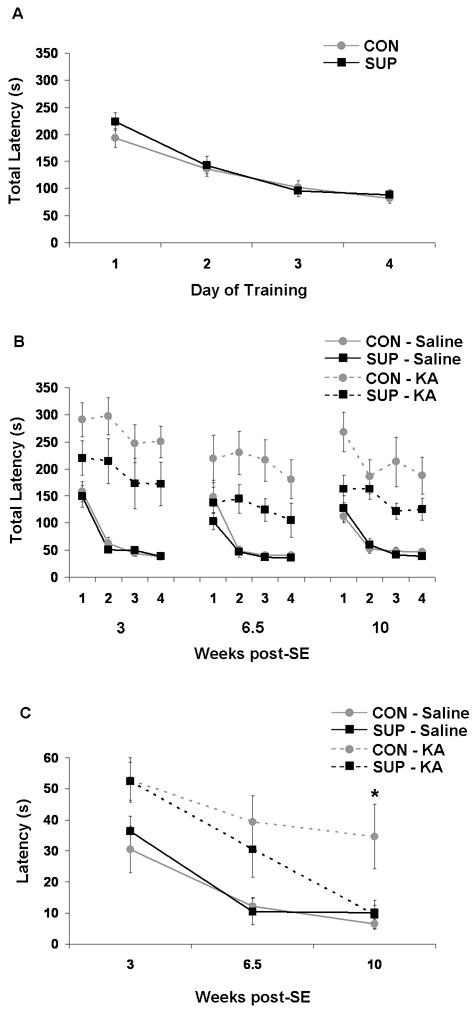
Water maze performance. (A) During pre-SE training, all CON (grey) and SUP (black) rats learned to locate the hidden platform with similar decreasing total latencies over 4 days of training. (B) Compared to saline-treated rats (solid lines), KA-treated (dashed lines) rats had significant impairments in spatial learning (higher total latencies) across 4 days during each post-SE session, which was attenuated in KA-treated SUP rats. (C) SE also impaired spatial memory over a 3-week retention interval, but KA-treated SUP rats showed a complete recovery of spatial memory retention by 10 weeks post-SE. * significantly different from all other groups at p < 0.05.
To assess the loss of spatial learning ability following SE and recovery over the next 10 weeks, we trained rats to learn a novel platform location in the water maze over three separate 4-day sessions at 3, 6.5, and 10 weeks after SE. KA-treated rats had significantly higher escape latencies than saline-treated rats at all post-SE sessions (ps < 0.01), confirming that KA-induced SE elicited significant deficits in spatial learning (Fig. 2B). To examine the extent of recovery in KA-treated CON vs, SUP rats, separate 2 (prenatal diet) × 4 (day of training) ANOVAs were performed for saline- and KA-treated rats for each session and these data are also presented in Figure 2B. Consistent with our predictions, prenatal choline supplementation did not affect spatial learning in saline-treated rats (Fig. 2B). Spatial learning on this reference memory water maze task is not difficult for an intact rat, likely because it does not impose many cognitive demands, which is when prenatal choline supplementation memory enhancing effects are observed (Meck and Williams, 1999; Meck and Williams, 2003; Wong-Goodrich et al., 2008a). Analyses of KA-treated rats revealed a significant main effect of day for all three sessions (all ps < 0.05), indicating that both KA-treated CON and KA-treated SUP rats learned the new platform location across four days of training and suggesting some cognitive recovery of spatial learning. However, analyses also revealed a significant main effect of prenatal diet at 10 weeks post-SE where KA-treated SUP rats had lower latencies than KA-treated CON rats, F(1,13) = 4.61, p = 0.05 (Fig. 2B). There was a similar trend at 3 weeks post-SE, F(1,14) = 2.74, p = 0.12, and at 6.5 weeks post-SE, F(1,14) = 3.64, p = 0.07. These data revealed that SUP rats showed an attenuated spatial learning deficit following KA-induced SE; KA-treated SUP rats appeared to locate the platform with more efficiency than the KA-treated CON rats.
To examine recovery of long-term memory retention, rats were tested at 3, 6.5 and 10 weeks post SE for their memory of a platform location learned 3 weeks prior to the test. Latency to locate the platform was recorded across each session and analyzed using separate repeated measures ANOVA for each treatment group to assess performance over time post-KA or -saline treatment. There was a significant effect of session for both saline-treated CON rats, F(2,18) = 8.41, p < 0.01, and saline-treated SUP rats, F(2,16) = 15.73, p < 0.001. Both groups demonstrated a significant decrease in latency from the 3-week to 6.5-week and 10 week post-SE sessions, indicating an improvement in spatial memory retention by the second session in saline-treated rats (ps < 0.05; Fig. 2C). Analyses did not reveal an effect of session for KA-treated CON rats, indicating a lack of improvement of memory retention across post-SE sessions. Planned comparisons also revealed that KA-treated CON rats had significantly higher escape latencies than saline-treated CON and SUP rats at all three post-SE sessions (ps < 0.05), confirming a persistent spatial memory deficit in KA-treated CON rats. Similar to saline-treated rats, there was also a significant effect of session for KA-treated SUP rats, F(2,14) = 9.68, p < 0.01, where KA-treated SUP rats exhibited a progressive decrease in escape latency with each post-SE water maze session (Fig. 2C). While KA-treated SUP rats had significantly higher latencies than saline-treated CON and SUP rats at 3 and 6.5 weeks post-SE (p < 0.05), KA-treated SUP rats’ memory retention was remarkably comparable to that of saline-treated CON and SUP rats by 10 weeks post-SE (Fig. 2C), suggesting complete recovery of spatial memory retention.
Analyses of pathlength data revealed a similar pattern of findings for spatial learning and memory retention testing (data not presented). In addition, group differences in latency to find the platform during spatial learning and memory retention tests were not due to differences in mean swim speeds (saline-treated CON = 0.26 ± 0.01 m/s; saline-treated SUP = 0.25 ± 0.02 m/s; KA-treated CON = 0.26 m/s; KA-treated SUP = 0.26 ± 0.02 m/s; ps = n.s.).
Hippocampal Lesion
We observed varying degrees of histopathology throughout the rostral-caudal extent of the hippocampi in all KA-treated rats. KA-induced SE led to moderate to considerable cell loss and disruption of cellular architecture in CA1, CA3, and the hilus of both CON and SUP rats, which can be seen in Figure 3. In contrast, there was mild to no cell loss in the GCL of the dentate gyrus, which is consistent with previous reports using the KA model of SE (Covolan et al., 2000). We used total hippocampal volume estimates to quantify hippocampal cell loss. Separate analyses of total hippocampal volume estimates for CON rats (saline-treated home cage = 69.59 mm3 ± 5.03; KA-treated home cage = 54.38 mm3 ± 16.73; saline-treated water maze = 79.15 mm3 ± 12.91; KA-treated water maze = 48.29 mm3 ± 41.54) and SUP rats (saline-treated home cage = 66.79 mm3 ± 5.03; KA-treated home cage = 52.02 mm3 ± 13.52; saline-treated water maze = 74.21 mm3 ± 11.19; KA-treated water maze = 55.91 mm3 ± 7.13) revealed a significant main effect of seizure for CON rats, F(1,15) = 6.68, p < 0.05, and a strong trend for a main effect of seizure for SUP rats, F(1,18) = 3.97, p = 0.06. KA-treated CON rats had a 31% overall decrease in hippocampal volume compared to saline-treated CON rats, while KA-treated SUP rats had a 23% overall decrease. While SE reduced total hippocampal volume in all rats, there were no significant differences in hippocampal volume between any group of KA-treated rats (all ps > 0.05), suggesting that our KA treatment produced similar levels of hippocampal cell loss across all treatment groups.
Figure 3.

Histopathology of the hippocampus 11 weeks following KA-induced SE from representative sections from the dorsal hippocampus of CON (B) and SUP (C) rats. Panel A depicts an intact hippocampus from a saline-treated CON rat. Saline-treated SUP rats also did not show any lesions (histology data not shown). Note significant cell loss in CA1, CA3, and hilus regions (indicated by arrows) while the dentate gyrus granule cell layer remained relatively intact. Patterns of lesion severity were similar across all KA-treated rats. Photomicrographs in each set were taken with a 4x objective. Bars indicate 250 μm. DG, dentate gyrus. H, hilus.
Hippocampal GAD65 and 67 mRNA and Protein
Disruption of hippocampal GABAergic function is a robust consequence of seizures (Obenaus et al., 1993; Morimoto et al., 2004; Brooks-Kayal et al., 2009). In our previous study, we reported that prenatal choline supplementation prevented the decrease in hippocampal GAD65 mRNA and protein expression observed 16 days after SE (Wong-Goodrich et al., 2008b). To investigate whether this protection against the loss of GABAergic function in the hippocampus persists for 11 weeks after SE, we measured mRNA and protein levels of GAD65 and 67, enzymes important for local GABA synthesis at synaptic (GAD65) and cytoplasmic (GAD67) sites (Erlander and Tobin, 1991; Esclapez et al., 1994) and for the packaging and release of GABA (Namchuk et al., 1997; Tian et al., 1999).
GAD65 and 67 mRNA and protein levels were expressed as percent of control values and were subjected to separate 2-way ANOVAs for CON and SUP rats with seizure (saline vs. KA) and experience (home cage vs. water maze) as between-subjects factors. We generally observed higher within-group variability for KA-treated groups, which we have also previously observed at 16 days after SE (Wong-Goodrich et al., 2008b). Analyses revealed that CON rats were significantly affected by KA treatment and/or post-SE experience on all GAD measures quantified. Interestingly, two distinct patterns emerged for mRNA versus protein for both GAD65 and 67 in CON rats (see Fig. 4). For GAD 65 mRNA, there was a reduction (~30%) in GAD65 mRNA in home cage CON rats following SE, with a trend toward recovery from the SE-induced loss of GAD65 mRNA in water maze trained rats (p = 0.10). As such, analyses revealed a significant main effect of experience, F(1,23) = 7.95, p = 0.01, where water maze trained rats showed elevated levels of GAD65 mRNA compared to home cage rats. There was no significant main effect of seizure or seizure × experience interaction (Fig. 4A). For GAD67 mRNA, there were no main effects of seizure or experience. However, a significant seizure × experience interaction, F(1,23) = 5.57, p < 0.05, confirmed that similar to the pattern of GAD65 mRNA in CON rats, KA-induced SE led to a significant 43% decrease in GAD67 mRNA levels in home cage CON rats (p < 0.05), but did not affect GAD67 mRNA levels in water maze CON rats (Fig. 4B). Together these analyses revealed that exposure to water maze training following KA-induced SE appears to rescue the decline in GAD mRNA in CON rats. Analyses of both GAD65 and 67 protein levels in CON rats revealed a significant main effect of seizure (GAD65: F(1,21) = 5.39, p < 0.05; GAD67: F(1,21) = 8.36, p < 0.01), but no main effect of experience or any seizure × experience interaction. In contrast to mRNA levels, KA-induced SE appeared to decrease GAD65 and 67 protein levels in CON rats regardless of post-SE experience (Fig. 4C, 4D).
Figure 4.

Mean (± SEM) percent of control levels for hippocampal GAD65 and 67 mRNA (A, B) and protein (C, D) for CON and SUP rats that were treated with saline (solid bars) or KA (hatched bars) and that remained in their home cage (grey) or received additional water maze training (black) following treatment. CON rats showed significant SE-induced reductions in GAD67 mRNA, GAD65 protein, and GAD67 protein. There was a trend for an SE-induced decrease in GAD65 mRNA (p = 0.10). Repeated water maze training rescued levels of GAD mRNA in KA-treated CON rats. There was little effect of SE on GAD levels in SUP rats. * significantly different at p < 0.05; main effect of seizure (**) or experience (#) revealed by a within diet 2-way ANOVA (seizure × experience), p < 0.05.
In contrast to CON rats, there was little effect of SE on hippocampal GAD levels in SUP rats. GAD65 and 67 mRNA levels and GAD65 protein levels in SUP rats were not affected by KA-induced SE in either home cage or water maze rats (Fig. 4). Compared saline-treated home cage SUP rats, there was a 20% reduction in GAD67 protein levels in KA-treated home cage SUP rats (p < 0.05), but this reduction was not evident in KA-treated water maze SUP rats (Fig. 4D). However, KA-treated home cage SUP rats and KA-treated water maze SUP rats were not significantly different.
Hippocampal GFAP mRNA and Protein
Reactive astrogliosis (proliferative and hypertrophic astrocytes) in the hippocampus, as measured by increased levels of GFAP, is also a robust consequence of SE that has been shown to persist for weeks to months following excitotoxic injury (Jorgensen et al., 1993; Niquet et al., 1994a; Aronica et al., 2000; Shapiro et al., 2008). We have previously shown that prenatal choline supplementation attenuates this seizure-induced increase in hippocampal mRNA and protein levels at 16 days following KA-induced SE (Wong-Goodrich et al., 2008b). To examine whether this attenuation persists beyond 16 days after seizures, we quantified mRNA and protein levels of hippocampal GFAP at 11 weeks post-SE. GFAP mRNA and protein levels were expressed as percent of control levels and were subjected to separate 2-way ANOVAs for CON and SUP rats with seizure and experience as between-subjects factors. For CON rats, analyses revealed a main effect of seizure for GFAP mRNA, F(1,23) = 21.52, p < 0.001, and protein, F(1,21) = 183.88, p < 0.001. There was also a main effect of experience, F(1,21) = 19.69, p < 0.001, and a seizure × experience interaction, F(1,21) = 20.78, p < 0.001, for GFAP protein in CON rats. While KA-induced SE led to a similar increase in hippocampal GFAP mRNA in KA-treated CON home cage rats and water maze rats, the SE-induced increase in hippocampal GFAP protein levels was significantly reduced in KA-treated CON rats that received additional water maze training after SE (p < 0.01; Fig. 5). These data revealed that elevated levels of hippocampal GFAP mRNA and protein persist for at least 11 weeks after KA-induced SE, but that elevated levels of GFAP protein are attenuated with water maze training.
Figure 5.
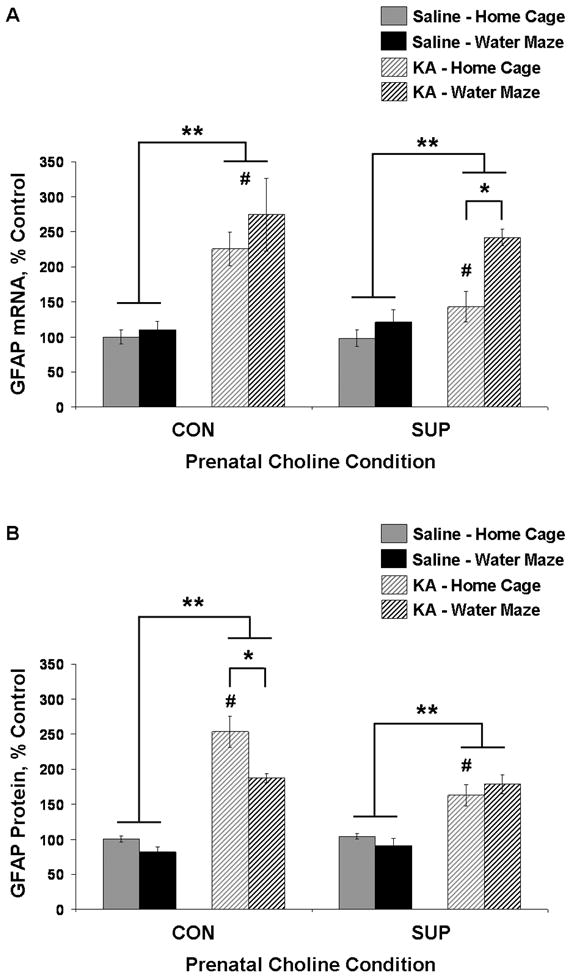
Mean (± SEM) percent of control levels for hippocampal GFAP mRNA (A) and protein (B) for CON and SUP rats that were treated with saline (solid bars) or KA (hatched bars) and that remained in their home cage (grey) or received additional water maze training (black) following treatment. Both CON and SUP rats showed a significant overall SE-induced increase in GFAP mRNA and protein expression, but this increase was attenuated in KA-treated home cage SUP rats. Repeated water maze training attenuated elevated GFAP protein levels in KA-treated CON rats, and further increased GFAP mRNA levels in KA-treated SUP rats. * significantly different at p < 0.05; ** main effect of seizure revealed by a within diet 2-way ANOVA (seizure × experience), p < 0.05; # KA-treated home cage SUP rats are significantly different from KA-treated home cage CON rats (A, B) and KA-treated water maze CON rats (A).
For SUP rats, analyses revealed a main effect of seizure for both GFAP mRNA, F(1,24) = 25.62, p < 0.001, and protein levels, F(1,24) = 37.87, p < 0.001. There was also a significant effect of experience, F(1,24) = 13.98, p < 0.01, and seizure × experience interaction, F(1,24) = 5.36, p < 0.05, for GFAP mRNA. Unexpectedly, the increase in hippocampal GFAP mRNA levels was significantly higher for KA-treated SUP rats that received additional water maze training (p < 0.01; Fig. 5A). Hippocampal GFAP protein levels, however, were increased similarly for KA-treated home cage and water maze SUP rats (Fig. 5B). Planned comparisons revealed that the seizure-induced increase in hippocampal GFAP mRNA and protein levels for KA-treated SUP rats was attenuated in comparison to KA-treated CON rats: home cage CON rats showed a 125% and 153% increase in GFAP mRNA and protein, respectively, whereas home cage SUP rats showed only a 46% and 56% increase (ps < 0.05; Fig. 5). These findings reveal that prenatal choline supplementation leads to a remarkable long-term attenuation in SE-induced increases in GFAP mRNA and protein levels that persists for 11 weeks following KA-induced SE.
Long-term Survival of Dentate Cells Born Shortly After Seizures
To examine the long-term (9–10.5 weeks) survival and migration of dentate cells born 5 to 15 days after KA-induced SE, we visualized and quantified the number of BrdU+ cells in the SGZ-GCL and hilus of all rats. Figure 6 shows photomicrographs of BrdU labeling in the hippocampus of representative sections from CON and SUP saline- and KA-treated rats (home cage and water maze). BrdU+ cells were expressed throughout the rostral-caudal extent of the dentate gyrus and in both the suprapyramidal and infrapyramidal blades. As can be seen in Figure 6, most BrdU+ cells that were found in the SGZ-GCL had a large and rounded BrdU-immunostained nuclei characteristic of mature granule cells. However, a considerable number of BrdU+ cells were much smaller in size with many cells exhibiting a more elongated oval-shaped nuclei than those found in the granule cell layer, suggesting that many of these hilar BrdU+ cells were putative glial or endothelial cells (Scott et al., 2000; Hellsten et al., 2004). Moreover, a vast majority of BrdU+ cells displayed morphological characteristics of normal non-pyknotic cells (e.g., round or oval nuclei that did not appear highly condensed). Only non-pyknotic cells were counted when quantifying numbers of BrdU+ cells. In KA-treated rats, more BrdU+ cells were present in the hilus in comparison to saline-treated rats (Fig. 6A–F).
Figure 6.

Long-term (9 to 10.5 weeks) survival of BrdU-immunopositive cells (i.e., newly generated cells) born 6 to 14 days after saline or KA treatment. (A–F) Photomicrographs of BrdU+ cells in the SGZ-GCL and hilus in saline treated rats (A, CON; B, SUP) and KA-treated rats who either remained in their home cage (C, CON; D, SUP) or received additional water maze training (E, CON; F, SUP) after SE. Most BrdU+ cells that were found in the GCL had large and rounded BrdU-immunostained nuclei characteristic of mature granule cells (large arrows), but many BrdU+ cells in the SGZ-GCL and hilus had morphological features characteristic of glial or endothelial cells (small arrows). KA-treated SUP rats exhibited more BrdU labeling in the SGZ-GCL than KA-treated CON rats. KA-induced SE increased the number of BrdU+ cells in the hilus of all KA-treated rats. (G, H) Bar graphs show mean (± SEM) numbers of BrdU+ cells detected in the SGZ-GCL (G) and hilus (H) of CON and SUP rats that were treated with saline (solid bars) or KA (hatched bars) and that remained in their home cage (grey) or received additional water maze training (black) following treatment. For the SGZ-GCL (G), CON rats showed a significant decrease in the number of BrdU+ cells, which was rescued by repeated water maze training. SUP rats showed an increase in the number of BrdU+ cells in the SGZ-GCL weeks after SE, regardless of post-SE experience. For the hilus (H), both CON and SUP rats showed a significant overall increase in the number of BrdU+ cells. Note that we did not directly compare numbers of BrdU+ cells between KA-treated CON and SUP rats because we have previously found that the amount of SGZ-GCL and hilar cell proliferation observed shortly after seizures is altered by prenatal choline availability (see text). Bars in photomicrographs indicate 50 μm. Photomicrographs in first and third columns were taken with a 10x objective and photomicrographs in the second and fourth columns were taken with a 40x objective. GCL, granule cell layer. SGZ, subgranular zone. H, hilus. * significantly different at p < 0.05; ** main effect of seizure revealed by a within diet 2-way ANOVA (seizure × experience), p < 0.05.
SE significantly reduced the overall size of hippocampus in both KA-treated CON and SUP rats (as previously stated), but did not appear to significantly alter the volume of the dentate GCL per se in either CON rats (saline-treated home cage = 0.67 ± 0.05 mm3; KA-treated home cage = 0.72 ± 0.07 mm3; saline-treated water maze = 0.70 ± 0.02 mm3; KA-treated water maze = 0.78 ± 0.06 mm3) or SUP rats (saline-treated home cage = 0.74 ± 0.04 mm3; KA-treated home cage = 0.86 ± 0.07 mm3; saline-treated water maze = 0.74 ± 0.01 mm3; KA-treated water maze = 0.77 ± 0.05 mm3). To investigate whether SE altered the overall density of granule cells within the dentate gyrus, we generated a granule cell density measure by estimating the total number of NeuN+ cells in the GCL using the optical fractionator and dividing this estimate by the total GCL volume estimate (number of NeuN+ cells/unit of volume). Importantly, there were no significant differences in granule cell density across treatment groups for CON rats (saline-treated home cage = 231,994 ± 7,928 cells/mm3; KA-treated home cage = 248,287 ± 27,946 cells/mm3; saline-treated water maze = 253,180 ± 9,648 cells/mm3; KA-treated water maze = 239,893 ± 12,450 cells/mm3) or SUP rats (saline-treated home cage = 234,181 ± 18,027 cells/mm3; KA-treated home cage = 224,488 ± 8,015 cells/mm3; saline-treated water maze = 259,980 ± 6.223 cells/mm3; KA-treated water maze = 245,031 ± 6,290 cells/mm3). Thus, we can conclude that differences in BrdU+ counts (and DCX+ counts; see next section) across treatment groups within each diet condition are due to differences in the proportion of all granule cells that are BrdU+ and DCX+. Taken together, these data are also consistent with our observation that the dentate GCL remained relatively intact compared to other cellular subfields in the hippocampus of KA-treated rats (Fig. 3), and in line with previous reports showing that dentate granule cells appear to be more resistant than other hippocampal neurons to KA-induced SE (Covolan et al., 2000; Hattiangady et al., 2004).
Interestingly, KA-treated home cage SUP rats had significantly more BrdU+ cells in the SGZ-GCL than KA-treated CON rats in either post-SE experience condition (ps < 0 .05; Fig. 6G). However, because CON and SUP rats show different rates of SGZ-GCL and hilar cell proliferation shortly after seizures (Wong-Goodrich et al., 2008b), we can not draw conclusions about the effects of prenatal choline supplementation on the long-term survival of granule cells born shortly after seizures by directly comparing numbers of BrdU+ cells (labeled 6 to 14 days after SE) between KA-treated CON and KA-treated SUP rats. We, therefore, analyzed stereological estimates of the number of surviving BrdU+ cells in the SGZ-GCL and hilus separately for CON and SUP rats, using separate 2 (seizure) × 2 (experience) ANOVAs. For CON rats, analyses of the number of BrdU+ cells in the SGZ-GCL revealed a significant main effect of experience, F (1,23) = 7.84, p = 0.01, and a significant treatment × experience interaction, F(1,23) = 4.57, p < 0.05). Post-hoc analyses revealed that KA-treated CON rats that remained in their home cage after seizures had significantly fewer BrdU+ cells (about a 34% reduction) in the SGZ-GCL than all other CON groups (ps < 0.05; Fig. 5G). There was no difference between KA-treated CON rats who received post-SE water maze experience and both saline-treated CON rat groups, indicating that post-SE water maze experience rescued the decrease in SGZ-GCL cell survival (Fig. 6G). In contrast, KA-treated SUP rats had significantly more BrdU+ cells in the SGZ-GCL than saline-treated SUP rats regardless of post-SE experience, which was confirmed by a main effect of seizure, F(1,24) = 8.40, p < 0.01; (Fig. 6G). There were no effects of experience or prenatal diet on the number of BrdU+ cells in the SGZ-GCL of saline-treated rats. Confocal analysis of BrdU+ cells revealed that the majority of BrdU+ cells (~72–90%) found in the SGZ-GCL of all treatment groups were also immunopositive for the mature neuronal marker, NeuN (Fig. 7; Table 1). For CON rats, a 2-way ANOVA revealed a main effect of experience for the percentage of BrdU+/NeuN+ neurons in the dentate gyrus, F(1,12) = 5.38, p < 0.05, where water maze experience decreased the proportion of surviving BrdU+ cells that differentiated into neurons in both saline- and KA-treated CON rats. Others have also reported decreases in granule cell survival by water maze training, particularly for cells that are in more advanced stages of neuronal development (Ambrogini et al., 2004; Ehninger and Kempermann, 2006; Mohapel et al., 2006). There was no effect of seizure or seizure × experience interaction for CON rats, and no main effects or interaction for SUP rats. Estimated numbers of new neurons in the SGZ-GCL revealed a similar pattern as that of total BrdU+ cells in the SGZ-GCL (Table 1).
Figure 7.
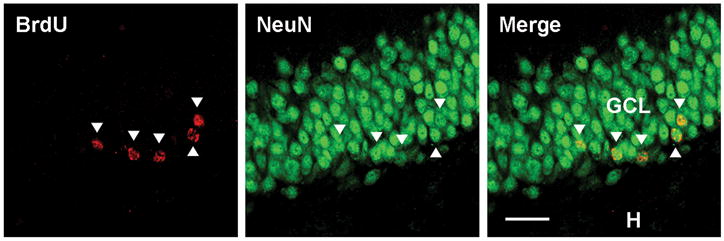
Confocal images of BrdU+ cells co-labeled with the mature neuronal marker NeuN (arrow heads). Bar indicates 25 μm. Images were taken with a 40x objective. GCL, granule cell layer. H, hilus.
Table 1.
Mean (± SEM) Percentage of BrdU+ Cells that Co-Expressed NeuN and Estimated Number of New Neurons as a Function of Prenatal Diet, KA-Induced SE, and Repeated Water Maze Experience
| % NeuN+/BrdU+ in SGZ-GCL | # New Neurons in SGZ-GCL | % NeuN+/BrdU+ in Hilus | # New Neurons in Hilus | |
|---|---|---|---|---|
| CON | ||||
| Saline - Home Cage | 85.58 (2.28) | 5305.52 (516.24) | ** | ** |
| Saline - Water Maze | 72.05 (2.86) | 5143.62 (554.68) | ** | ** |
| KA - Home Cage | 85.09 (5.40) | 3715.62 (883.20) | 53.73 (18.05) | 1670.18 (357.33) |
| KA - Water Maze | 78.06 (3.04) | 5547.80 (680.28) | 46.05 (7.88) | 2481.88 (880.87) |
| SUP | ||||
| Saline - Home Cage | 85.84 (6.57) | 6048.74 (352.82) | ** | ** |
| Saline - Water Maze | 80.72 (5.75) | 6135.98 (339.33) | ** | ** |
| KA - Home Cage | 90.47 (2.05) | 11,311.40 (957.84) | 69.27 (4.91) | 3571.22 (528.88) |
| KA - Water Maze | 86.58 (2.52) | 11, 475.02 (2482.57) | 51.55 (14.37) | 1597.88 (739.64) |
BrdU+ cells labeled 6 to 14 days following saline or KA treatment were allowed to survive for 9–10.5 weeks. A fraction of BrdU+ cells were analyzed for co-expression of NeuN (n = 4 brains per group). Water maze experience decreased the percentage of BrdU+/NeuN+ neurons in the dentate gyrus for saline- and KA-treated CON rats (main effect of seizure, F(1, 12) = 5.38, p < 0.05). All treatment groups of SUP rats showed similar percentages of BrdU+/NeuN+ neurons in the dentate gyrus. Estimated number of new neurons in the SGZ-GCL revealed a similar pattern of findings as total BrdU+ cells.
Saline-treated rats had very few BrdU+ cells in the hilus and were thus not included in the analysis. There were no significant differences between KA-treated CON and KA-treated SUP rats in percentages of BrdU+/NeuN+ neurons in the hilus, and no significant effect of water maze experience on estimated number of new neurons in the hilus for either KA-treated CON or SUP rats.
The pattern of BrdU labeling in the hilus showed that both CON and SUP KA-treated rats had more BrdU+ cells in the hilus than that of CON and SUP saline-treated rats (Fig. 6H). Separate analyses of the total number of BrdU+ cells in the hilus for CON and SUP rats revealed a main effect of seizure for both CON rats, F(1,23) = 22.22, p < 0.001, and SUP rats, F(1,24) = 20.19, p < 0.001, but no significant effect of experience or seizure × experience interaction for either prenatal diet group. KA-induced SE led to a significant increase in the number of BrdU+ cells that were observed in the hilus for all treatment groups, with no significant differences between any KA-treated groups (Fig. 6H). In contrast to the pattern of BrdU labeling in the SGZ-GCL, these data reveal that neither prenatal choline supplementation nor water maze training affected the long-term survival of BrdU+ cells born 5–16 days after SE that were observed in the hilus. We also assessed relative levels of hilar migration of newborn neurons in KA-treated rats and these data are also presented in Table 1. Saline-treated rats had very few BrdU+ cells in the hilus and were not included in the analysis. A 2 (prenatal diet) × 2 (experience) ANOVA did not reveal any effect of prenatal diet or experience on the percentage of BrdU+/NeuN+ neurons in the hilus. More than half of the hilar BrdU+ cells we analyzed in KA-treated CON and KA-treated SUP rats coexpressed NeuN (Table 1). Water maze experience did not significantly alter the estimated number of new neurons in the hilus of KA-treated CON rats, and only a trend toward a reduction in the estimated number of new neurons in the hilus was observed for water maze KA-treated SUP rats (p = 0.07; Table 1). We did, however, observe much greater within-group variability in percentage of Brdu+/NeuN+ neurons and estimated new neurons in the hilus vs. the SGZ-GCL.
Neurogenesis 11 Weeks After KA-induced SE
To examine the neurogenic capacity of the injured hippocampus at 11 weeks following SE, we immunostained adjacent tissue sections to visualize cells immunopositive for the microtubule-associated phosphoprotein, doublecortin (DCX), that is transiently expressed in newly-born neurons that are still in the process of migrating and differentiating (Brown et al., 2003; Rao and Shetty, 2004). Others have confirmed that DCX expression is a reliable indicator of ongoing neurogenesis in the adult brain (Couillard-Despres et al., 2005). DCX+ cells with processes in various stages of development were evident along the SGZ and GCL in all rats (Fig. 8). In all KA-treated rats, DCX+ cells were also present in the hilus, which is consistent with a previous study demonstrating persistent hilar migration of DCX+ neurons months following KA-induced SE (Hattiangady et al., 2004). In comparison to saline-treated rats, many of the DCX+ neurons in KA-treated rats also appeared displaced and exhibited abnormal morphological features, such as horizontally oriented cell bodies and processes (Fig. 8C–F). SUP rats tended to display overall more DCX-labeling than CON rats.
Figure 8.

Hippocampal neurogenesis at 11 weeks after saline or KA treatment. (A–F) Photomicrographs of DCX-immunopositive neurons (i.e., newly generated neurons) in the SGZ-GCL and hilus in saline treated rats (A, CON; B, SUP) and KA-treated rats who either remained in their home cage (C, CON; D, SUP) or received additional water maze training (E, CON; F, SUP) after SE. In KA-treated rats of both prenatal diet groups (C–F), DCX+ neurons were aberrantly located in the hilus and exhibited abnormal morphological features, such as horizontally oriented cell bodies and processes (arrows). SUP rats exhibited more overall DCX labeling than CON rats. (G, H) Bar graphs show mean (± SEM) numbers of DCX+ cells detected in the SGZ-GCL (G) and mean percentage of DCX+ found in the hilus (H) of CON and SUP rats that were treated with saline (solid bars) or KA (hatched bars) and that remained in their home cage (grey) or received additional water maze training (black) following treatment. (G) CON rats showed a significant decrease in the number of DCX+ cells in the SGZ-GCL, which was rescued by repeated water maze training. KA-treated SUP rats showed preserved levels of DCX+ cells in the SGZ-GCL, regardless of post-SE experience. Saline-treated SUP rats also had a higher number of DCX+ neurons overall than saline-treated CON rats. (H) Both KA-treated CON and SUP rats generated a proportion of DCX+ cells that migrated to the hilus. KA-treated water maze SUP rats had a significantly lower percentage of DCX+ neurons in the hilus than KA-treated home cage and water maze rats (#, p < 0.05). A–F, Bars in photomicrographs indicate 50 μm. Photomicrographs in first and third columns were taken with a 10x objective and photomicrographs in the second and fourth columns were taken with a 40x objective. GCL, granule cell layer. SGZ, subgranular zone. H, hilus. * significantly different at p < 0.05; ** main effect of seizure revealed by a within diet 2-way ANOVA (seizure × experience), p < 0.05; ## main effect of prenatal diet revealed by a within diet 2-way ANOVA (diet × experience), p < 0.05.
Stereological estimates of the number of DCX+ neurons in the SGZ-GCL were generated for each rat and are shown in Figure 8. Separate 2-way ANOVAs were performed for CON and SUP rats. For CON rats, analyses revealed a significant main effect of seizure, F(1,23) = 4.12, p < 0.04, experience, F(1,23) = 4.16, p < 0.05, and a significant seizure × experience interaction, F(1,23) = 5.00, p < 0.05. KA-treated CON rats that remained in their home cage after seizures had significantly fewer DCX+ neurons in the SGZ-GCL than saline-treated CON rats (p = 0.01; Fig. 8G), which is consistent with a previous report showing decreased DCX+ neurons at 5 months following KA-induced SE (Hattiangady et al., 2004). As can be seen in Figure 8G, 11 weeks after KA-induced seizures, SUP rats showed no difference in the number of DCX+ neurons in the SGZ-GCL compared to saline-treated rats (no significant main effects or seizure × experience interaction for SUP rats, Fs < 1). Consistent with our previous report (Glenn et al., 2007), saline-treated SUP rats also exhibited overall higher numbers of DCX+ neurons than saline-treated CON rats, F(1,26) = 4.80, p < 0.05.
To assess relative levels of hilar migration of DCX+ neurons in KA-treated rats, the number of DCX+ neurons in the hilus was expressed as a percentage of the total number of DCX+ cells (in both the dentate gyrus and hilus), and this measure was subjected to a 2-way ANOVA with prenatal diet and experience as between-subjects factors. Analyses did not reveal a significant effect of experience or prenatal diet × experience interaction, though there was a trend for KA-treated SUP rats to show a lower percentage of DCX+ neurons in the hilus than KA-treated CON rats, F(1,21) = 3.55, p = 0.07. Planned comparisons revealed that KA-treated SUP rats that received post-SE water maze experience had a lower percentage of DCX+ in the hilus than both KA-treated home cage CON rats and KA-treated water maze CON rats (p < 0.05; Fig. 8H). There was no difference between KA-treated CON and SUP rats that remained in their home cage. Similar to the estimates of BrdU+ neurons that migrated to the hilus (Table 1), there was a trend for KA-treated water maze SUP rats to show a lower percentage of hilar migration of DCX+ neurons when compared to KA-treated home cage SUP rats (p = 0.07).
Sustained SE-Induced Increase in Hippocampal BDNF Protein
Prolonged seizures produce dramatic changes in the hippocampal microenvironment, including altered levels of various neurotrophic/growth factors (Gall, 1993; Rocamora et al., 1994; Marcinkiewicz et al., 1997; Shetty et al., 2003). We analyzed protein expression levels of BDNF in the hippocampus of each rat and the data are presented in Figure 9. Analyses revealed a main effect of seizure for both CON rats, F(1,23) = 15.30, p < 0.01, and SUP rats, F(1,23) = 25.79, p < 0.001, where KA-treated rats showed elevated levels of BDNF protein compared to saline-treated rats. There was no effect of experience or seizure × experience interaction for either CON or SUP rats, but there was a trend for SE to induce a greater increase in BDNF in home cage SUP rats than home cage CON rats (p = 0.09; Fig. 9). Consistent with our previous report (Glenn et al., 2007; Wong-Goodrich et al., 2008b), analysis of saline-treated rats revealed that SUP rats exhibited overall higher levels of BDNF protein (about 30% more) than CON rats, F(1,26) = 5.06, p < 0.05.
Figure 9.
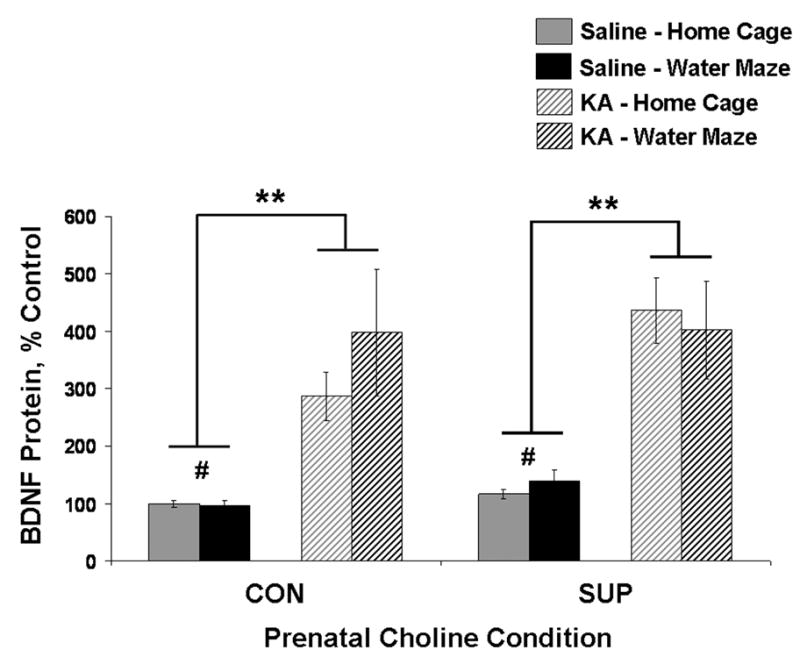
Mean (± SEM) percent of control levels for hippocampal BDNF protein for CON and SUP rats that were treated with saline (solid bars) or KA (hatched bars) and that remained in their home cage (grey) or received additional water maze training (black) following treatment. Both CON and SUP rats showed a significant overall SE-induced increase in BDNF protein. Saline-treated SUP rats also had a higher levels of BDNF protein overall than saline-treated CON rats. * significantly different at p < 0.05; ** main effect of seizure revealed by a within diet 2-way ANOVA (seizure × experience), p < 0.05; # main effect of prenatal diet revealed by a within diet 2-way ANOVA (diet × experience), p < 0.05.
Discussion
The present study revealed that prenatal choline supplemented rats that had undergone kainic acid-induced status epilepticus in young adulthood had attenuated spatial learning deficits and a complete recovery of spatial memory retention by 10 weeks after SE. This prophylactic dietary manipulation also prevented or mitigated long-term alterations in GABAergic function, reactive astrogliosis, and hippocampal neurogenesis that were present nearly 3 months after SE and that are hypothesized to contribute to the development of chronic epilepsy and ensuing learning and memory impairments. These data add to the growing literature that point to the important role of perinatal choline intake in neural protection (Guo-Ross et al., 2002; Holmes et al., 2002; Guo-Ross et al., 2003; Thomas et al., 2004; Nag and Berger-Sweeney, 2007; Thomas et al., 2007; Glenn et al., 2008; Meck et al., 2008; Thomas et al., 2009). We also report, for the first time, that repeatedly engaging in a hippocampally-mediated water-maze task following SE is beneficial for long-term hippocampal recovery. For example, repeated post-SE water maze training rescued SE-induced declines in GAD mRNA and dentate neurogenesis, and attenuated elevated GFAP protein levels. Consistent with previous reports (Glenn et al., 2007; Wong-Goodrich et al., 2008b), prenatal choline supplemented rats had a larger baseline pool of hippocampal BDNF and increased hippocampal neurogenesis. Enhanced adult hippocampal plasticity and trophic support may have provided a more permissive hippocampal microenvironment that enabled the prenatal choline supplemented adult hippocampus to better withstand the long-term consequences of SE, and possibly respond more effectively to the rehabilitative effects of post-SE water maze training. We discuss the long-term consequences of KA-induced SE on hippocampal and cognitive function, how our rehabilitative (water maze training) and prophylactic (prenatal choline supplementation) treatments altered the long-term effects of SE, and the potential implications of these altered outcomes for cognitive and neural recovery.
Long-Term Effects of KA-Induced SE in the Adult Hippocampus
Although widespread neurodegeneration and altered plasticity of the hippocampus is observed months following excitotoxic injury-induced SE, GABAergic neurons in the hippocampus are especially vulnerable. SE can lead to a loss of some types of inhibitory neurons, changes in GAD content, and altered GABA receptor expression, all of which are thought contribute to epileptogenesis (Sloviter, 1987; Davenport et al., 1990; Kash et al., 1997; Esclapez and Houser, 1999; Morimoto et al., 2004; Sperk et al., 2004; Brooks-Kayal et al., 2009). Our finding that hippocampal GAD65/67 mRNA and protein levels were decreased at nearly 3 months after KA-induced SE is consistent with previous reports that have shown a significant reduction in GAD65- and GAD67-containing neurons (cell bodies and terminals) in all cellular subfields of the hippocampus at 1 to 6 months after excitotoxic injury/seizures (Houser and Esclapez, 1996; Esclapez and Houser, 1999; Shetty and Turner, 2001; Dinocourt et al., 2003; Shetty et al., 2009). Using in situ hybridization and immunohistochemical techniques, it has also been demonstrated that the remaining GAD-containing neurons in the hippocampus, including interneurons and dentate granule cells, show continued upregulation of GAD65 and 67 mRNA and immunoreactivity months after injury (Feldblum et al., 1990; Houser and Esclapez, 1996; Sloviter et al., 1996; Esclapez and Houser, 1999; Knopp et al., 2008). The present study quantified total hippocampal GAD65 and 67 mRNA and protein content, but our techniques did not allow us to localize the distribution of GAD mRNA and protein. Thus, it is possible that the decrease in total GAD content that we observed in KA-treated CON rats masked a selective upregulation of GAD in the remaining GABAergic neurons that were preserved months after SE.
Increased hippocampal levels of GFAP may also play a role in hyperexcitability and structural reorganization of the hippocampus after SE that may underlie development of epilepsy. In addition to altered membrane channel, receptor, and transporter properties of astrocytes following SE that might add to neuronal hyperexcitability (Jabs et al., 2008), GFAP-expressing reactive astrocytes express neurite growth-promoting genes thought to support aberrant mossy fiber excitatory innervation of the dentate gyrus (Represa et al., 1993; Niquet et al., 1994a; Niquet et al., 1994b; Khrestchatisky et al., 1995; Stringer, 1996), which occurs prior to the onset of spontaneous motor seizures and may generate recurrent excitation underlying the development of seizures (Koyama and Ikegaya, 2004; Sharma et al., 2008). In the present study, we report that elevated hippocampal GFAP mRNA and protein persist for almost 3 months after KA-induced SE, which is consistent with elevated hippocampal GFAP immunoreactivity at 1 to 3 months after experimental SE (Aronica et al., 2000; Bendotti et al., 2000; Borges et al., 2006) and elevated GFAP in association with mossy fiber sprouting in the human epileptic hippocampus (Proper et al., 2000).
It is well established that seizures stimulate a vast proliferation of adult neural progenitors in the dentate gyrus within 1–3 days of SE (Bengzon et al., 1997; Parent et al., 1997; Gray and Sundstrom, 1998; Parent et al., 1999; Jessberger et al., 2005; Steiner et al., 2008), many of which become mature neurons and survive for weeks to months after SE (Parent et al., 1997; Madsen et al., 2000; Bonde et al., 2006); a profound number undergo apoptotic cell death by at least 4 weeks after cell division (Ekdahl et al., 2001; Ekdahl et al., 2002). Beyond 4 weeks, little is known about the survival rate of seizure-induced dentate granule cells born just 1–2 weeks after experimental SE. In the current study, we report a decrease in the ~2.5 month survival of BrdU+ cells in the SGZ-GCL that were labeled 6 to 14 days after SE, compared to the survival of SGZ-GCL BrdU+ cells from saline-treated rats, with a majority of these cells expressing a mature neuronal phenotype. In contrast, we observed that SE increased BrdU labeling in the dentate hilus, which suggests that the survival rates of newborn dentate cells born shortly after SE that migrate to the hilus versus GCL may operate via different mechanisms. Indeed, prior work has demonstrated that a loss in reelin following SE (via death of reelin-producing hilar interneurons) leads to aberrant hilar migration of dentate granule cell precursors (Gong et al., 2007). Moreover, blocking caspase-mediated cell death 7 days after SE selectively increases the survival of newborn dentate cells born shortly after seizures that migrate to the SGZ-GCL, but not to the hilus (Ekdahl et al., 2002), perhaps due to a savings in hilar reelin levels (Gong et al., 2007). Our findings are also consistent with a previous study showing that SE reduced the number of BrdU+/NeuN+ granule cells that were born 1 week after SE and observed in the SGZ-GCL 4 weeks later, despite an increase in the number of BrdU+ cells present in the hilus (Yang et al., 2008). However, others have also reported an increase in the survival of SGZ-GCL BrdU+/NeuN+ neurons born shortly after experimental SE (Ekdahl et al., 2003; Mohapel et al., 2004; Bonde et al., 2006; Jessberger et al., 2007a). Our findings suggest that there may be continued cell death of seizure-induced dentate granule cells in the GCL-SGZ beyond 4 weeks in this injury model. Notably, more severe, fully convulsive SE (as produced in our present study) has been shown to reduce the long-term survival of dentate granule cells born 1 week after SE in comparison to granule cells born in response to more mild seizures (Mohapel et al., 2004; Yang et al., 2008). Thus, differences in models of SE may account for the disparate findings reported in the present study.
Similar to the pattern of BrdU labeling found in the present study, we also found that at nearly 3 months after SE, there was a decrease in the number of DCX+ neurons in the SGZ-GCL with a concomitant appearance of DCX+ neurons in the hilus. Long-term declines in dentate neurogenesis in models of temporal lobe epilepsy have been previously reported (Hattiangady et al., 2004; Kralic et al., 2005; Heinrich et al., 2006; Hattiangady and Shetty, 2009), though others have also reported no decline (Jessberger et al., 2007b). Our findings are also supported by similar declines found in hippocampus tissue ressected from temporal lobe epilepsy patients (Mathern et al., 2002; Parent et al., 2006; Fahrner et al., 2007). Despite the reduction in dentate neurogenesis at nearly 3 months post-SE, we observed a sustained increase in hippocampal BDNF protein levels, which others have reported weeks to months after experimental SE (Vezzani et al., 1999; Revuelta et al., 2001; Scharfman et al., 2002; Sartori et al., 2009) as well as in the human epileptic hippocampus (Mathern et al., 1997; Takahashi et al., 1999; Murray et al., 2000). Future studies are needed to clarify whether or not elevated hippocampal BDNF following SE and in the epileptic brain is neuroprotective or detrimental to functional recovery, as accumulating evidence has provided support for both theories (Binder et al., 2001; Koyama and Ikegaya, 2005; Hattiangady and Shetty, 2008b).
Repeated Water Maze Testing and Prenatal Choline Supplementation Aid Recovery from SE
We report that despite no significant differences in seizure severity and progression during SE induction across KA-treated groups and similar putative levels of hippocampal damage, repeated water maze testing and prenatal choline supplementation differentially promoted the long-term hippocampal recovery from KA-induced SE. While many others have assessed cognitive function during a specific time point after experimental SE and have found that cognition is compromised weeks to months following SE (Stafstrom et al., 1993; Liu et al., 1994; Sarkisian et al., 1997; Hort et al., 1999; Mikati et al., 2001; Kemppainen and Pitkanen, 2004; McKay and Persinger, 2004; Detour et al., 2005; Lin et al., 2009; Sartori et al., 2009), the present study is the first to examine whether repeatedly engaging the hippocampus in a cognitive task would promote the long-term recovery from SE. Spatial learning in the water maze offers an experience that engages the hippocampus (Jenkins et al., 2004; Kee et al., 2007) and provides animals with a learning, enrichment, and exercise component all of which have been shown to enhance hippocampal plasticity and cognitive function in the intact and/or injured brain (Gould et al., 1999; van Praag et al., 2000; Gobbo and O’Mara, 2004; Gobbo and O’Mara, 2005; Cotman et al., 2007; Sartori et al., 2009), suggesting that this experience may be a good candidate for facilitating some aspects of recovery from SE. It is important to note that after SE, our water maze trained CON rats still showed poor spatial learning and no evidence of memory retention recovery over 3 weeks. However, water maze-trained rats in the current study only received a total of 15 days of intermittent testing in a span of 11 weeks after SE. Perhaps more prolonged and uninterrupted exposure to cognitive testing after SE would have facilitated a better cognitive recovery. It is also possible that we rehabilitated spatial learning and memory retention to some degree with repeated water maze testing in KA-treated CON rats, as post-excitotoxic injury enrichment and exercise has been shown to only attenuate, not completely abolish, cognitive deficits (Gobbo and O’Mara, 2004; Gobbo and O’Mara, 2005; Sartori et al., 2009). As such, the cognitive deficits observed at 10 weeks post-SE might have been further exacerbated in KA-treated CON rats that remained in their home cage (though we did not test home cage rats because we wanted to preserve a true post-SE home cage condition for comparison of brain measures).
Despite poor cognitive performance, the experience of water maze training in KA-treated CON rats prevented declines in hippocampal GAD mRNA, attenuated the SE-induced increase in GFAP protein, and preserved levels of dentate neurogenesis. A lack of obvious behavioral benefit from repeated water maze experience despite restorative effects on the hippocampus may also suggest that these specific neural outcomes may not be sufficient for restoring spatial learning and memory function following SE. For example, there is evidence that hippocampal neurogenesis appears to be important for some, but not all types of spatial learning and memory function (Shors et al., 2002; Madsen et al., 2003; Snyder et al., 2005; Saxe et al., 2006; Deng et al., 2009), and that neurogenesis in the epileptic brain may actually be detrimental (Scharfman and Gray, 2007). Future studies using SE models are needed to determine whether the recovery of newborn granule cells in an epileptic brain directly contribute to recovery of cognitive function. Alternately, other extrahippocampal areas that have been implicated in aspects of learning and memory are also severely damaged following KA-induced SE, such as the pyriform cortex, entorhinal cortex, septum, and amygdala (Schwob et al., 1980; Heggli et al., 1981; Sperk et al., 1983). We did not examine these areas in the current study, but we cannot exclude the possibility that SE-induced changes in these extrahippocampal areas may contribute to cognitive outcomes.
The present study is also the first to investigate whether the protective effects of prenatal choline supplementation against the sequelae of seizures are long-lasting. We found that prenatal choline supplementation alone mitigated the long-term consequences of SE to an even greater extent than that of water maze experience: SUP rats that remained in their home cage after SE were protected against losses in GAD mRNA and GAD65 protein, showed attenuated seizure-induced elevations in both GFAP mRNA and protein, and did not show deficits in dentate neurogenesis. That we found preserved levels of GAD and attenuated GFAP expression in our KA-treated SUP rats at nearly 3 months after SE extends our initial findings from our first report that revealed a similar pattern of protection in KA-treated SUP rats at just 16 days after SE (Wong-Goodrich et al., 2008b), indicating that prenatal choline supplementation’s protective effects on these measures are remarkably long-lasting. In the present study, we also discovered that our KA-treated SUP rats showed an increase in the number of BrdU+ granule cells in the SGZ-GCL compared to saline-treated SUP rats, which was surprising given that there was no such increase in water maze KA-treated CON rats and that our previous study demonstrated an attenuation of BrdU+ cells in the SGZ-GCL at just 16 days after SE in KA-treated SUP rats (Wong-Goodrich et al., 2008b). It is difficult to directly compare numbers of BrdU+ cells between these two studies, given the differences in BrdU regimens. However, taken together, our previous and current findings suggest that 1) many of these newly generated cells labeled 6 to 14 days after SE likely undergo cell death 2.5 months later, and 2) in addition to attenuated dentate cell proliferation shortly after seizures (Wong-Goodrich et al., 2008b), prenatal choline supplementation may also lessen the magnitude of cell death occurring weeks to months after KA-induced SE. Prenatal choline supplementation has been found to reduce levels of apoptotic cell death in the fetal hippocampus (Albright et al., 1999; Craciunescu et al., 2003), and we have recently discovered that levels of apoptotic cell death in the adult hippocampus are reduced in aged prenatal choline supplemented rats (unpublished observations, M.J., Glenn, M.D. Niculescu, C.L. Williams, and S.H. Zeisel). It is therefore possible that prenatal choline supplementation also reduced the amount of dentate granule cell death following seizures, although this remains to be determined.
Protection (or recovery) from post-SE disruptions in hippocampal GAD, GFAP, and granule cell survival may have implications for ameliorating the epileptogenic outcome of SE. Preventing losses in GAD (via protecting against the death of GABAergic neurons and/or increasing expression of GAD in remaining GABAergic neurons) may restore GABAergic function in the injured hippocampus (Houser and Esclapez, 1996; Esclapez and Houser, 1999; Shetty and Turner, 2001; Dinocourt et al., 2003; Shetty et al., 2009). Attenuated hippocampal GFAP expression following SE may also help reduce hyperexcitability. Indeed, the magnitude of reactive gliosis may be a reliable predictor of epileptic seizure severity after experimental SE (Somera-Molina et al., 2007) and in human epilepsy patients (Guerreiro et al., 2003). Interestingly, the protective effects of repeated water maze experience and prenatal choline supplementation on the survival of granule cells born shortly after SE appear to be specific to dentate granule cells that migrate to the GCL. This finding is compelling because dentate granule cells born 1 week after SE that migrate properly to the GCL show reduced excitability compared to mature granule cells, and may therefore serve a restorative function in counteracting the hyperexcitability that underlies epilepsy (Jakubs et al., 2006). Thus, enhanced granule cell survival in the GCL in particular may be especially beneficial to recovery from SE. The number of spontaneous motor seizures that are observed following SE using the model employed in the current study are apparent as early as one month after SE induction, and become increasingly more apparent during the third through sixth month after SE (Hellier et al., 1998; Hattiangady et al., 2004; Williams et al., 2009). We observed spontaneous motor seizure activity in very few of our KA-treated rats in the present study that were euthanized shortly before 3 months post-SE, but we did not perform extensive observations of spontaneous seizure activity, or monitor EEG activity. It is possible that we underestimated the number of rats having spontaneous seizures, or that epileptic seizures would have manifested at a later time point, which may also make interpreting some of these findings difficult. For example, spontaneous epileptic seizures has been shown to enhance BDNF immunoreactivity in the hippocampus (Vezzani et al., 1999), and a higher frequency of spontaneous seizures has been linked with a more pronounced decline in neurogenesis months after SE (Hattiangady et al., 2004). Whether the protective effects of water maze testing and prenatal choline supplementation on brain and behavior pertain to the development of epileptic seizures, however, remains to be determined.
The present study also showed that both repeated water maze experience and prenatal choline supplementation also rescued the decline in the number of newborn neurons present at nearly 3 months after KA-induced SE. While it is not clear whether aberrantly high levels of dentate neurogenesis directly contribute to cognitive deficits observed shortly after SE, it has been suggested that the subsequent downregulation of dentate neurogenesis weeks to months after the initial injury may contribute to cognitive deficits associated with epilepsy (Mikati et al., 2001; Elger et al., 2004; Hattiangady et al., 2004; Dhanushkodi and Shetty, 2008; Hattiangady and Shetty, 2008b). Remarkably, we discovered that prenatal choline supplementation attenuated the spatial learning deficits elicited by SE and completely restored spatial memory retention ability to that of saline-treated rats by 10 weeks after SE. Our findings suggest that the protection of spatial learning and memory function conferred by prenatal choline supplementation extends much further beyond 1–2 weeks after SE (Yang et al., 2000; Holmes et al., 2002). Recovery of neurogenesis in SUP rats months after seizures also appeared to be independent of post-SE water maze training, as home cage KA-treated SUP rats also showed a preservation of DCX+ neurons. However, it has been suggested that aberrant migration of newborn neurons to the hilus may contribute to epileptogenesis and cognitive deficits after SE (Parent et al., 1997; Scharfman et al., 2000; Scharfman and Gray, 2007; Scharfman and Hen, 2007). Although neither repeated water maze testing nor prenatal choline supplementation significantly altered the number of ectopic hilar granule cells born shortly after SE, there was a trend for SUP rats who received additional water maze training after SE to show reduced BrdU+ neurons in the hilus compared to SUP rats that remained in their home cage after SE. A similar trend was observed on the percentage of DCX+ neurons that migrated to the hilus, but it is still not definitive whether the effects of prenatal choline and water maze experience are additive. When comparing KA-treated CON vs. SUP rats that received post-SE water maze experience, hilar migration of newborn DCX+ neurons at 3 months post-SE was significantly reduced in our SUP rats. Given that these two groups both showed a similar recovery of the number of DCX+ neurons at 3 months after SE, perhaps it is the recovery in migratory behavior of newborn dentate granule that may have contributed to the improved spatial learning and memory retention ability observed in KA-treated SUP rats. Future studies will further explore whether prenatal choline supplementation can enable the injured adult hippocampus to more effectively respond to the rehabilitative effects of cognitive enrichment.
Protective Effects of Prenatal Choline Supplementation
We report that increased availability of a single nutrient, choline, during early development produced a lasting change in the adult brain that mitigated the long-term effects of KA-induced SE on the adult hippocampus and spatial learning and memory retention. Prenatal and early postnatal choline supplementation has also been shown to protect against impairments in performance on hippocampal-dependent tasks caused by aging (Meck et al., 1988; Meck et al., 1989; Meck and Williams, 2003; McCann et al., 2006; Meck et al., 2008) and neonatal alcohol exposure (Thomas et al., 2004; Wagner and Hunt, 2006; Thomas et al., 2007), as well as protect against neurotoxicity-induced cortical neurodegeneration in the adult rat (Guo-Ross et al., 2002; Guo-Ross et al., 2003).
Enhanced adult hippocampal plasticity and neurotrophic milieu prior to injury may underlie prenatal choline supplementation’s cognitive enhancing effects and neuroprotection of the adult hippocampus. That our KA-treated CON rats did not show recovery of spatial learning and memory may further support the notion that factors that affect the hippocampal microenvironment prior to injury may be extremely important predictors of cognitive recovery. The present study found that prenatal choline supplementation increased the number of dentate DCX+ neurons and hippocampal BDNF protein in saline-treated adult rats, which we have previously demonstrated elsewhere (Glenn et al., 2007; Wong-Goodrich et al., 2008b). BDNF may be especially relevant for the long-term neural recovery following SE, including protection of GABAergic interneurons (Prince et al., 2009), reducing hippocampal neuronal damage and spontaneous seizures, and rescuing dentate neurogenesis (Paradiso et al., 2009). Prenatal choline supplementation also upregulates basal levels of other neurotrophic/growth factors in the adult hippocampus with putative neuroprotective properties in the context of brain injury, including nerve growth factor (NGF), insulin growth-like factor-1 (IGF-1), and vascular endothelial growth factor (VEGF) (Saatman et al., 1997; Sofroniew et al., 2001; Hennigan et al., 2007; Glenn et al., 2008; Nicoletti et al., 2008; Vezzani, 2008; Wong-Goodrich et al., 2008a; Wong-Goodrich et al., 2008b). In addition to dentate neurogenesis, prenatal choline supplementation also enhances other features of adult hippocampal plasticity implicated in learning and memory process, including LTP (Pyapali et al., 1998; Jones et al., 1999), MAPK and CREB activation (Mellott et al., 2004), and increased dendritic spine density in the CA1 and dentate gyrus (Meck et al., 2008). The mechanism by which prenatal choline availability early in development produces long-lasting alterations in hippocampal plasticity and cognitive function in the intact and injured brain is still not fully understood, but one mechanism might be via choline’s conversion to betaine, which serves as a methyl donor that can influence the regulation of gene expression, including specific genes relevant for hippocampal plasticity (Blusztajn and Wurtman, 1983; Blusztajn, 1998). Indeed, evidence supports the view that variations in the prenatal supply of choline causes multiple modifications in the developmental patterns of expression of genes known to influence learning and memory and provide molecular correlates for the cognitive changes evoked by altered availability of choline in utero (Mellott et al., 2007). Lifelong enhanced hippocampal plasticity and trophic support may render the hippocampus better able to withstand or recover from a neural insult. Interventions that have been shown to enhance hippocampal plasticity, such as cognitive enrichment and early choline availability, may provide helpful insights into the development of treatments for the long-term consequences of SE and epilepsy.
Acknowledgments
We would like to thank Becky Hurt, Sophia Cao, Verne Tsang, and Lauren Williamson for assistance with data collection. This work was supported by AG009525 to C.L.W. and J.K.B.
Grant sponsor: NIA
Grant number: AG09525
References
- Albright CD, Tsai AY, Friedrich CB, Mar MH, Zeisel SH. Choline availability alters embryonic development of the hippocampus and septum in the rat. Brain Res Dev Brain Res. 1999;113:13–20. doi: 10.1016/s0165-3806(98)00183-7. [DOI] [PubMed] [Google Scholar]
- Ambrogini P, Orsini L, Mancini C, Ferri P, Ciaroni S, Cuppini R. Learning may reduce neurogenesis in adult rat dentate gyrus. Neurosci Lett. 2004;359:13–16. doi: 10.1016/j.neulet.2003.12.123. [DOI] [PubMed] [Google Scholar]
- Arida RM, Scorza FA, Scorza CA, Cavalheiro EA. Is physical activity beneficial for recovery in temporal lobe epilepsy? Evidences from animal studies. Neurosci Biobehav Rev. 2009;33:422–431. doi: 10.1016/j.neubiorev.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. 2000;12:2333–2344. doi: 10.1046/j.1460-9568.2000.00131.x. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Represa A. Brief seizure episodes induce long-term potentiation and mossy fibre sprouting in the hippocampus. Trends Neurosci. 1990;13:312–318. doi: 10.1016/0166-2236(90)90135-w. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Guglielmetti F, Tortarolo M, Samanin R, Hirst WD. Differential expression of S100beta and glial fibrillary acidic protein in the hippocampus after kainic acid-induced lesions and mossy fiber sprouting in adult rat. Exp Neurol. 2000;161:317–329. doi: 10.1006/exnr.1999.7262. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmer E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci U S A. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EH, Cornett J. The ontogeny of seizures in a rat model of limbic epilepsy: evidence for a kindling process in the development of chronic spontaneous seizures. Brain Res. 1993;625:295–300. doi: 10.1016/0006-8993(93)91071-y. [DOI] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 2001;24:47–53. doi: 10.1016/s0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK. Choline, a vital amine. Science. 1998;281:794–795. doi: 10.1126/science.281.5378.794. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Wurtman RJ. Choline and cholinergic neurons. Science. 1983;221:614–620. doi: 10.1126/science.6867732. [DOI] [PubMed] [Google Scholar]
- Bonde S, Ekdahl CT, Lindvall O. Long-term neuronal replacement in adult rat hippocampus after status epilepticus despite chronic inflammation. Eur J Neurosci. 2006;23:965–974. doi: 10.1111/j.1460-9568.2006.04635.x. [DOI] [PubMed] [Google Scholar]
- Borges K, McDermott D, Irier H, Smith Y, Dingledine R. Degeneration and proliferation of astrocytes in the mouse dentate gyrus after pilocarpine-induced status epilepticus. Exp Neurol. 2006;201:416–427. doi: 10.1016/j.expneurol.2006.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Raol YH, Russek SJ. Alteration of epileptogenesis genes. Neurotherapeutics. 2009;6:312–318. doi: 10.1016/j.nurt.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14:3106–3121. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30:464–472. doi: 10.1016/j.tins.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Covolan L, Ribeiro LT, Longo BM, Mello LE. Cell damage and neurogenesis in the dentate granule cell layer of adult rats after pilocarpine- or kainate-induced status epilepticus. Hippocampus. 2000;10:169–180. doi: 10.1002/(SICI)1098-1063(2000)10:2<169::AID-HIPO6>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Craciunescu CN, Albright CD, Mar MH, Song J, Zeisel SH. Choline availability during embryonic development alters progenitor cell mitosis in developing mouse hippocampus. J Nutr. 2003;133:3614–3618. doi: 10.1093/jn/133.11.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport CJ, Brown WJ, Babb TL. GABAergic neurons are spared after intrahippocampal kainate in the rat. Epilepsy Res. 1990;5:28–42. doi: 10.1016/0920-1211(90)90063-2. [DOI] [PubMed] [Google Scholar]
- Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–13542. doi: 10.1523/JNEUROSCI.3362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats. Epilepsia. 2005;46:499–508. doi: 10.1111/j.0013-9580.2005.38704.x. [DOI] [PubMed] [Google Scholar]
- Dhanushkodi A, Shetty AK. Is exposure to enriched environment beneficial for functional post-lesional recovery in temporal lobe epilepsy? Neurosci Biobehav Rev. 2008;32:657–674. doi: 10.1016/j.neubiorev.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinocourt C, Petanjek Z, Freund TF, Ben-Ari Y, Esclapez M. Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J Comp Neurol. 2003;459:407–425. doi: 10.1002/cne.10622. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Kempermann G. Paradoxical effects of learning the Morris water maze on adult hippocampal neurogenesis in mice may be explained by a combination of stress and physical activity. Genes Brain Behav. 2006;5:29–39. doi: 10.1111/j.1601-183X.2005.00129.x. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Mohapel P, Elmer E, Lindvall O. Caspase inhibitors increase short-term survival of progenitor-cell progeny in the adult rat dentate gyrus following status epilepticus. Eur J Neurosci. 2001;14:937–945. doi: 10.1046/j.0953-816x.2001.01713.x. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Mohapel P, Weber E, Bahr B, Blomgren K, Lindvall O. Caspase-mediated death of newly formed neurons in the adult rat dentate gyrus following status epilepticus. Eur J Neurosci. 2002;16:1463–1471. doi: 10.1046/j.1460-9568.2002.02202.x. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Zhu C, Bonde S, Bahr BA, Blomgren K, Lindvall O. Death mechanisms in status epilepticus-generated neurons and effects of additional seizures on their survival. Neurobiol Dis. 2003;14:513–523. doi: 10.1016/j.nbd.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. 2004;3:663–672. doi: 10.1016/S1474-4422(04)00906-8. [DOI] [PubMed] [Google Scholar]
- Erlander MG, Tobin AJ. The structural and functional heterogeneity of glutamic acid decarboxylase: a review. Neurochem Res. 1991;16:215–226. doi: 10.1007/BF00966084. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci. 1994;14:1834–1855. doi: 10.1523/JNEUROSCI.14-03-01834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner A, Kann G, Flubacher A, Heinrich C, Freiman TM, Zentner J, Frotscher M, Haas CA. Granule cell dispersion is not accompanied by enhanced neurogenesis in temporal lobe epilepsy patients. Exp Neurol. 2007;203:320–332. doi: 10.1016/j.expneurol.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Faverjon S, Silveira DC, Fu DD, Cha BH, Akman C, Hu Y, Holmes GL. Beneficial effects of enriched environment following status epilepticus in immature rats. Neurology. 2002;59:1356–1364. doi: 10.1212/01.wnl.0000033588.59005.55. [DOI] [PubMed] [Google Scholar]
- Feldblum S, Ackermann RF, Tobin AJ. Long-term increase of glutamate decarboxylase mRNA in a rat model of temporal lobe epilepsy. Neuron. 1990;5:361–371. doi: 10.1016/0896-6273(90)90172-c. [DOI] [PubMed] [Google Scholar]
- Gall CM. Seizure-induced changes in neurotrophin expression: implications for epilepsy. Exp Neurol. 1993;124:150–166. doi: 10.1006/exnr.1993.1186. [DOI] [PubMed] [Google Scholar]
- Glenn MJ, Gibson EM, Kirby ED, Mellott TJ, Blusztajn JK, Williams CL. Prenatal choline availability modulates hippocampal neurogenesis and neurogenic responses to enriching experiences in adult female rats. Eur J Neurosci. 2007;25:2473–2482. doi: 10.1111/j.1460-9568.2007.05505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn MJ, Kirby ED, Gibson EM, Wong-Goodrich SJ, Mellott TJ, Blusztajn JK, Williams CL. Age-related declines in exploratory behavior and markers of hippocampal plasticity are attenuated by prenatal choline supplementation in rats. Brain Res. 2008;1237:110–123. doi: 10.1016/j.brainres.2008.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbo OL, O’Mara SM. Impact of enriched-environment housing on brain-derived neurotrophic factor and on cognitive performance after a transient global ischemia. Behav Brain Res. 2004;152:231–241. doi: 10.1016/j.bbr.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Gobbo OL, O’Mara SM. Exercise, but not environmental enrichment, improves learning after kainic acid-induced hippocampal neurodegeneration in association with an increase in brain-derived neurotrophic factor. Behav Brain Res. 2005;159:21–26. doi: 10.1016/j.bbr.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Golden GT, Smith GG, Ferraro TN, Reyes PF. Rat strain and age differences in kainic acid induced seizures. Epilepsy Res. 1995;20:151–159. doi: 10.1016/0920-1211(94)00079-c. [DOI] [PubMed] [Google Scholar]
- Golden GT, Smith GG, Ferraro TN, Reyes PF, Kulp JK, Fariello RG. Strain differences in convulsive response to the excitotoxin kainic acid. Neuroreport. 1991;2:141–144. doi: 10.1097/00001756-199103000-00008. [DOI] [PubMed] [Google Scholar]
- Gong C, Wang TW, Huang HS, Parent JM. Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus. J Neurosci. 2007;27:1803–1811. doi: 10.1523/JNEUROSCI.3111-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, Goncalves Pereira PM, van Vliet EA, Aronica E, Lopes da Silva FH, Lucassen PJ. Neuronal cell death in a rat model for mesial temporal lobe epilepsy is induced by the initial status epilepticus and not by later repeated spontaneous seizures. Epilepsia. 2003;44:647–658. doi: 10.1046/j.1528-1157.2003.53902.x. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Gray WP, Sundstrom LE. Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res. 1998;790:52–59. doi: 10.1016/s0006-8993(98)00030-4. [DOI] [PubMed] [Google Scholar]
- Guerreiro MM, Quesney LF, Salanova V, Snipes GJ. Continuous electrocorticogram epileptiform discharges due to brain gliosis. J Clin Neurophysiol. 2003;20:239–242. doi: 10.1097/00004691-200307000-00002. [DOI] [PubMed] [Google Scholar]
- Guo-Ross SX, Clark S, Montoya DA, Jones KH, Obernier J, Shetty AK, White AM, Blusztajn JK, Wilson WA, Swartzwelder HS. Prenatal choline supplementation protects against postnatal neurotoxicity. J Neurosci. 2002;22:RC195. doi: 10.1523/JNEUROSCI.22-01-j0005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo-Ross SX, Jones KH, Shetty AK, Wilson WA, Swartzwelder HS. Prenatal dietary choline availability alters postnatal neurotoxic vulnerability in the adult rat. Neurosci Lett. 2003;341:161–163. doi: 10.1016/s0304-3940(03)00119-8. [DOI] [PubMed] [Google Scholar]
- Haas KZ, Sperber EF, Opanashuk LA, Stanton PK, Moshe SL. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus. 2001;11:615–625. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol Dis. 2004;17:473–490. doi: 10.1016/j.nbd.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Aging does not alter the number or phenotype of putative stem/progenitor cells in the neurogenic region of the hippocampus. Neurobiol Aging. 2008a;29:129–147. doi: 10.1016/j.neurobiolaging.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia. 2008b;49(Suppl 5):26–41. doi: 10.1111/j.1528-1167.2008.01635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Decreased neuronal differentiation of newly generated cells underlies reduced hippocampal neurogenesis in chronic temporal lobe epilepsy. Hippocampus. 2009 doi: 10.1002/hipo.20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggli DE, Aamodt A, Malthe-Sorenssen D. Kainic acid neurotoxicity; effect of systemic injection on neurotransmitter markers in different brain regions. Brain Res. 1981;230:253–262. doi: 10.1016/0006-8993(81)90405-4. [DOI] [PubMed] [Google Scholar]
- Heinrich C, Nitta N, Flubacher A, Muller M, Fahrner A, Kirsch M, Freiman T, Suzuki F, Depaulis A, Frotscher M, et al. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J Neurosci. 2006;26:4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellier JL, Dudek FE. Spontaneous motor seizures of rats with kainate–induced epilepsy: effect of time of day and activity state. Epilepsy Res. 1999;35:47–57. doi: 10.1016/s0920-1211(98)00127-2. [DOI] [PubMed] [Google Scholar]
- Hellier JL, Dudek FE. Chemoconvulsant model of chronic spontaneous seizures. Curr Protoc Neurosci. 2005;Chapter 9(Unit 9):19. doi: 10.1002/0471142301.ns0919s31. [DOI] [PubMed] [Google Scholar]
- Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE. Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res. 1998;31:73–84. doi: 10.1016/s0920-1211(98)00017-5. [DOI] [PubMed] [Google Scholar]
- Hellsten J, Wennstrom M, Bengzon J, Mohapel P, Tingstrom A. Electroconvulsive seizures induce endothelial cell proliferation in adult rat hippocampus. Biol Psychiatry. 2004;55:420–427. doi: 10.1016/j.biopsych.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Hennigan A, O’Callaghan RM, Kelly AM. Neurotrophins and their receptors: roles in plasticity, neurodegeneration and neuroprotection. Biochem Soc Trans. 2007;35:424–427. doi: 10.1042/BST0350424. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Yang Y, Liu Z, Cermak JM, Sarkisian MR, Stafstrom CE, Neill JC, Blusztajn JK. Seizure-induced memory impairment is reduced by choline supplementation before or after status epilepticus. Epilepsy Res. 2002;48:3–13. doi: 10.1016/s0920-1211(01)00321-7. [DOI] [PubMed] [Google Scholar]
- Hort J, Brozek G, Mares P, Langmeier M, Komarek V. Cognitive functions after pilocarpine-induced status epilepticus: changes during silent period precede appearance of spontaneous recurrent seizures. Epilepsia. 1999;40:1177–1183. doi: 10.1111/j.1528-1157.1999.tb00845.x. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. Epilepsy Res. 1996;26:207–218. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- Jabs R, Seifert G, Steinhauser C. Astrocytic function and its alteration in the epileptic brain. Epilepsia. 2008;49(Suppl 2):3–12. doi: 10.1111/j.1528-1167.2008.01488.x. [DOI] [PubMed] [Google Scholar]
- Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron. 2006;52:1047–1059. doi: 10.1016/j.neuron.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Jenkins TA, Amin E, Pearce JM, Brown MW, Aggleton JP. Novel spatial arrangements of familiar visual stimuli promote activity in the rat hippocampal formation but not the parahippocampal cortices: a c-fos expression study. Neuroscience. 2004;124:43–52. doi: 10.1016/j.neuroscience.2003.11.024. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Nakashima K, Clemenson GD, Jr, Mejia E, Mathews E, Ure K, Ogawa S, Sinton CM, Gage FH, Hsieh J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J Neurosci. 2007a;27:5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Romer B, Babu H, Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol. 2005;196:342–351. doi: 10.1016/j.expneurol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Zhao C, Toni N, Clemenson GD, Jr, Li Y, Gage FH. Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J Neurosci. 2007b;27:9400–9407. doi: 10.1523/JNEUROSCI.2002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JP, Meck WH, Williams CL, Wilson WA, Swartzwelder HS. Choline availability to the developing rat fetus alters adult hippocampal long-term potentiation. Brain Res Dev Brain Res. 1999;118:159–167. doi: 10.1016/s0165-3806(99)00103-0. [DOI] [PubMed] [Google Scholar]
- Jorgensen MB, Finsen BR, Jensen MB, Castellano B, Diemer NH, Zimmer J. Microglial and astroglial reactions to ischemic and kainic acid-induced lesions of the adult rat hippocampus. Exp Neurol. 1993;120:70–88. doi: 10.1006/exnr.1993.1041. [DOI] [PubMed] [Google Scholar]
- Kang TC, Kim DS, Kwak SE, Kim JE, Won MH, Kim DW, Choi SY, Kwon OS. Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia. 2006;54:258–271. doi: 10.1002/glia.20380. [DOI] [PubMed] [Google Scholar]
- Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Teixeira CM, Wang AH, Frankland PW. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci. 2007;10:355–362. doi: 10.1038/nn1847. [DOI] [PubMed] [Google Scholar]
- Kemppainen S, Pitkanen A. Damage to the amygdalo-hippocampal projection in temporal lobe epilepsy: a tract-tracing study in chronic epileptic rats. Neuroscience. 2004;126:485–501. doi: 10.1016/j.neuroscience.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Khrestchatisky M, Ferhat L, Charton G, Bernard A, Pollard H, Represa A, Ben-Ari Y. Molecular correlates between reactive and developmental plasticity in the rat hippocampus. J Neurobiol. 1995;26:426–436. doi: 10.1002/neu.480260314. [DOI] [PubMed] [Google Scholar]
- Knopp A, Frahm C, Fidzinski P, Witte OW, Behr J. Loss of GABAergic neurons in the subiculum and its functional implications in temporal lobe epilepsy. Brain. 2008;131:1516–1527. doi: 10.1093/brain/awn095. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. Mossy fiber sprouting as a potential therapeutic target for epilepsy. Curr Neurovasc Res. 2004;1:3–10. doi: 10.2174/1567202043480242. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus. Neuroscientist. 2005;11:282–287. doi: 10.1177/1073858405278266. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Ledergerber DA, Fritschy JM. Disruption of the neurogenic potential of the dentate gyrus in a mouse model of temporal lobe epilepsy with focal seizures. Eur J Neurosci. 2005;22:1916–1927. doi: 10.1111/j.1460-9568.2005.04386.x. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Holmes GL, Kubie JL, Muller RU. Recurrent seizures induce a reversible impairment in a spatial hidden goal task. Hippocampus. 2009 doi: 10.1002/hipo.20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gatt A, Werner SJ, Mikati MA, Holmes GL. Long-term behavioral deficits following pilocarpine seizures in immature rats. Epilepsy Res. 1994;19:191–204. doi: 10.1016/0920-1211(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Kristjansen PE, Bolwig TG, Wortwein G. Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neuroscience. 2003;119:635–642. doi: 10.1016/s0306-4522(03)00199-4. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry. 2000;47:1043–1049. doi: 10.1016/s0006-3223(00)00228-6. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz M, Nagao T, Day R, Seidah NG, Chretien M, Avoli M. Pilocarpine-induced seizures are accompanied by a transient elevation in the messenger RNA expression of the prohormone convertase PC1 in rat hippocampus: comparison with nerve growth factor and brain-derived neurotrophic factor expression. Neuroscience. 1997;76:425–439. doi: 10.1016/s0306-4522(96)00318-1. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Micevych PE, Blanco CE, Pretorius JK. Granule cell mRNA levels for BDNF, NGF, and NT-3 correlate with neuron losses or supragranular mossy fiber sprouting in the chronically damaged and epileptic human hippocampus. Mol Chem Neuropathol. 1997;30:53–76. doi: 10.1007/BF02815150. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Leiphart JL, De Vera A, Adelson PD, Seki T, Neder L, Leite JP. Seizures decrease postnatal neurogenesis and granule cell development in the human fascia dentata. Epilepsia. 2002;43(Suppl 5):68–73. doi: 10.1046/j.1528-1157.43.s.5.28.x. [DOI] [PubMed] [Google Scholar]
- McCann JC, Hudes M, Ames BN. An overview of evidence for a causal relationship between dietary availability of choline during development and cognitive function in offspring. Neurosci Biobehav Rev. 2006;30:696–712. doi: 10.1016/j.neubiorev.2005.12.003. [DOI] [PubMed] [Google Scholar]
- McKay BE, Persinger MA. Normal spatial and contextual learning for ketamine-treated rats in the pilocarpine epilepsy model. Pharmacol Biochem Behav. 2004;78:111–119. doi: 10.1016/j.pbb.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Meck WH, Smith RA, Williams CL. Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev Psychobiol. 1988;21:339–353. doi: 10.1002/dev.420210405. [DOI] [PubMed] [Google Scholar]
- Meck WH, Smith RA, Williams CL. Organizational changes in cholinergic activity and enhanced visuospatial memory as a function of choline administered prenatally or postnatally or both. Behav Neurosci. 1989;103:1234–1241. doi: 10.1037//0735-7044.103.6.1234. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Characterization of the facilitative effects of perinatal choline supplementation on timing and temporal memory. Neuroreport. 1997a;8:2831–2835. doi: 10.1097/00001756-199709080-00005. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Perinatal choline supplementation increases the threshold for chunking in spatial memory. Neuroreport. 1997b;8:3053–3059. doi: 10.1097/00001756-199709290-00010. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Simultaneous temporal processing is sensitive to prenatal choline availability in mature and aged rats. Neuroreport. 1997c;8:3045–3051. doi: 10.1097/00001756-199709290-00009. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Choline supplementation during prenatal development reduces proactive interference in spatial memory. Brain Res Dev Brain Res. 1999;118:51–59. doi: 10.1016/s0165-3806(99)00105-4. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Metabolic imprinting of choline by its availability during gestation: implications for memory and attentional processing across the lifespan. Neurosci Biobehav Rev. 2003;27:385–399. doi: 10.1016/s0149-7634(03)00069-1. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL, Cermak JM, Blusztajn JK. Developmental periods of choline sensitivity provide an ontogenetic mechanism for regulating memory capacity and age-related dementia. Frontiers In Integrative Neuroscience. 2008;1:1–11. doi: 10.3389/neuro.07.007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellott TJ, Follettie MT, Diesl V, Hill AA, Lopez-Coviella I, Blusztajn JK. Prenatal choline availability modulates hippocampal and cerebral cortical gene expression. Faseb J. 2007;21:1311–1323. doi: 10.1096/fj.06-6597com. [DOI] [PubMed] [Google Scholar]
- Mellott TJ, Williams CL, Meck WH, Blusztajn JK. Prenatal choline supplementation advances hippocampal development and enhances MAPK and CREB activation. Faseb J. 2004;18:545–547. doi: 10.1096/fj.03-0877fje. [DOI] [PubMed] [Google Scholar]
- Mikati MA, Tarif S, Lteif L, Jawad MA. Time sequence and types of memory deficits after experimental status epilepticus. Epilepsy Res. 2001;43:97–101. doi: 10.1016/s0920-1211(00)00187-x. [DOI] [PubMed] [Google Scholar]
- Mirescu C, Peters JD, Noiman L, Gould E. Sleep deprivation inhibits adult neurogenesis in the hippocampus by elevating glucocorticoids. Proc Natl Acad Sci U S A. 2006;103:19170–19175. doi: 10.1073/pnas.0608644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapel P, Ekdahl CT, Lindvall O. Status epilepticus severity influences the long-term outcome of neurogenesis in the adult dentate gyrus. Neurobiol Dis. 2004;15:196–205. doi: 10.1016/j.nbd.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Mundt-Petersen K, Brundin P, Frielingsdorf H. Working memory training decreases hippocampal neurogenesis. Neuroscience. 2006;142:609–613. doi: 10.1016/j.neuroscience.2006.07.033. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Mouton P. Principles and practices of unbiased stereology. Baltimore: The Johns Hopkins University Press; 2002. [Google Scholar]
- Murray KD, Isackson PJ, Eskin TA, King MA, Montesinos SP, Abraham LA, Roper SN. Altered mRNA expression for brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase in the hippocampus of patients with intractable temporal lobe epilepsy. J Comp Neurol. 2000;418:411–422. doi: 10.1002/(sici)1096-9861(20000320)418:4<411::aid-cne4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Nag N, Berger-Sweeney JE. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol Dis. 2007;26:473–480. doi: 10.1016/j.nbd.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Namchuk M, Lindsay L, Turck CW, Kanaani J, Baekkeskov S. Phosphorylation of serine residues 3, 6, 10, and 13 distinguishes membrane anchored from soluble glutamic acid decarboxylase 65 and is restricted to glutamic acid decarboxylase 65alpha. J Biol Chem. 1997;272:1548–1557. doi: 10.1074/jbc.272.3.1548. [DOI] [PubMed] [Google Scholar]
- Napoli I, Blusztajn JK, Mellott TJ. Prenatal choline supplementation in rats increases the expression of IGF2 and its receptor IGF2R and enhances IGF2–induced acetylcholine release in hippocampus and frontal cortex. Brain Res. 2008;1237:124–135. doi: 10.1016/j.brainres.2008.08.046. [DOI] [PubMed] [Google Scholar]
- Nicoletti JN, Shah SK, McCloskey DP, Goodman JH, Elkady A, Atassi H, Hylton D, Rudge JS, Scharfman HE, Croll SD. Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience. 2008;151:232–241. doi: 10.1061/j.neuroscience.2007.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, Ben-Ari Y, Represa A. Glial reaction after seizure induced hippocampal lesion: immunohistochemical characterization of proliferating glial cells. J Neurocytol. 1994a;23:641–656. doi: 10.1007/BF01191558. [DOI] [PubMed] [Google Scholar]
- Niquet J, Jorquera I, Ben-Ari Y, Represa A. Proliferative astrocytes may express fibronectin-like protein in the hippocampus of epileptic rats. Neurosci Lett. 1994b;180:13–16. doi: 10.1016/0304-3940(94)90902-4. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA-containing neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci. 1993;13:4470–4485. doi: 10.1523/JNEUROSCI.13-10-04470.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso B, Marconi P, Zucchini S, Berto E, Binaschi A, Bozac A, Buzzi A, Mazzuferi M, Magri E, Navarro Mora G, et al. Localized delivery of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model. Proc Natl Acad Sci U S A. 2009;106:7191–7196. doi: 10.1073/pnas.0810710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol. 2006;59:81–91. doi: 10.1002/ana.20699. [DOI] [PubMed] [Google Scholar]
- Parent JM, Tada E, Fike JR, Lowenstein DH. Inhibition of dentate granule cell neurogenesis with brain irradiation does not prevent seizure-induced mossy fiber synaptic reorganization in the rat. J Neurosci. 1999;19:4508–4519. doi: 10.1523/JNEUROSCI.19-11-04508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JP, Melton J, Punsoni M, McCloskey DP, Scharfman HE. Mossy fibers are the primary source of afferent input to ectopic granule cells that are born after pilocarpine-induced seizures. Exp Neurol. 2005;196:316–331. doi: 10.1016/j.expneurol.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Kharatishvili I, Karhunen H, Lukasiuk K, Immonen R, Nairismagi J, Grohn O, Nissinen J. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl 2):13–20. doi: 10.1111/j.1528-1167.2007.01063.x. [DOI] [PubMed] [Google Scholar]
- Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50(Suppl 2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proper EA, Oestreicher AB, Jansen GH, Veelen CW, van Rijen PC, Gispen WH, de Graan PN. Immunohistochemical characterization of mossy fibre sprouting in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2000;123 ( Pt 1):19–30. doi: 10.1093/brain/123.1.19. [DOI] [PubMed] [Google Scholar]
- Pyapali GK, Turner DA, Williams CL, Meck WH, Swartzwelder HS. Prenatal dietary choline supplementation decreases the threshold for induction of long-term potentiation in young adult rats. J Neurophysiol. 1998;79:1790–1796. doi: 10.1152/jn.1998.79.4.1790. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Raedt R, Van Dycke A, Van Melkebeke D, De Smedt T, Claeys P, Wyckhuys T, Vonck K, Wadman W, Boon P. Seizures in the intrahippocampal kainic acid epilepsy model: characterization using long-term video-EEG monitoring in the rat. Acta Neurol Scand. 2009;119:293–303. doi: 10.1111/j.1600-0404.2008.01108.x. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Represa A, Niquet J, Charriaut-Marlangue C, Ben-Ari Y. Reactive astrocytes in the kainic acid-damage hippocampus have the phenotypic features of type-2 astrocytes. J Neurocytol. 1993;22:299–310. doi: 10.1007/BF01187128. [DOI] [PubMed] [Google Scholar]
- Revuelta M, Castano A, Venero JL, Machado A, Cano J. Long-lasting induction of brain-derived neurotrophic factor is restricted to resistant cell populations in an animal model of status epilepticus. Neuroscience. 2001;103:955–969. doi: 10.1016/s0306-4522(01)00032-x. [DOI] [PubMed] [Google Scholar]
- Ricceri L, Berger-Sweeney J. Postnatal choline supplementation in preweanling mice: sexually dimorphic behavioral and neurochemical effects. Behav Neurosci. 1998;112:1387–1392. [PubMed] [Google Scholar]
- Rocamora N, Massieu L, Boddeke HW, Palacios JM, Mengod G. Differential regulation of the expression of nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 mRNAs in adult rat brain after intrahippocampal injection of quinolinic acid. Brain Res Mol Brain Res. 1994;26:89–98. doi: 10.1016/0169-328x(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Rutten A, van Albada M, Silveira DC, Cha BH, Liu X, Hu YN, Cilio MR, Holmes GL. Memory impairment following status epilepticus in immature rats: time-course and environmental effects. Eur J Neurosci. 2002;16:501–513. doi: 10.1046/j.1460-9568.2002.02103.x. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, Sanderson KL, Voddi M, McIntosh TK. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol. 1997;147:418–427. doi: 10.1006/exnr.1997.6629. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Habor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sandstrom NJ, Loy R, Williams CL. Prenatal choline supplementation increases NGF levels in the hippocampus and frontal cortex of young and adult rats. Brain Res. 2002;947:9–16. doi: 10.1016/s0006-8993(02)02900-1. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR, Tandon P, Liu Z, Yang Y, Hori A, Holmes GL, Stafstrom CE. Multiple kainic acid seizures in the immature and adult brain: ictal manifestations and long-term effects on learning and memory. Epilepsia. 1997;38:1157–1166. doi: 10.1111/j.1528-1157.1997.tb01211.x. [DOI] [PubMed] [Google Scholar]
- Sartori CR, Pelagio FC, Teixeira SA, Valentinuzzi VS, Nascimento AL, Rogerio F, Muscara MN, Ferrari EA, Langone F. Effects of voluntary running on spatial memory and mature brain-derived neurotrophic factor expression in mice hippocampus after status epilepticus. Behav Brain Res. 2009 doi: 10.1016/j.bbr.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci. 2000;20:6144–6158. doi: 10.1523/JNEUROSCI.20-16-06144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Gray WP. Relevance of seizure-induced neurogenesis in animal models of epilepsy to the etiology of temporal lobe epilepsy. Epilepsia. 2007;48(Suppl 2):33–41. doi: 10.1111/j.1528-1167.2007.01065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Hen R. Neuroscience. Is more neurogenesis always better? Science. 2007;315:336–338. doi: 10.1126/science.1138711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Smith KL, Jackson MB, Goodman JH. Structural and functional asymmetry in the normal and epileptic rat dentate gyrus. J Comp Neurol. 2002;454:424–439. doi: 10.1002/cne.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- Scott BW, Wojtowicz JM, Burnham WM. Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp Neurol. 2000;165:231–236. doi: 10.1006/exnr.2000.7458. [DOI] [PubMed] [Google Scholar]
- Segi-Nishida E, Warner-Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc Natl Acad Sci U S A. 2008;105:11352–11357. doi: 10.1073/pnas.0710858105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LA, Wang L, Ribak CE. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia. 2008;49(Suppl 2):33–41. doi: 10.1111/j.1528-1167.2008.01491.x. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Jordan WH, Reams RY, Hall DG, Snyder PW. Temporal profile of clinical signs and histopathologic changes in an F-344 rat model of kainic acid–induced mesial temporal lobe epilepsy. Toxicol Pathol. 2008;36:932–943. doi: 10.1177/0192623308326093. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol. 2007;35:984–999. doi: 10.1080/01926230701748305. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B, Rao MS. Vulnerability of Hippocampal GABA-ergic Interneurons to Kainate Induced Excitotoxic Injury During Old Age. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2008.00675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Glutamic acid decarboxylase-67-positive hippocampal interneurons undergo a permanent reduction in number following kainic acid-induced degeneration of ca3 pyramidal neurons. Exp Neurol. 2001;169:276–297. doi: 10.1006/exnr.2001.7668. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Shetty GA. Hippocampal neurotrophin levels in a kainate model of temporal lobe epilepsy: a lack of correlation between brain-derived neurotrophic factor content and progression of aberrant dentate mossy fiber sprouting. J Neurochem. 2003;87:147–159. doi: 10.1046/j.1471-4159.2003.01979.x. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dichter MA, Rachinsky TL, Dean E, Goodman JH, Sollas AL, Martin DL. Basal expression and induction of glutamate decarboxylase and GABA in excitatory granule cells of the rat and monkey hippocampal dentate gyrus. J Comp Neurol. 1996;373:593–618. doi: 10.1002/(SICI)1096-9861(19960930)373:4<593::AID-CNE8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Smith PD, McLean KJ, Murphy MA, Turnley AM, Cook MJ. Seizures, not hippocampal neuronal death, provoke neurogenesis in a mouse rapid electrical amygdala kindling model of seizures. Neuroscience. 2005;136:405–415. doi: 10.1016/j.neuroscience.2005.07.055. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Hong NS, McDonald RJ, Wojtowicz JM. A role for adult neurogenesis in spatial long-term memory. Neuroscience. 2005;130:843–852. doi: 10.1016/j.neuroscience.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Somera-Molina KC, Robin B, Somera CA, Anderson C, Stine C, Koh S, Behanna HA, Van Eldik LJ, Watterson DM, Wainwright MS. Glial activation links early-life seizures and long-term neurologic dysfunction: evidence using a small molecule inhibitor of proinflammatory cytokine upregulation. Epilepsia. 2007;48:1785–1800. doi: 10.1111/j.1528-1167.2007.01135.x. [DOI] [PubMed] [Google Scholar]
- Sperk G, Furtinger S, Schwarzer C, Pirker S. GABA and its receptors in epilepsy. Adv Exp Med Biol. 2004;548:92–103. doi: 10.1007/978-1-4757-6376-8_7. [DOI] [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O. Kainic acid induced seizures: neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL. Age-dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420–432. doi: 10.1111/j.1528-1157.1993.tb02582.x. [DOI] [PubMed] [Google Scholar]
- Steiner B, Zurborg S, Horster H, Fabel K, Kempermann G. Differential 24 h responsiveness of Prox1-expressing precursor cells in adult hippocampal neurogenesis to physical activity, environmental enrichment, and kainic acid-induced seizures. Neuroscience. 2008;154:521–529. doi: 10.1016/j.neuroscience.2008.04.023. [DOI] [PubMed] [Google Scholar]
- Stringer JL. Repeated seizures increase GFAP and vimentin in the hippocampus. Brain Res. 1996;717:147–153. doi: 10.1016/0006-8993(96)00059-5. [DOI] [PubMed] [Google Scholar]
- Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–1150. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Hayashi S, Kakita A, Wakabayashi K, Fukuda M, Kameyama S, Tanaka R, Takahashi H, Nawa H. Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res. 1999;818:579–582. doi: 10.1016/s0006-8993(98)01355-9. [DOI] [PubMed] [Google Scholar]
- Tees RC, Mohammadi E. The effects of neonatal choline dietary supplementation on adult spatial and configural learning and memory in rats. Dev Psychobiol. 1999;35:226–240. [PubMed] [Google Scholar]
- Thomas JD, Abou EJ, Dominguez HD. Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicol Teratol. 2009;31:303–311. doi: 10.1016/j.ntt.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Biane JS, O’Bryan KA, O’Neill TM, Dominguez HD. Choline supplementation following third–trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behav Neurosci. 2007;121:120–130. doi: 10.1037/0735-7044.121.1.120. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garrison M, O’Neill TM. Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicol Teratol. 2004;26:35–45. doi: 10.1016/j.ntt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooyama I, Bellier JP, Park M, Minnasch P, Uemura S, Hisano T, Iwami M, Aimi Y, Yasuhara O, Kimura H. Morphologic study of neuronal death, glial activation, and progenitor cell division in the hippocampus of rat models of epilepsy. Epilepsia. 2002;43(Suppl 9):39–43. doi: 10.1046/j.1528-1157.43.s.9.10.x. [DOI] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci. 2000;1:191–198. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- Vezzani A. VEGF as a target for neuroprotection. Epilepsy Curr. 2008;8:135–137. doi: 10.1111/j.1535-7511.2008.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Ravizza T, Moneta D, Conti M, Borroni A, Rizzi M, Samanin R, Maj R. Brain-derived neurotrophic factor immunoreactivity in the limbic system of rats after acute seizures and during spontaneous convulsions: temporal evolution of changes as compared to neuropeptide Y. Neuroscience. 1999;90:1445–1461. doi: 10.1016/s0306-4522(98)00553-3. [DOI] [PubMed] [Google Scholar]
- Wagner AF, Hunt PS. Impaired trace fear conditioning following neonatal ethanol: reversal by choline. Behav Neurosci. 2006;120:482–487. doi: 10.1037/0735-7044.120.2.482. [DOI] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- Williams CL, Meck WH, Heyer DD, Loy R. Hypertrophy of basal forebrain neurons and enhanced visuospatial memory in perinatally choline-supplemented rats. Brain Res. 1998;794:225–238. doi: 10.1016/s0006-8993(98)00229-7. [DOI] [PubMed] [Google Scholar]
- Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, Dudek FE. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Goodrich SJ, Glenn MJ, Mellott TJ, Blusztajn JK, Meck WH, Williams CL. Spatial memory and hippocampal plasticity are differentially sensitive to the availability of choline in adulthood as a function of choline supply in utero. Brain Res. 2008a;1237:153–166. doi: 10.1016/j.brainres.2008.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Goodrich SJ, Mellott TJ, Glenn MJ, Blusztajn JK, Williams CL. Prenatal choline supplementation attenuates neuropathological response to status epilepticus in the adult rat hippocampus. Neurobiol Dis. 2008b;30:255–269. doi: 10.1016/j.nbd.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Wang JC, Han JL, Zhao G, Jiang W. Different effects of mild and severe seizures on hippocampal neurogenesis in adult rats. Hippocampus. 2008;18:460–468. doi: 10.1002/hipo.20409. [DOI] [PubMed] [Google Scholar]
- Yang Y, Liu Z, Cermak JM, Tandon P, Sarkisian MR, Stafstrom CE, Neill JC, Blusztajn JK, Holmes GL. Protective effects of prenatal choline supplementation on seizure-induced memory impairment. J Neurosci. 2000;20:RC109. doi: 10.1523/JNEUROSCI.20-22-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D, Lawlor PA, Leone P, Dragunow M, During MJ. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]
- Zeisel SH. Nutritional importance of choline for brain development. J Am Coll Nutr. 2004;23:621S–626S. doi: 10.1080/07315724.2004.10719433. [DOI] [PubMed] [Google Scholar]
- Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]