

Abstract
4-Hydroxy-2-trans-nonenal (HNE) is a lipid peroxidation product that contributes to the pathophysiology of several diseases with components of oxidative stress. The electrophilic nature of HNE results in covalent adduct formation with proteins, fatty acids and DNA. However, it remains unclear whether enzymes that metabolize HNE avoid inactivation by it. Glutathione transferase A4-4 (GST A4-4) plays a significant role in the elimination of HNE by conjugating it with glutathione (GSH), with catalytic activity toward HNE that is dramatically higher than the homologous GST A1-1 or distantly related GSTs. To determine whether enzymes that metabolize HNE resist its covalent adduction, the rates of adduction of these GST isoforms were compared and the functional effects of adduction on catalytic properties were determined. Although GST A4-4 and GST A1-1 have striking structural similarity, GST A4-4 was insensitive to adduction by HNE under conditions that yield modest adduction of GST A1-1 and extensive adduction of GST P1-1. Furthermore, adduction of GST P1-1 by HNE eliminated its activity toward the substrates 1-chloro- 2,4-dinitrobenzene (CDNB) and toward HNE itself. HNE effects on GST A4-4 and A1-1 were less significant. The results indicate that enzymes that metabolize HNE may have evolved structurally to resist covalent adduction by it.
Keywords: HNE, Oxidative stress, Protein adduct, Lipid peroxidation
Introduction
HNE1 is a highly reactive lipid peroxidation product of oxidative stress, and it plays a causal role in diseases involving progressive oxidative damage, including neurodegenerative diseases, atherosclerosis, cataracts, cancer, and rheumatoid arthritis [1,2]. HNE levels in blood plasma may rise from 0.3 to 0.7 μM under normal physiological conditions to 10 μM or higher during oxidative stress [3–5]. Importantly, in addition to its role in toxic processes, HNE is normally present in tissues and acts as a signaling molecule in signal transduction pathways [3,6]. As an electrophilic α–β unsaturated aldehyde, HNE readily forms covalent adducts with the nucleophilic protein residues cysteine, histidine and lysine [7,8]. HNE adducts with numerous proteins have been identified in in vitro models and uncertainty remains concerning which adducts are relevant to the progression of disease. In fact, it is clear that nearly every protein is chemically competent for HNE adduct formation to some degree, which makes it challenging to determine which protein targets are most important for the toxic effects of HNE [9–11].
The ability of HNE to adduct so many proteins reflects its lack of selectivity for the various nucleophiles present on their surfaces. Although the thiol nucleophile of Cys residues are modestly more reactive [8], it is clear that Lys and His side chains also attack the C3 carbon and the C1 aldehydic center of HNE [10–14]. In fact, after adduction of HNE at one electrophilic center, intramolecular ring closure may occur or a second protein nucleophile may react, resulting in a complex array of chemically distinct HNE adducts [10]. Importantly, even proteins with only a few or no cysteine residues are often good candidates for adduction by HNE.
The lack of specificity observed for HNE reactivity raises an interesting but underappreciated question regarding HNE protein targets: Do some proteins that participate in clearance of, or regulation by, HNE avoid adduction by it? We hypothesize that evolutionary pressures to combat the effects of oxidative stress could modify protein sequences, removing nucleophilic sites of HNE adduction. This would occur particularly in proteins where function is either most critical or is most efficiently destroyed by HNE. Proteins that participate in the detoxification of HNE would reasonably be expected to have evolved to avoid facile inactivation by HNE, and to test this hypothesis we have examined a series of homologous glutathione transferases (GSTs) to determine whether they exhibit functionally relevant differences in sensitivity to HNE adduction.
A major route of HNE elimination is GST-catalyzed adduction to glutathione (GSH), and the GST A4-4 isoform exhibits remarkable selectivity for and high catalytic efficiency toward HNE [15,16]. In contrast, the structurally homologous GST A1-1 isoform has relatively low catalytic activity toward HNE, but GST A1-1 is expressed at extremely high levels in hepatic tissue, where HNE levels tend to be high [17]. Other GST isoforms, such as GST P1-1, also show activity toward HNE but are not typically found in tissues exposed to high levels of oxidative stress [18]. An earlier study indicated that GST P1-1 loses catalytic activity upon adduction with HNE [19]. However, no previous studies have directly compared specific human GST isoforms and their kinetic susceptibility towards HNE adducts formation, thus precluding a detailed analysis of sequence differences that may have contributed to differences in sensitivity to HNE. Here, we examine in greater detail the differential reactivity of GST isoforms toward HNE adduct formation to examine whether those with highest catalytic activity may have evolved HNE “resistance”.
Materials and methods
Chemicals
HNE was purchased from Cayman Chemicals (Ann Arbor, MI). All other chemicals were purchased from Sigma–Aldrich (St. Louis, MO).
Protein expression, purification and quantification
The genes expressing human GST A1-1, A4-4 and P1-1 were subcloned into the vectors pKK233-3, pRB269 and pET17b, respectively, and used to transform E. coli for heterologous expression. GST A4-4 was expressed as a fusion protein with ubiquitin, and the gene for ubiquitinase, which was subcloned into the vector pRB173, was used to co-transform cells expressing GST A4-4. Protein was purified via GSH-agarose affinity chromatography as described previously [15], and purified protein underwent extensive dialysis against 10 mM Hepes pH 7.0 to remove GSH. Protein was ≥98% pure as determined by gel electrophoresis and whole-protein liquid chromatography mass spectrometry (LCMS). Protein concentration was determined using the Edelhoch method [20–22], and the molar absorptivity coefficients used for the folded dimer of protein were 46,147 ± 1.3% M−1 cm−1 for GST A1-1, 34,300 ± 0.9% M−1 cm−1 for GST A4-4, and 63,412 ± 1.6% M−1 cm−1 for GST P1-1.
Adduction of GSTs by HNE
To determine the time course of HNE adduction, the following solutions were combined and allowed to incubate up to 48 h at 22 °C (concentrations listed are final): 25 μM enzyme (concentrations given are for the dimer unless otherwise specified), 200 μM HNE, 1% EtOH, 10 mM Hepes, pH 7. At several time points, aliquots of the solution were removed and remaining HNE quenched with 10 mM cysteine. Positive controls for HNE adduction for all three isoforms were generated, and contained the following: 6 M GdnHCl, 5 mM HNE, 25% EtOH, 25 μM enzyme, 50 mM KPi, pH ~7. Control reactions were allowed to sit at room temperature for 24 h before quenching with 10 mM cysteine.
Samples for analyses of the effect of HNE adduction on metabolism were made by combining the following solutions and allowing to incubate at 22 °C (concentrations given are final): 25 μM enzyme, 200 μM HNE, 1% EtOH, 10 mM Hepes, pH 7. Control samples contained 1% EtOH but no HNE. After 3 h, remaining HNE was quenched with 10 mM cysteine. This concentration of cysteine was shown to inhibit further time-dependent adduction of the GSTs, based on MS (data not shown). Immediately after incubation, solutions were dialyzed against 100 mM potassium phosphate buffer (KPi), pH 6.5, using 10,000 Da molecular-weight-cut-off Slide-A-Lyzer dialysis cassettes (ThermoScientific, Waltham, MA) to remove cysteine, EtOH and HNE. After dialysis, all samples were centrifuged in a micro-centrifuge at 4 °C at 14,000 rpm for 10 min, and any precipitated protein was discarded. The concentration of each sample was determined by monitoring the absorbance at 280 nm using an Olis Modernized Aminco DW-2 spectrophotometer (Olis, Inc., Bogart, GA).
Quantitation of adduction by whole-protein LCMS
To determine the number of HNE adducts, adducted protein samples were injected onto a POROS R2 20 μm column (Applied Biosystems, Foster City, CA) and analyzed by LCMS using the continuum format of data collection on a Waters Micromass Synaption-mobility time-of-flight mass spectrometer (Waters Corporation, Milford, MA). The instrument scanned from 500 to 2200 m/z with a scan time of 0.5 s and a cone voltage of 20 V. A Waters Alliance 2690 high-pressure liquid chromatograph (HPLC, Waters Corporation, Milford, MA) provided a flow rate of 0.3 mL/min using 70% A (water, 0.1% TFA) and 30% B (ACN, 0.1% TFA) for the first 3 min of the run followed by a linear gradient over 3.5 min to 65% B, a linear gradient over 0.5 min to 100% B, equilibration at 100% B for 2 min, and a linear gradient to 30% B over 0.1 min with the run ending at 10.5 min.
The LCMS chromatogram for each sample in the time course was integrated over the same portion of the single chromatographic peak to generate the mass spectrum. Additionally, to account for differences in ionization efficiency between runs, the chromatographic peak area was used to normalize the deconvoluted spectrum. Mass spectra were deconvoluted from 800 to 2000 m/z using the Max-Ent I algorithm within the Waters Mass- Lynx v4.1 software package (Micromass Ltd., Milford, MA). The parameters for deconvolution were: 1 Da/channel resolution, uniform Gaussian damage model, 1 Da peak width at half height, iterated to convergence.
The deconvoluted peak areas for unadducted species were integrated, summed (all enzymes exist partly in the N-terminal methionine-cleaved form, so peak areas due to full-length and methionine-cleaved enzyme were added), normalized to 1 at t0, and fit to the following exponential equation:
| (1) |
where y = peak area; span = the peak area at t0 minus the minimum peak area; k = the rate of decrease; t = incubation time in hours and plateau = the minimum peak area.
Sample digestion and location of HNE adducts
GST A4-4 and P1-1 samples used for the determination of HNE adduct locations were prepared by incubating 100 μM GST (dimer) and 200 μM HNE in 10 mM Hepes, pH 7.3, 3% EtOH for 3 h at room temperature. A similar control sample lacking HNE was also prepared. The GST A4-4 sample used for determining the location of HNE adducts was prepared by incubating 200 μM GST A4-4 with 800 μM HNE in 10 mM HEPES, pH 7.3, 1.25% EtOH at −80 °C for at least two weeks. A similar control sample lacking HNE was also prepared. The samples were dialyzed against NaPi buffer as previously described.
GST A4-4 and P1-1 were reduced, alkylated and subjected to proteolytic digestion using an established protocol as described previously [23]. Briefly, the following were combined and allowed to incubate for 1 h at 37 °C: 200 μL of 0.5 mg/mL protein solution in 25 mMNH4HCO3, 70 mg urea, 15 μL of 1.5 M Tris–HCl (pH 8.8), and 5 μL of 200 mM tris-(2-carboxylethyl)phosphine. Next, 40 μL of 200 mM iodoacetamide to modify free cysteines were added, and the mixtures reacted for 1 h in darkness at room temperature. To quench all unreacted IAM, 40 μL of 200 mM dithiothreitol were added and the solutions incubated for 1 h at room temperature. 600 μL of 25mM NH4HCO3 were added to dilute the urea before addition of 200 μL of LCMS-grade methanol (MeOH). Sequencegrade trypsin (Promega, Madison, WI) was added for a final (w/w) protease:GST ratio of 1:50. The samples were incubated overnight at room temperature. The following morning, digested samples were evaporated to near dryness in a SpeedVac concentrator. To reduce the amount of NH4HCO3 in the sample, 200 μL H2O were added three times and evaporated. Desalting was performed using a MacroSpin C18 column (The Nest Group, Inc., Southborough, MA); digests were re-suspended in 50% ACN and 0.1% trifluoroacetic acid (TFA) and stored at −80 °C until analysis.
Peptide digests were analyzed by ESI + on a LTQ Orbitrap MS (Thermo Fisher, San Jose, CA) equipped with a nanoflow HPLC system (NanoAcquity; Waters Corporation, Milford, MA). A 5 μL injection of unmodified or modified peptides (0.1 μg/μL) was loaded onto a homemade pre-column (100 μm × 18 mm, 200 Ǻ, 5 μm; C18AQ; Michrom Bioresources, Inc., Auburn, CA) in water:ACN (95:5) with 0.1% (v/v) FA and eluted from a homemade analytical column (75 μm × 150 mm, 100 Ǻ, 5 μm; C18AQ; Michrom Bioresources, Inc.) at a flow rate of 250 nL/min using 95% A (water, 0.1% FA) 5% B (ACN, 0.1% FA) at the time of injection followed by a linear gradient over 55 min to 35% B, a linear gradient over 5 min to 85% B, a linear gradient over 5 min to 95% B, a linear gradient over 10 min to 5% B, and equilibration at 5% with the run ending at 90 min. Ion source conditions were optimized using the tuning and calibration solution recommended by the instrument provider. The analyses were performed using MS survey scans from m/z 400 to 2000 at a resolution of 60,000 (m/z 400) and ion populations of 5 × 105. For tandem mass spectrometry, ion population was set to 2 × 105, resolution to 7500, and a precursor isolation width of 4 m/z units. LTQ collision energy values of 40% were used. MS survey scans were used prior to data-dependent selection of 2+, 3+ and 4+ precursor masses. Data redundancy was minimized using dynamic exclusion where previously selected precursor ions (−0.1/+1.1 Da) were excluded for 45 s before additional fragmentation.
Acquired MS/MS spectra were searched against the International Protein Index (IPI) human protein database (v3.53) using SEQUEST [24,25] with the following search parameters: parent and fragment peptide mass tolerance of 1.5 and 1.0, respectively; trypsin digestion with C-terminal cleavage after residues K and R (except when followed by P), maximum number of internal cleavage sites = 2. The high mass accuracy of the LTQ Orbitrap was used during the search with the following residue modifications specified as variables: #1: 15.999 M (oxidized Met), 79.7663 STY (−PO3), 156.115 CHK (HNE Michael adduct); #2: 15.999 M, 156.115 HK, 57.02 C (alkylated); #3: 15.999 M, 57.02 C, 138.11 HK (Schiff-base) #4: 15.999 M, 57.02 C, 120.09 CHK (pyrrole adduct). PeptideProphet and ProteinProphet were used for statistical analysis of the SEQUEST results [24]. PeptideProphet probability filter of 0.9 was used corresponding to an error rate of <0.3%; ProteinProphet probability filter of 0.9 yielded an error rate of <0.02%.
Kinetics of CDNB metabolism
The kinetic parameters for CDNB metabolism were determined as described previously [26,27], and reactions were conducted in 100 mM KPi, pH 6.5 at 25 °C. GST concentrations were chosen such that 10% or less of the substrate was depleted over the course of the reaction.
Kinetics of HNE metabolism
To determine the kinetics of HNE metabolism by LCMS, reactions containing 10–150 μM HNE, 1 mM GSH and enzyme (or without enzyme to control for the spontaneous reaction) in 100 mM KPi, pH 6.5 were allowed to proceed for 30 s at 30 °C before quenching with 10 mM cysteine (quench solution also contained 0.5 μM midazolam as an internal standard). All reactions were performed in duplicate. To control for the spontaneous reaction, the amount of GSHNE formed spontaneously was subtracted from the amount of GSHNE formed enzymatically at each substrate concentration.
GST concentrations were chosen such that 10% or less of the substrate was depleted over the course of the reaction. For each sample, 50 μL of the quenched reaction mixture were injected onto an Altima C18 column (150 × 2.1 mm, Alltech Corp., Deerfield, IL) equipped with a C18 guard column (4 × 3 mm) and analyzed by single-ion recording LCMS using either a Waters Micromass Platform LCZ quadrupole or a Waters Micromass Quattro Micro API quadrupole (Waters Corporation, Milford, MA). Inline separations were performed with a Waters Alliance 2690 HPLC (Waters Corporation, Milford, MA) at 56% A (water, 0.1%) and 44% B (MeOH, 0.1% FA) for the first 3.5 min of the run followed by a linear gradient over 0.5 min to 65% B, a linear gradient over 0.1 min to 44% B, and equilibration at 44% B for the remaining 2.9 min of the run. The ionization mode was positive electrospray using a cone voltage of 20 V and a dwell time of 0.1 s for each of the two single-ions recorded: midazolam (m/z = 326.08) and GSHNE (m/z = 464.20). The source temperature was 120 °C, and the desolvation temperature was 350 °C. Data were analyzed using Waters MassLynx 4.0 (Micromass Ltd., Milford, MA). For each reaction, the amount of GSHNE formed was determined by integrating the area under the 464.20 m/z peaks and comparing that area to a standard curve generated by runs with 2, 4, 6, 8 and 10 μM purified GSHNE.
GSHNE synthesis, purification and quantitation
GSHNE for use as a standard was synthesized by incubating concentrated HNE with GSH in a 1:1 M ratio. An aliquot of HNE in 100% EtOH was dried on a rotovap, and a stoichiometric amount of 100 mM GSH was added and allowed to incubate for several hours. After synthesis, the product was purified from the starting material by reverse-phase HPLC using an Altima C18 column (150 ×2.1 mm, Alltech Corp., Deerfield, IL) and a Shimadzu Prominence HPLC (Columbia, MD). Since the molar absorptivity coefficient of GSHNE is small, fractions were collected based on the time they eluted from the column as determined by LCMS. Fractions were rotovapped to remove organic solvent. Quantification was performed using 2,4,6-trinitrobenzenesulfonic acid (TNBS), which reacts with primary amines to form a complex that absorbs at 335 nm [15,28]. Dilutions of the stock GSHNE solution as well as several known concentrations of GSH were derivatized with TNBS by incubating for 2 h at 42 °C with 0.00833% TNBS in the presence of 1.3% NaHCO3. Reactions were quenched with 0.11 M HCl. The absorbance was monitored using a Tecan Infinite M200 plate reader (Tecan Group Ltd., Männedorf, Switzerland), and the concentrations of the primary amines in the GSHNE solutions – and thus the concentration of GSHNE since it contains one primary amine – were determined by linear regression and comparison to the GSH standard curve.
Calculation of root-mean-square distances and electrostatic potential surfaces
The three-dimensional molecular viewer PyMOL (DeLano Scientific, LLC) was used to generate all crystal structure images shown. The program’s “align” function was used to calculate root-mean-square distances (RMSD) between crystal structures. PyMOL was also used to generate electrostatic potential surfaces.
Results
Quantitation of adduction by whole-protein LCMS
As shown in Fig. 1, denaturing GST A4-4 followed by the addition of molar excess HNE eliminates mass spectral peaks due to unadducted enzyme and produces peaks due to multiply adducted species. GSTs A1-1 and P1-1 also become multiply adducted when denatured and incubated with molar excess HNE (data not shown). This serves as proof of principle that HNE adducts all three isoforms of GST when nucleophilic residues are solvent exposed. As expected, all three isoforms are chemically competent for HNE adduction.
Fig. 1.

Deconvoluted spectra of natively folded GST A4-4 (dotted line) and after being denatured and incubated with 100-fold molar excess HNE (solid line). Mass shown is of the monomer. The major peaks are annotated according to the species they represent. The A4-4 isoform exists as both the N-terminal methionine-cleaved form, which is labeled “A4-Met,” and the full-length form, which is labeled “A4”.
To compare the relative susceptibility of GST isoforms to HNE adduction, native (i.e. folded) 25 μM GST A1-1, GST A4-4, and GST P1-1 were incubated in separate reactions with 200 μM HNE, and the mass spectral peak due to the unadducted enzyme disappeared with increasing incubation time as shown in Fig. 2. The apparent pseudo first-order rate constants and extent of adduction, based on the fits of the data to Eq. (1), are summarized in Table 1.
Fig. 2.
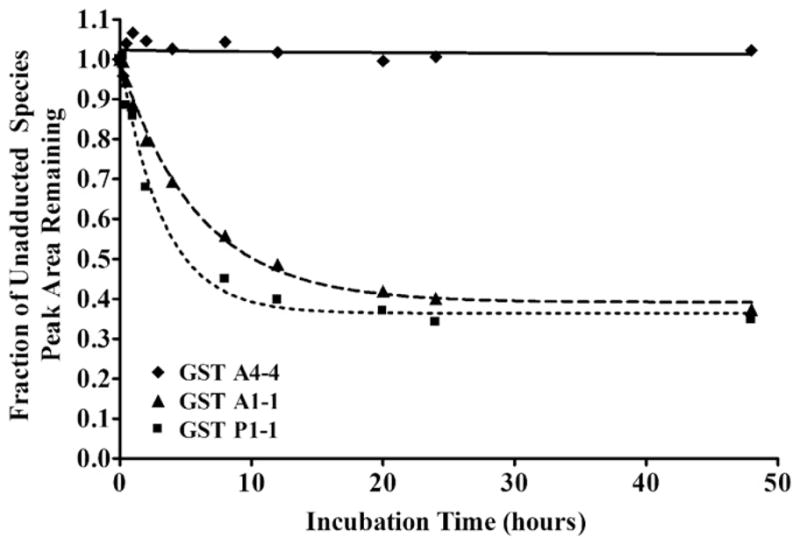
Change in peak areas of unadducted species upon incubation with HNE. Data for GST A4-4 (diamonds, solid line), GST A1-1 (triangles, dashed line) and GST P1-1 (squares, dotted line) were fit to Eq. (1). To normalize data, the peak areas due to the full-length and N-terminal methionine-cleaved unadducted forms of each enzyme were summed and set to 1 at t0.
Table 1.
Comparison of rates of HNE adduction.
| Isoform | Rate of disappearance of unadducted species (k, hour−1) | Minimum peak area of unadducted species (fraction of unadducted species remaining) |
|---|---|---|
| GST A4-4 | –a | –a |
| GST A1-1 | 0.17 ± 0.01 | 0.39 ± 0.01 |
| GST P1-1 | 0.31 ± 0.04 | 0.36 ± 0.02 |
Peak area did not decrease.
The unadducted species’ minimum peak area is not zero due to HNE depletion. Incubation conditions were optimized for a fast rate of reaction, which requires high concentrations of both enzyme and HNE, the solubility of HNE (the maximum concentration completely soluble in buffer with 1% ethanol is ~200 μM), a need for molar excess HNE to monitor possible multiple GST adductions, and the desirability of LCMS injections of ≥500 pmol monomer enzyme in an injection volume of not more than approximately 25 μL for good signal intensity and chromatography. For these reasons, the concentrations selected were 50 μM GST monomer (25 μM GST dimer) and 200 μM HNE. Since HNE may adduct a single molecule of enzyme more than once, HNE was depleted more rapidly than enzyme.
Several types of adducts were observed. HNE forms a Michael addition type of adduct that rapidly cyclizes when the nucleophilic attack occurs on C3, yielding a mass increase of 156 Da, and it forms a Schiff-base, yielding a mass increase of 138 Da, when the nucleophilic attack is by lysine or histidine and occurs on the carbonyl carbon [11,12,29]. Also, the Schiff-base form slowly and spontaneously rearranges to a pyrrole with a mass increase of 119 Da (Fig. 3) [13,14]. Additionally, since unreacted HNE was quenched with 10 mM cysteine for each time point, solvent exposed cysteine residues were able to form disulfide bonds with free cysteine from the quench solution, a point that will be further discussed later. GST P1-1 in particular showed peaks due to multiple adducts, and, because of the numerous permutations of adducts possible, peaks frequently were indistinguishable from one another. For example, the peak labeled “i” in Fig. 4C could arise from full-length GST P1 with one Schiff-base HNE adduct and one cysteine adduct (mass = 23,615 Da), from N-terminal methionine-cleaved GST P1 + 1 Michael addition HNE adduct + 2 pyrrole HNE adducts (mass = 23,619 Da), from N-terminal methionine-cleaved GST P1 + 2 Schiff-base HNE adducts + 1 pyrrole adduct (mass = 23,620 - Da) or from N-terminal methionine-cleaved GST P1 + 2 Schiff-base HNE adducts + 1 cysteine adduct (mass = 23,622 Da); the spectral resolution prevents differentiation of these possibilities.
Fig. 3.

The three forms of Michael adducts possible. “Nu” = the nucleophile of a cysteine, histidine or lysine residue. Schiff-base and pyrrole adducts only take place on lysines.
Fig. 4.
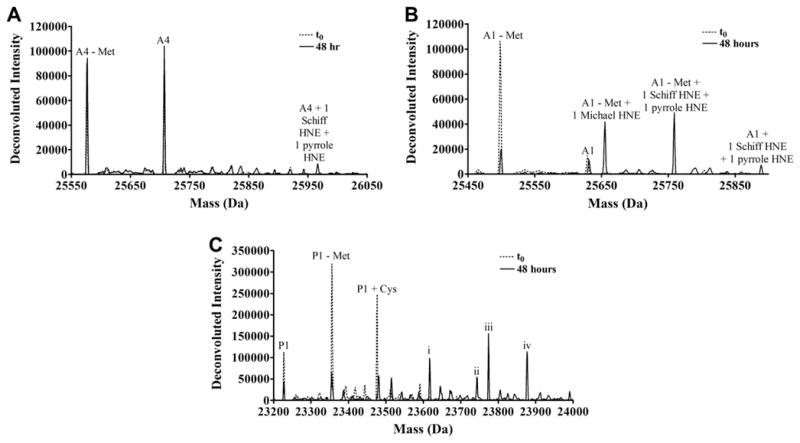
Deconvoluted spectra of (A) GST A4-4, (B) GST A1-1 or (C) GST P1-1 incubated with HNE at t0 (blue) and t = 48 h (black). All masses shown are for the monomer. The peaks due to a few of the most abundant species are labeled. GST P1-1 peaks i through iv could result from more than one type of adduct as described in the text.
GST A4-4 yielded the smallest number of peaks due to HNE adducts (three), GST A1-1 yielded five peaks, and GST P1-1 generated the largest number of peaks due to adducts (≥11), further suggesting that P1-1 is more susceptible to HNE adduction than GST A4-4.
Quantitative analysis of the rate at which specific HNE adducts formed based on the peak areas of the adducted species was not possible for several reasons. First, all adducted species likely do not ionize with identical efficiency, especially since the residues getting adducted are also the residues likely to carry a positive charge. It was not possible to generate standards for the various types of adduct because chromatographic separation was not possible. With 23, 28 and 18 possible sites of adduction for each monomer of GST A4-4, A1-1 and P1-1 respectively (Table 2), the permutations of possible adducts was massive. Second, adduction by more than one residue could result in a single, identical mass, e.g. nucleophilic attack on HNE to form a Michael addition adduct by any of the 24 lysines, three histidines or one cysteine in the monomer of GST A1 yields the same mass increase, but their absolute rates of adduction are unlikely to be identical.
Table 2.
Number of nucleophilic residues per monomer in GST A4-4, A1-1 and P1-1.
| Isoform | # His | # Lys | # Cys | Total nucleophilic residues |
|---|---|---|---|---|
| GST A4-4 | 6 | 17 | 0 | 23 |
| GST A1-1 | 3 | 24 | 1 | 28 |
| GST P1-1 | 2 | 12 | 4 | 18 |
However, despite these limitations, comparing the types of adducts observed throughout the time course is nonetheless qualitatively informative. For the A-class GSTs, the fraction of the overall peak area at each time point is greater for peaks due to a Schiff-base or pyrrole form of HNE adduct than for a Michael addition form of adduct. Since Schiff-base and pyrrole adducts can only form on lysine residues, lysine adducts constitute a larger percentage of the adducts present in a sample than cysteine or histidine adducts. In all three isoforms, the number of lysines is greater than the number cysteines or histidines (Table 2), and many lie on the surface of the protein.
For the P-class GST, the whole-protein LCMS data for the adducted species are inconclusive. Of the 11 peaks in the spectrum due to adducted species, only four are clearly due to only one type of adducted species, and all have at least one Michael adduct, so the rate of adduction on histidines and lysines compared to cysteines cannot be determined.
Location of HNE adducts
To identify sites of adduction, proteins were incubated with HNE, proteolytically digested and analyzed by LC–MS/MS, as described in the methods section. For GST A4-4, Michael addition forms of HNE adducts were found at His49, Lys64, His73 and Lys78. Again, it is essential to note that these were formed exceedingly slowly compared to adducts of GST P1-1 of GST A1-1.
For GST P1-1, Michael addition of HNE was found on Lys30, Lys55, Lys103 and Lys128, and Schiff-base HNE adducts were found on Lys30, Lys128, Lys191. Peptides unambiguously identified in the peptide maps included Cys48 and Cys102 (our numbering), residues that have been identified previously as sites of adduction [19], but found no adducts on those residues. Presumably these Cys residues were adducted to some degree but the adducted peptides were not recovered.
Attempts to identify the location of HNE adducts of GST A1-1 were unsuccessful for unknown reasons.
Kinetics of CDNB metabolism
To determine whether HNE adduction altered functional properties, steady-state catalytic experiments were performed with two substrates: CDNB and HNE itself.
Nearly all GSTs catalyze the aromatic substitution of glutathione for chlorine on CDNB, and thus it is used as the prototypical GST substrate for comparing catalytic efficiencies. For this reason, it was selected as one substrate to measure the effect of HNE adduction on catalysis. Normally, the efficiency with which the isoforms of interest catalyze this reaction is GST A1-1 >GST P1-1 »GST A4-4. HNE adduction affected the rate of metabolism of CDNB to differing extents for the three isoforms of GST. In terms of catalytic efficiency, GST P1-1 and A1-1 were most affected by HNE adduction, and GST A4-4 was affected the least. It should be stressed that the comparison in Table 3 is between GST isoforms subjected to identical exposure to HNE rather than between isoforms with identical degree (mol adduct/mol protein) of HNE adduction. It is not possible to prepare different GST isoforms with identical degrees of HNE adduction because of both sequence differences and the extreme differences in relative reactivity to HNE Table 4.
Table 3.
Kinetic parameters for CDNB metabolism.
| Isoform | Treatment | KM (mM) | Vmax (μmol min−1 mg−1) | kcat (s−1) | Catalytic efficiency (kcat/KM, s−1 mM−1) |
|---|---|---|---|---|---|
| GST A1-1 | Control | 0.31 ± 0.06 | 41 ± 3 | 35 ± 2 | 113 ± 24 |
| +HNE | 0.5 ± 0.2 | 31 ± 4 | 27 ± 4 | 58 ± 23 | |
| GST A4-4 | Control | 1.2 ± 0.1 | 6.3 ± 0.4 | 5.4 ± 0.4 | 4.4 ± 0.6 |
| +HNE | 1.4 ± 0.5 | 6 ± 1 | 5 ± 1 | 3.5 ± 1.4 | |
| GST P1-1 | Control | 1.7 ± 0.2 | 95 ± 8 | 74 ± 6 | 43 ± 7 |
| +HNE | 1.0 ± 0.2 | 30 ± 3 | 23 ± 2 | 24 ± 5 |
Table 4.
Kinetic parameters for HNE metabolism.
| Isoform | Treatment | KM (μM) | Vmax (μmol min−1 mg−1) | kcat (s−1) | Catalytic efficiency (kcat/KM, s−1 mM−1) |
|---|---|---|---|---|---|
| GST A1-1 | Control | 43 ± 25 | 14 ± 2 | 12 ± 2 | 270 ± 170 |
| +HNE | 24 ± 14 | 10 ± 2 | 9 ± 1 | 400 ± 200 | |
| GST A4-4 | Control | 7 ± 6 | 44 ± 7 | 37 ± 6 | 5000 ± 4500 |
| +HNE | 14 ± 8 | 28 ± 4 | 24 ± 3 | 1700 ± 1000 | |
| GST P1-1 | Control | 80 ± 10 | 21 ± 1 | 16 ± 1 | 200 ± 30 |
| +HNE | 14 ± 8 | 0.21 ± 0.03 | 0.16 ± 0.02 | 11 ± 6 |
Kinetics of HNE metabolism
For the two A-class GSTs, HNE adduction did not decrease the velocity of HNE metabolism to the same extent as for GST P1-1, which was greatly affected.
Discussion
It is well established that HNE forms covalent adducts in vitro with nearly any protein to which it is exposed, and in vivo studies have also shown the presence of HNE adducts on a large number of proteins as well [7,9,10,30]. This nonselective reactivity prompted us to hypothesize that enzymes that have evolved specifically to metabolize or transport HNE would be less susceptible to adduction in both the degree of adduction and the metabolic consequences. This possibility is particularly interesting in the context of GSTs, wherein isoforms that share the same canonical fold and are nearly identical when superimposed have dramatically different activities toward HNE. GST A4-4, which is often presumed to have evolved to clear long-chain lipid peroxidation products such as HNE, has a polypeptide backbone that deviates from that of GST A1-1 by less than 1 Å, yet GST A1-1 shows a catalytic efficiency with HNE that is an order of magnitude lower. Although GST A1-1 has relatively low activity toward HNE, it is expressed at very high levels in relevant tissues and could play a role in HNE clearance [17,31]. GST P1-1 also has comparatively lower activity with HNE than GST A4-4 and was used as a non-A-class control.
The differences in HNE adduction susceptibility were as striking as the differences in catalytic substrate selectivity. GST A4-4 is dramatically less susceptible to HNE adduction than either GST A1-1, to which it has a nearly identical three-dimensional structure, or GST P1-1, which shares the same GST canonical fold. Even after 48 h of incubation with HNE, GST A4-4 shows little adduction while GST A1-1 and especially GST P1-1 show a large number of adducts, most or all of the species being doubly or more adducted.
This difference in susceptibility to HNE adduction demands consideration of the molecular basis, especially for the case of GST A4-4 and GST A1-1. The difference is not simply due to a decrease in the number of nucleophilic sites, as all three isoforms compared herein contain multiple nucleophilic residues (Table 2), and an electrostatic surface for all three isoforms shows no striking difference (Fig. 5). However, an examination of the surface of each isoform (Fig. 6) shows that GST A4-4 appears to have a smaller number of nucleophilic residues present on its surface than GST A1-1. This may explain the difference in reactivity between these two isoforms. In general, the total number of nucleophilic residues for each isoform is similar, but for GSTA4-4, they appear to be less accessible than for GSTA1-1. An additional factor is likely to be the global protein dynamics, which are known to be different: GST A1-1 is globally more dynamic than GST A4-4, based on H/D-exchange mass spectrometry [27]. The greater solvent exposure of GST A1-1 likely allows for greater accessibility to HNE. In contrast, GST P1-1 does not have as many solvent exposed nucleophiles as GST A1-1, but it is clearly the most easily adducted. H/D-exchange data for GST P1-1 are not available, so it is impossible to compare the protein dynamics with the A-class isoforms on this basis. Importantly, these data on the differential dynamics of GSTA1-1 vs. GSTA4-4 are for the ligand free proteins. It is known from X-ray crystallography that GSTA4-4 is a rigid scaffold that does not undergo conformational rearrangement upon GSH binding, whereas GSTA1-1 exhibits local dynamic changes at its C-terminus. Thus, the very slight adduction of GSTA4-4 that we observed is unlikely to be reduced further in the presence of GSH. Possibly, GSTA1-1 is more resistant to HNE adduction with GSH bound than without. Thus, the degree of adduction we observed represents the upper limit of adduction expected in the presence of GSH. We found it impossible to directly determine the degree of HNE adduction in the presence of GSH because at the solubility limits of HNE and with GSH concentrations sufficiently high to saturate the enzyme (~100 μM), the GSH and HNE react and the HNE is depleted before any adducts are observed. In effect the GSH resulted in depletion of HNE.
Fig. 5.
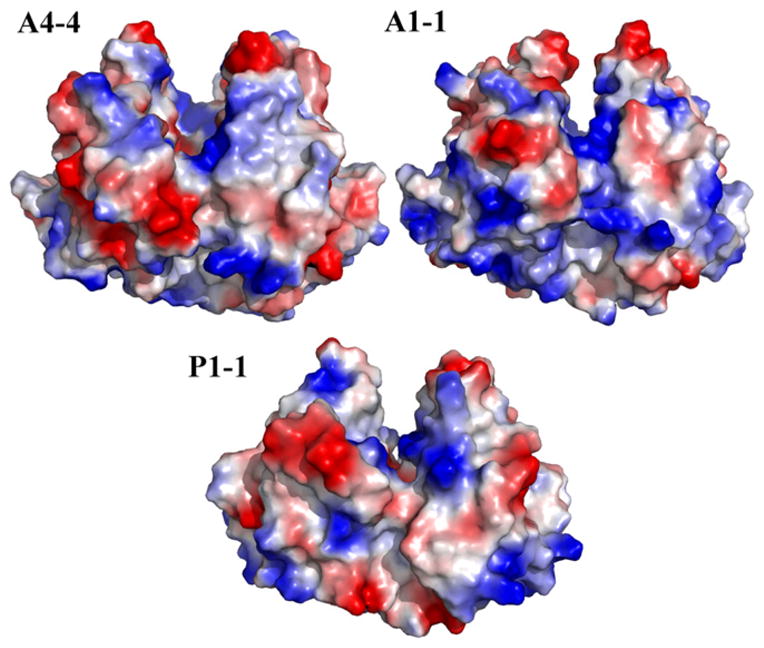
Electrostatic potential maps of GST A4-4, GST A1-1 and GST P1-1 with blue indicating the most positive electrostatic potential and red indicating the most negative electrostatic potential.
Fig. 6.

Surface representation of the crystal structures of GSTs A4-4 (PDB ID: 1GUL), A1-1 (PDB ID: 1K3L), and P1-1 (PDB ID: 9GSS) showing locations of nucleophilic residues cysteine (yellow), lysine (red with nitrogen atoms colored blue) and histidine (green with nitrogen atoms colored blue).
The molecular bases for the differences in catalytic effects due to HNE adduction are clearer. Work by others showed that HNE adduction of GST P1-1 Cys48 inhibited CDNB catalysis [19]. Cys48 lies directly beneath the highly flexible helix 2, and its adduction of thiol-reactive agents including HNE likely causes helix 2 to move into the glutathione-binding site (Fig. 7) [32]. In the same study, Cys102 was also found to be adducted but did not affect catalysis. The HNE adducts we found were all on lysine residues: Lys30, Lys103 and Lys128, and all were more distant from the active site than Cys48. The basis for the difference in detected adducts is not known, but it is clear that adduction of GST P1-1 has deleterious effects on catalytic activity. In contrast to GSTP1-1, the A-class GSTs have no reactive Cys residues near the active site, and in general their active sites are lined with non-nucleophilic hydrophobic residues. It is unlikely, therefore, that adduction within the HNE binding site would occur. One notable exception is the catalytic Tyr-9, which is well established to assist catalysis by deprotonating GSH. In the A-class GSTs Tyr-9 has a low pka and the resulting tyrosinate is nucleophilic. However, we found no evidence for adduction of this residue in either GSTA1-1 or A4-4, based on the insensitivity of their catalytic function to HNE exposure.
Fig. 7.

Ribbon schematic of one monomer from the crystal structures of GST A4-4 (PDB ID: 1GUL, left) and GST P1-1 (PDB ID: 9GSS, right) showing the location of HNE adducts identified in this study (red with side chains shown as sticks). GSH is shown as spheres (dark gray for C, red for O, blue for N, yellow for S). The catalytic tyrosine residues are shown in green with their side chains depicted as sticks. Portions of the sequence for which there was no coverage are shown in light blue.
In this work, cysteine-protein adducts, which were due to the cysteine used in the quench buffer forming disulfide bonds with cysteine residues on the protein, were found by whole-protein LCMS with GST P1-1 but not with the other two isoforms, showing that P1-1 has solvent-accessible cysteines while the A-class GSTs, under the conditions described here, do not. (Cysteine was added to quench unreacted HNE at each time point, so it did not compete with HNE for possible sites of adduction during the incubations). Studies with model peptides found that the rate of HNE adduction of nucleophilic residues was Cys » His > Lys [8], so the lack of cysteines in GST A4-4 may constitute an evolutionary protection against HNE adduction. While GST A1-1 does contain one cysteine per monomer, it is partially buried and apparently does not easily form disulfide bonds with free cysteine in solution, as evidenced by the lack of peaks due to a cysteine adduct. It is also likely to be less accessible to HNE for adduction than the solvent exposed cysteines in GST P1-1.
Whereas a previous study demonstrated that GST P1-1 is adducted by HNE with concomitant loss of catalytic activity, a direct comparison of the relative susceptibility of GST isoforms and the effects of HNE adduction on activity had not been reported. Differences in sequence and solvent accessibility prevent adduction of the three enzymes to identical extents and in identical locations, so a quantitative comparison of the effects of HNE adduction on catalytic function is impossible. However, this work demonstrates that upon exposure to HNE under identical conditions, the effect on catalysis is greatest for GST P1-1 and smallest for the A-class enzymes. These results suggest the possibility that enzymes that play a role in metabolism of HNE could evolve to resist HNE adduction without significant changes in overall structure. The dramatic difference in susceptibility to adduction of GST A1-1 and GST A4-4 provides a benchmark for the range of susceptibility even within a conserved canonical protein fold.
Footnotes
This work was supported by NIHGM 62284 (WMA) and T32GM 07752.
Abbreviations used: ACN, acetonitrile; CDNB, 1-chloro-2,4-dinitrobenzene; EtOH, ethanol; FA, formic acid; GSH, glutathione; GST, glutathione transferase; HEPES, 4-(2- hydroxyethyl)-1-piperazineethanesulfonic acid; HNE, 4-hydroxy-2-trans-nonenal; KPi, potassium phosphate; MeOH, methanol; NaPi, sodium phosphate; TFA, trifluoroacetic acid.
References
- 1.Poli G, Schaur RJ. IUBMB Life. 2000;50:315–321. doi: 10.1080/713803726. [DOI] [PubMed] [Google Scholar]
- 2.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. Med Res Rev. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 3.Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. Arch Biochem Biophys. 2008;477:183–195. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esterbauer H, Schaur RJ, Zollner H. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 5.Awasthi YC, Sharma R, Sharma A, Yadav S, Singhal SS, Chaudhary P, Awasthi S. Free Radic Biol Med. 2008;45:111–118. doi: 10.1016/j.freeradbiomed.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zarkovic N. Mol Aspects Med. 2003;24:281–291. doi: 10.1016/s0098-2997(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 7.LoPachin RM, Gavin T, Petersen DR, Barber DS. Chem Res Toxicol. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doorn JA, Petersen DR. Chem Res Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 9.Poli G, Biasi F, Leonarduzzi G. Mol Aspects Med. 2008;29:67–71. doi: 10.1016/j.mam.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 11.Guichardant M, Taibi-Tronche P, Fay LB, Lagarde M. Free Radic Biol Med. 1998;25:1049–1056. doi: 10.1016/s0891-5849(98)00149-x. [DOI] [PubMed] [Google Scholar]
- 12.Schaur RJ. Mol Aspects Med. 2003;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 13.Nadkarni DV, Sayre LM. Chem Res Toxicol. 1995;8:284–291. doi: 10.1021/tx00044a014. [DOI] [PubMed] [Google Scholar]
- 14.Sayre LM, Arora PK, Iyer RS, Salomon RG. Chem Res Toxicol. 1993;6:19–22. doi: 10.1021/tx00031a002. [DOI] [PubMed] [Google Scholar]
- 15.Balogh LM, Roberts AG, Shireman LM, Greene RJ, Atkins WM. J Biol Chem. 2008;283:16702–16710. doi: 10.1074/jbc.M801725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubatsch I, Ridderstrom M. Biochem J. 1998;330(Pt. 1):175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coles BF, Kadlubar FF. Methods Enzymol. 2005;401:9–42. doi: 10.1016/S0076-6879(05)01002-5. [DOI] [PubMed] [Google Scholar]
- 18.Hayes JD, Pulford DJ. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 19.van Iersel ML, Ploemen JP. Chem Biol Interact. 1997;108:67–78. doi: 10.1016/s0009-2797(97)00096-3. [DOI] [PubMed] [Google Scholar]
- 20.Edelhoch H. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 21.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 23.Nunn BL, Shaffer SA, Scherl A, Gallis B, Wu M, Miller SI, Goodlett DR. Brief Funct Genomic Proteomic. 2006;5:154–168. doi: 10.1093/bfgp/ell024. [DOI] [PubMed] [Google Scholar]
- 24.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 25.Eng JK, Fischer B, Grossmann J, Maccoss MJ. J Proteome Res. 2008;7:4598–4602. doi: 10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- 26.Habig WH, Pabst MJ, Jakoby WB. J Biol Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 27.Hou L, Honaker MT, Shireman LM, Balogh LM, Roberts AG, Ng KC, Nath A, Atkins WM. J Biol Chem. 2007;282:23264–23274. doi: 10.1074/jbc.M700868200. [DOI] [PubMed] [Google Scholar]
- 28.Sashidhar RB, Capoor AK, Ramana D, Immunol J. Methods. 1994;167:121–127. doi: 10.1016/0022-1759(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 29.Xu G, Liu Y, Sayre LM. J Org Chem. 1999;64:5732–5745. [Google Scholar]
- 30.Carini M, Aldini G, Facino RM. Mass Spectrom Rev. 2004;23:281–305. doi: 10.1002/mas.10076. [DOI] [PubMed] [Google Scholar]
- 31.Liang FQ, Alssadi R, Morehead P, Awasthi YC, Godley BF. Exp Eye Res. 2005;80:113–119. doi: 10.1016/j.exer.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 32.Wilce MCJ, Parker MW. Acta. 1994:1–18. doi: 10.1016/0167-4838(94)90086-8. [DOI] [PubMed] [Google Scholar]