

Abstract
Neural stem cells are a multipotent population of tissue-specific stem cells with a broad but limited differentiation potential. However, recent studies have shown that over-expression of the pluripotency gene, Oct4, alone is sufficient to initiate a process by which these can form ‘induced pluripotent stem cells’ (iPS cells) with the same broad potential as embryonic stem cells. This led us to examine the expression of Oct4 in endogenous neural stem cells, as data regarding its expression in neural stem cells in vivo are contradictory and incomplete. In this study we have therefore analysed the expression of Oct4 and other genes associated with pluripotency throughout development of the mouse CNS and in neural stem cells grown in vitro. We find that Oct4 is still expressed in the CNS by E8.5, but that this expression declines rapidly until it is undetectable by E15.5. This decline is coincident with the gradual methylation of the Oct4 promoter and proximal enhancer. Immunostaining suggests that the Oct4 protein is predominantly cytoplasmic in location. We also found that neural stem cells from all ages expressed the pluripotency associated genes, Sox2, c-Myc, Klf4 and Nanog. These data provide an explanation for the varying behaviour of cells from the early neuroepithelium at different stages of development. The expression of these genes also provides an indication of why Oct4 alone is sufficient to induce iPS formation in neural stem cells at later stages.
Keywords: methylation, Nanog, neural stem cells, Oct4, pluripotency
Introduction
Neural stem cells (NSCs) are a class of tissue-specific somatic stem cell. During normal development they produce all the cell types of the brain including numerous subtypes of neurons and glial cells. As stem cells, they also self-renew to maintain a population of progenitors capable of giving rise to a range of cell types throughout development and adulthood. However, it has become clear that the phenotype of these cells varies as the nervous system matures (Merkle & Alvarez-Buylla, 2006). Initially, the neuroepithelial cells of the neural tube and early brain can self-renew and are capable of forming all cell types of the CNS. By late foetal stages, only a smaller population of radial glial cells, derived from the earlier neuroepithelial cells, retain this capacity. Finally, in mature animals, astroglial cells, concentrated in the subventricular region of the lateral ventricles and the subgranular region of the hippocampus, are the main NSCs of the brain. Although these three stages of NSCs differ significantly in their morphology and gene expression, they all share a common lineage and the ability to self-renew and to give rise to multiple cell types. Despite these similarities, the types of cells produced when these NSCs give rise to later progenitors changes with time and there is evidence that this is due to cell autonomous changes in the NSCs themselves (Falk & Sommer, 2009; Kriegstein & Alvarez-Buylla, 2009). In addition, during early embryogenesis, the cells of the neuroepithelium undergo significant changes, most evident in their different growth requirements in vitro. When the neuroepithelium first forms, the cells cannot be isolated as neurospheres under standard culture conditions. At these stages cells can be grown as spheres, but like ES cells, they require Leukemia inhibitory factor (LIF). These ‘primitive NSCs’ not only exhibit features of NSCs, but also share other markers in common with embryonic stem cells (Hitoshi et al., 2004). Between E7.5 and E8.5, the cells of the neuroepithelium switch to a definitive NSC state when they can no longer grow in LIF but, like later NSCs, they now require fibroblast growth factor (FGF). There is then another transition to epidermal growth factor (EGF) dependence. It has been suggested that these sequential changes are, in part at least, under epigenetic control (Allen, 2008).
Recent studies have shown that pluripotency can be induced in many different somatic cell types by the over-expression of very few ‘pluripotency’ genes, in particular Sox2, Oct4 (Pou5f1), c-Myc and Klf4, to produce induced pluripotent stem cells (iPS) (Takahashi & Yamanaka, 2006; Wernig et al., 2007). NSCs are unusual in this respect, as they can be reprogrammed to iPS cells by the over-expression of Oct4 alone (Kim et al., 2009a,b;). It seems that active expression of these ‘pluripotency’ genes is central to the conversion of any cell to an iPS cell. To date, it appears that endogenous NSCs do, in fact, express Sox2 and c-Myc (Eminli et al., 2008). Indeed, there is evidence that they also express Klf4 (Kim et al., 2008, 2009b). However, there are few reports in which Oct4 expression has been analysed in NSCs and these provide conflicting evidence (Tropepe et al., 2001; Okuda et al., 2004; Lengner et al., 2007; Akamatsu et al., 2009; Chin et al., 2009; Takehara et al., 2009). It remains unclear, therefore, to what extent Oct4 is expressed in the developing CNS and in NSCs in particular.
In this paper, we have carried out a detailed analysis of the expression and methylation of Oct4 in NSCs derived from developing mouse embryos. We have also analysed the expression of the other key genes associated with pluripotency (Sox2, Klf4, c-Myc and Nanog).
Experimental procedures
Cell culture
Primary neurosphere cultures were established from the forebrain regions of CD1 mice (Charles Rivers, UK) and were maintained in growth medium comprising; Neurobasal medium (Gibco, Invitrogen, http://www.invitrogen.com), DMEM F12 (Gibco, Invitrogen), Penicillin/Streptomycin (Sigma, http://www.sigmaaldrich.com), B27 (1×) and N2 (1×) supplements (Gibco, Invitrogen), FGF (20 ng mL−1) and EGF (20 ng mL−1) (Invitrogen). All expression and methylation analyses were carried out on neurospheres at passages 3–5. The mouse embryonic stem cell line E14 IV was maintained in GMEM medium containing foetal calf serum (10%), non-essential amino acids (1×), LIF (1×), mercaptoethanol (0.1 mm), glutamine (2 mm) and sodium pyruvate (1 mm). The human neural stem/progenitor cell line ReN VM (Millipore, http://www.millipore.com) was cultured as neurospheres in growth medium or adherently on laminin (15 ng mL−1)-coated flasks in complete ReNcell NSC Maintenance Medium (Millipore), containing FGF (20 ng mL−1) and EGF (20 ng mL−1).
Reverse transcription PCR analysis
Total RNA was extracted from cell pellets using TRI reagent (Sigma) and digested with DNAse1 (Invitrogen) to prevent genomic DNA contamination. cDNA synthesis was carried out using random primers (Promega, http://www.promega.com) with SuperScript III Reverse Transcriptase (Invitrogen). PCR was carried out using Platinum Taq polymerase (0.04 U μL−1, Invitrogen), forward primer (1 μm), reverse primer (1 μm), PCR buffer (1×), MgCl2 (1.5 mm) and dNTP (0.8 mm). QPCR was carried out using Brilliant Sybr Green master mix (Stratagene, http://www.stratagene.com) on the MxPro 3005XP system. The QPCR reaction contained master mix (1×), Rox reference dye (300 nm), forward and reverse primers (150 nm each). Minus RT controls were routinely performed. Specific primers used are shown in Table 1. Beta actin was used as the loading control for all QPCR analysis.
Table 1.
RT-PCR primer sequences.
| Target gene/ref | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Oct4 primary PCR | TAGGTGAGCCGTCTTTCCAC | CTCGAACCACATCCTTCTCT |
| Oct4 secondary PCR | GTGAGCCGTCTTTCCACCAGG | TGATTGGCGATGTGAGTGAT |
| Oct4 QPCR | GTGAGCCGTCTTTCCACCAGG | GGGTGAGAAGGCGAAGTCTG |
| Nanog (Robertson et al., 2006) | ATGAAGTGCAAGCGGTGGCAGAAA | CCTGGTGGAGTCACAGAGTAGTTC |
| Sox2 (Takahashi & Yamanaka, 2006) | AAGTACACGCTTCCCGGAGGCTTG | AGTGGGAGGAAGAGGTAACCAC |
| c-Myc (Takahashi & Yamanaka, 2006) | GAGTGCATTGACCCCTCAGT | AATTCAGGGATCTGGTCACG |
| Gapdh | GGGTGGAGCCAAACGGGTC | GGAGTTGCTGTTGAAGTCGCA |
| Beta Actin | GTATGCCTCGGTCGTACCA | CTTCTGCATCCTGTCAGCAA |
| Clathrin | GACAGTGCCATCATGAATCC | TTTGTGCTTCTGGAGGAAAGAA |
Whole-mount in situ hybridization
Whole-mount in situ hybridization on mouse embryos was carried out using dioxygenin (DIG)-labelled riboprobes. Riboprobes were synthesized from plasmids that contained the target gene sequence using DIG-labelled nucleotides (Roche, http://www.roche.com) and RNA polymerase (Promega). The riboprobes were purified through G50 columns (GE healthcare, http://www4.gelifesciences.com). For hybridization, embryos were firstly treated with prewarmed 10 μg μL−1 proteinase K/PBST [phosphate-buffered saline (PBS) containing 0.1% Tween-20] at 37 °C with varying incubation times (15–30 min) depending on the size and the age of embryos and then fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. Embryos were prehybridized at 65 °C for at least 1 h and then the prehybridization solution was replaced with hybridization solution containing 1 : 200 to 1 : 600 dilution of riboprobes and incubated overnight at 65 °C. The embryos were sequentially washed in each diluted mix of hybridization buffer (75, 50 and 25% in 2× SSC) for 10 min at 65 °C. Further washes were carried out with 2× SSC containing 0.1% Tween-20 at 65 °C and 0.2× SSC containing 0.1% Tween-20 for 15 min at 65 °C (×4). Embryos were then sequentially washed at room temperature in dilutions (25, 50 and 75%) of MABT (100 mm maleic acid pH 7.5, 150 mm NaCl, 0.1% Tween-20) with 0.2× SSC. Secondary antibodies were preabsorbed by diluting in 2% Boehringer Blocking Regent™ and left at 4 °C with occasional shaking. The embryos were blocked with 2% Boehringer Blocking Regent™ (Roche) in MAB (100 mm maleic acid pH 7.5, 150 mm NaCl) at room temperature for at least 60∼90 min with gentle shaking. The blocking reagent was then replaced by 2% Boehringer Blocking Reagent™ containing diluted secondary anti-DIG-AP antibodies. All embryos were incubated at 4 °C overnight. The antibody solution was removed and embryos were washed eight times with MABT at room temperature for 15 min. Embryos were then stained with BM purple (Roche), until colour developed. The colour reactions were stopped by washing embryos in PBST with 20 mm EDTA (3×) at room temperature. Embryos were fixed in 4% paraformaldehyde in PBS for 20 min at room temperature and stored in 80% glycerol for imaging.
Immunocytochemistry
Immunohistochemical analysis was carried out using standard protocols and following manufacturer's recommendations. Cultured cells were grown on chamber slides, fixed in 4% paraformaldehyde-PBS for 15 min at room temperature and permeabilized in 0.2% Triton X-100-PBS (Sigma). Frozen sections of 12 μm were cut, placed onto slides and dried for 30 min at room temperature. Primary antibodies were mouse anti-OCT4 (SC-5279, Santa Cruz, http://www.scbt.com), rabbit anti-NANOG (ab21603, Abcam, http://www.abcam.com) and mouse anti-NESTIN (611658, BD Biosciences, http://www.bdbiosciences.com). Cells were incubated for 30–60 min in either 3% bovine serum albumin (BSA), 0.1% Triton X-100 or 10% sheep serum blocking solutions at room temperature and then incubated in 3% BSA with 1 : 100 primary antibody overnight at 4 °C. Cells were washed in PBS (3× 15 min each) and incubated in 3% BSA with 1 : 100 secondary antibody fluorescein-conjugated secondary antibody (Fl-1000 or Fl-2000, Vector, http://www.vectorlabs.com) for 1 h at room temperature. A final wash step in PBS (3× 15 min each) was carried out before mounting with 4,6-diamidino-2-phenylindole (DAPI) Vectashield (Vector).
Oct4 methylation analysis
Methylation analysis of the Oct4 promoter and proximal region was carried out using bisulphite sequencing and pyrosequencing. Genomic DNA was bisulphate-converted using the EZ DNA methylation kit (Zymo, http://www.zymoresearch.com). PCR was carried out as stated in Hattori et al. (2004), with the following modification: 200 nm forward primer (ProF 5′-TGG GTT GAA ATA TTG GGT TTA TTT), 200 nm reverse primer (Pro R 5′-CTA AAA CCA AAT ATC CAA CCA TA), 0.8 mm dNTP, 1.5 mm MgCl2, 1× PCR buffer and 0.05 U μL−1 Platinum Taq polymerase (Invitrogen). The PCR cycling condition was 94 °C for 10 min, followed by 45 cycles of 94 °C for 60 s, 55 °C for 60 s and 72 °C for 40 s, with a final 5-min extension at 72 °C. An amplicon of 530 bp was purified and cloned into pCRII TA cloning vector (Invitrogen) and sequenced. The Oct4 bisulphite PCR was modified for pyrosequencing. The target amplicon of 376 bp was amplified using forward primer (ProF1 5′-AGA GGG TGT AGT GTT AAT AGT T) and a biotinylated reverse primer (Pro R 5′-CTA AAA CCA AAT ATC CAA CCA TA). The biotin-labelled strand of the amplicon was isolated and pyrosequencing was carried out using three sequencing primers, F1 (SEQF1-5′-GTG TTA ATA GTT TTT GTG G), F2 (SEQF2-5′-AAG GGT TGT TTT GTT TAG A) and F3 (SEQF3-5′-GAG GGA GAG GTG AAA T). Pyrosequencing was carried out at Wolfson Sequencing services, University College London, UK.
Results
Oct4 gene expression in Mouse NSCs
As a first step in determining whether Oct4 is expressed during brain development, we used RT-PCR to analyse mouse brain tissue of different ages. Several pseudogenes and splice variants exist for Oct4 (Pou5F1) (Takeda et al., 1992; Pain et al., 2005; Mizuno & Kosaka, 2008) and therefore it was necessary to ensure that PCR products were amplified from the splice variant that has been shown to code for the full-length version of the Oct4 protein responsible for ‘stemness’ properties (Cauffman et al., 2006; Liedtke et al., 2007; Atlasi et al., 2008; Mizuno & Kosaka, 2008). All primers were therefore designed to include exon 1 and to span introns, and all PCR products were sequenced to verify that they were not derived from a pseudogene (the relationship of the primers used to the exons of Oct4 is shown in Fig. 1A). Of the tissues examined, Oct4 was only detected in brain tissue obtained from embryonic day 8.5 (E8.5) embryos, even when a second round of PCR cycles using nested primers was applied (Fig. 1B). As NSCs will progressively represent a smaller proportion of the cells present as the brain grows, the absence of PCR products when analysing older brain samples might be explained by the gradual decline in the proportion of NSCs in the samples being analysed. To determine whether Oct4 expression was indeed in the NSC population, we isolated NSCs from these various ages of brain and screened these using the same RT-PCR approach. The earliest stage we could analyse was E9, the first stage when cells of the CNS will grow under standard neurosphere culture conditions. This semi-quantitative RT-PCR showed Oct4 was robustly expressed in NSCs derived from embryos at E9, and was expressed at a much lower level, requiring a second round of PCR cycles with nested primers for its detection in NSCs derived from E13.5 embryos (Fig. 1C). By E15.5, expression was barely detectable and by P3, no product was detected. These data show that Oct4 is expressed at a readily detectable level at E8.5 but that expression in NSCs rapidly declines between E9 and E15.5. To verify this decline in expression and to gain insight into the level of Oct4 expression relative to ES cells, we next applied qRT-PCR. After relative quantification, the Oct4 expression in E9 NSCs was found to be 105-fold lower than that in ESCs. Thus, it seems that Oct4, a key marker of pluripotent cells, is expressed in neural stem cells during development but this expression decreases rapidly as the embryo matures.
Fig. 1.
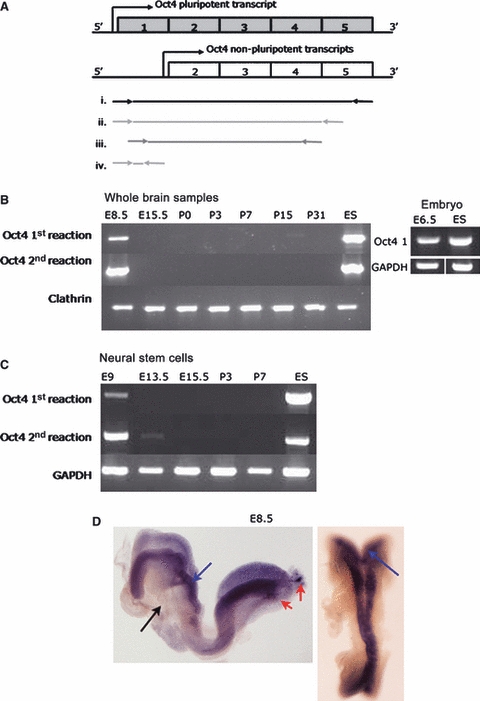
Oct4 expression analysis at embryonic and postnatal stages of brain and neural stem cell development. (A) Schematic representation of Oct4 transcripts. Only the transcript including exon 1 has been shown to play a central role in pluripotency. Primers shown below were therefore designed incorporating exon1. (i) Amplification of full length Oct4 transcript (used in E); (ii) Primary RT-PCR (used in B,C); (iii) Secondary nested RT-PCR (used in B,C); and (iv) QPCR primer locations. (B) RT-PCR from whole brain samples, except E6.5 which were whole embryos and ES cells as positive control. Products are shown from first PCR reaction and after a second nested PCR reaction. Clathrin was included as loading control for the amount of RNA in the reactions. (C) RT-PCR from neural stem cells, all populations used at passage 3–6, ES cells included as positive control. GAPDH included as loading control. (D) Whole mount in situ hybridization using a full length Oct4 riboprobe on E8.5 embyros. Oct4 expression identified in the neural plate (blue arrows) and PGC (red arrows), not in the non-neural tissue (black arrow). E, embryonic stage; P, postnatal stage.
To gain more detailed insight into the location of populations of cells expressing Oct4, whole-mount in situ hybridization was carried out on early mouse embryos. Expression was only detected at E8.5 when it was seen throughout the entire CNS (Fig. 1D). There was no striking variation in expression either rostrocaudally or mediolaterally, implying that, at this stage, all cells of the neuroepithelium expressed Oct4 to a similar extent.
Immunohistochemical analysis of Oct4 protein in mouse NSCs
To verify the expression of Oct4 in NSCs and because its subcellular distribution is critical to its function, we carried out immunofluorescence analysis on sections from embryos and neurospheres derived from different ages and regions of mouse brains. The Oct4 antibody used has been shown to bind to the region of Oct4 encoded by exon 1. This therefore provides an assay of the full-length protein that is involved in pluripotency and it is widely used in studies of Oct4 (Cauffman et al., 2006; Atlasi et al., 2008; Liedtke et al., 2008). Analysis of cryo-sections of embryos showed that, in addition to strong expression in the primordial germ cells (PGCs) of the genital ridge, expression of Oct4 could be weakly detected in the cytoplasm of neural plate cells in E8.5 and E9.5 embryos, consistent with RNA levels determined by RT-PCR and in situ hybridization (Fig. 2). We next investigated the expression and subcellular distribution of Oct4 in cultured neurospheres isolated from mouse brains of various ages. Oct4 protein was barely detectable in NSCs using immunofluorescence at E9.5, and was not observed in NSCs isolated from later developmental stages (data not shown). E8.5 neurospheres were not tested as these cells do not readily grow as neurospheres.
Fig. 2.
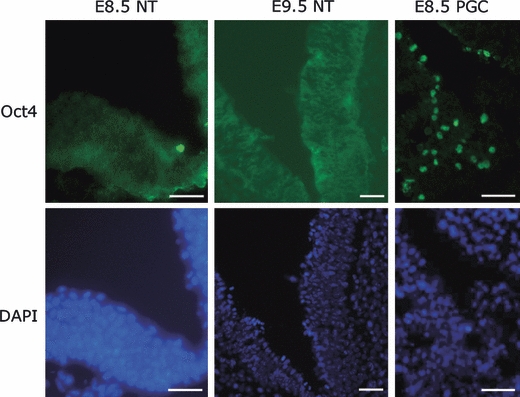
Oct4 immunoreactivity in mouse embyronic cranial neuroepithelium. Oct4 was identified in the E8.5 and E9.5 neural plate. Although this was predominantly cytoplasmic, some staining also seen in the nuclei. Staining in PGCs was predominantly nuclear.
Oct4 gene methylation in NSCs
Oct4 expression is known to be repressed by methylation of CpG dinucleotides within its promoter, the proximal enhancer and distal enhancer regions (Feldman et al., 2006; Li et al., 2007). It has also been shown previously that Oct4 expression can be induced by treatment of adult NSCs with the DNA methyltransferase inhibitor, 5-azacytidine and histone deacetylase inhibitor, TSA (Ruau et al., 2008). We hypothesized that the expression of Oct4 at very early stages of CNS development might be due to a reduced level of methylation and that loss of expression as the embryo matures would reflect gradual methylation. We therefore carried out a combination of bisulphite sequence analysis and pyrosequencing of CpGs within the promoter and proximal enhancer (shown schematically in Fig. 3A). ES cells were analysed as a control in which methylation is known to be generally absent.
Fig. 3.
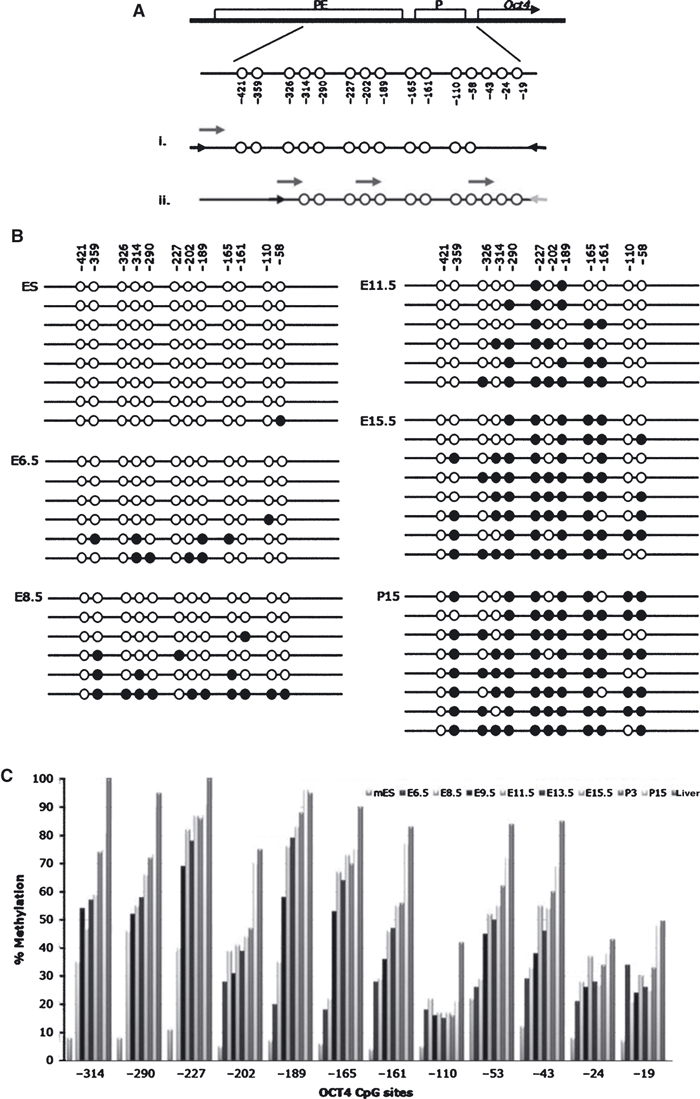
Oct4 DNA methylation analysis of neural stem cells at embryonic and postnatal stages. (A) Schematic representation of the CpG sites within the Oct4 promoter (P) and proximal enhancer (PE). CpGs covered by (i) bisulphite sequencing and (ii) pyrosequencing (locations of primers shown by arrows, PCR primers in black and short arrows above the product represents the sequencing primers). (B) Bisulphite sequencing results on ES cells, E6.5 head, E8.5 head, E11.5 NSCs, E15.5 NSCs and P15 NSCs. Open circles represent unmethylated CpGs, closed circle represent methylated CpGs, with each line representing one sequenced clone. (C) Pyrosequencing results showing percentage methylation at each CpG site from E6.5 head, E8.5 head to embryonic and postnatal NSCs. ES cells and liver used as controls for unmethylated and highly methylated, respectively.
Bisulphite sequencing of individual clones identified a gradual methylation of CpGs, from the unmethylated pattern in ES cells to the highly methylated pattern in P15 NSCs (Fig. 3B). This was confirmed by pyrosequencing, which allows a larger number of amplicons to be analysed, providing a reliable quantitative value for each sample (Fig. 3C). Both pyrosequencing and sequencing of individual bisulphate-treated clones revealed a similar gradual methylation from when Oct4 expression was detectable until it was absent by postnatal stages. It is interesting that both bisulphite and pyrosequencing analysis show that there is only a small increase in methylation after E8.5, but the level of Oct4 expression falls dramatically in NSCs from later stages, even though most CpG sites at the embryonic stages are still unmethylated. However, the changes in methylation are not uniform across all CpGs. The CpG sites at −165, −189 and −227 in particular are highly methylated in NSCs after embryonic stage E8.5.
The expression of other ‘pluripotency genes’ in mouse NSCs
In recent years a number of genes have been shown to be central to the ability of cells to behave as pluripotent stem cells. Other than Oct4, these include Sox2, c-Myc, Klf4 and Nanog. The expression of Sox2 is well established as a marker for NSCs, and c-Myc and Klf4 expression has been reported in NSCs (Kim et al., 2008, 2009b). However, Nanog expression has been reported to be negative in NSCs (Chambers et al., 2003; Kim et al., 2008, 2009b). We have shown that Oct4 was expressed in NSCs in early embryos and therefore we reanalysed the expression of these four genes in NSCs during mouse development. As anticipated, Sox2 and c-Myc and Klf4 all showed robust expression at all stages analysed (Fig. 4).
Fig. 4.
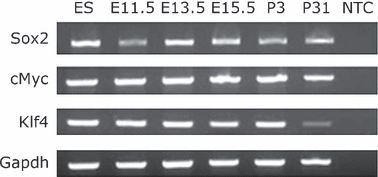
Sox2, c-Myc and Klf4 expression in embryonic and postnatal brain. RT-PCR results for brain samples, with positive control ES and NTC (no template control). GAPDH used as loading control for the amount of RNA in the reactions.
Analysis of Nanog is complicated by the presence of a number of pseudogenes and retrogenes. Therefore, to test whether Nanog was expressed in NSCs, a method of RT-PCR (Robertson et al., 2006) was utilized to discriminate between the pseudogenes (no amplification), retrogenes (band size shift and RsaI digestion) and the genuine transcript, with all PCR products sequenced. Nanog was shown to be expressed consistently in whole brain tissue at all developmental stages analysed and the retrogene NanogPd was also detected in samples of E15.5, P7 and P15 (Fig. 5A). Nanog was also detected in NSCs derived from embryonic, postnatal and mature mice (Fig. 5B). However, no expression of NanogPd was observed in any of the NSC samples.
Fig. 5.
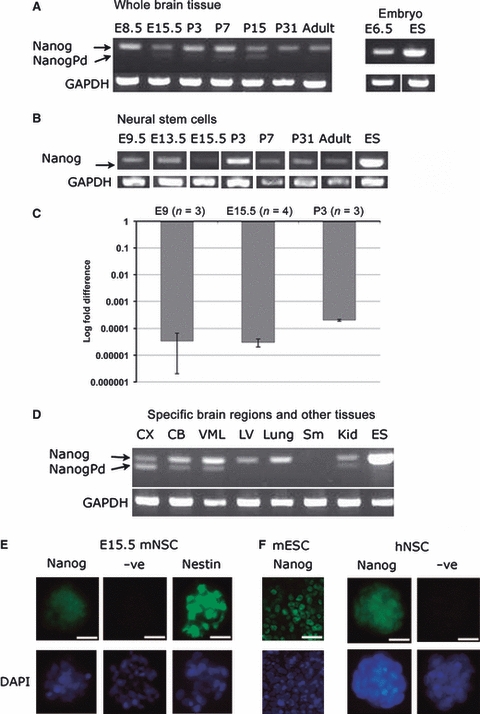
Analysis of Nanog expression and protein localization in neural stem cells. (A) RT-PCR results for whole brain samples showing amplification of Nanog (upper band) and NanogPd pseudogene (lower band). (B) RT-PCR amplification of Nanog and NanogPd showed only Nanog transcript was expressed in mouse neural stem cells. (C) Relative fold change of NANOG expression in neural stem cells compared to ES cells. Each bar represents the average fold change of three independent cultures of cells, with error bars showing SD. (D) RT-PCR results for specific brain regions and other tissues. In all RT-PCR reactions, ES cells were used as the positive control and GAPDH as a loading control. (E) Immunostaining of mouse E15.5 NSC with Nanog, negative control of secondary antibody only and Nestin as a positive control. (F) Nanog immunostaining of human NSCs (hNSCs) with mouse ES (mESC) cells as positive control for nanog expression.
We next used qRT-PCR to determine the level of Nanog transcripts present. Although we were able to detect Nanog RNA in all the tissues we analysed, the level was 104 lower relative to ES cells (Fig. 5C).
Because the above data suggested that Nanog transcripts were present in NSCs at a similar, but very low, level throughout development and adulthood, we also analysed a range of other tissues to determine whether this low level was a general feature of many tissues. Nanog was indeed detected in lung and kidney tissue, but not in stomach (Fig. 5D). Kidney also expressed NanogPd. More specific adult brain regions were also analysed and both Nanog and NanogPd were expressed in all regions apart from the lateral ventricles, where only Nanog was expressed (Fig. 5D). RsaI digests of the Nanog PCR product confirmed that another retrogene, NanogPc, was not detected in any of the samples.
Immunohistochemical analysis of Nanog in NSCs
To determine whether the very low levels of Nanog transcripts we detected might be of significance, we analysed NSCs for the presence of the Nanog protein. The Nanog antibody used in this study has been shown by other groups to act as a marker for pluripotent cells (Storm et al., 2007; Shi et al., 2008; Singh et al., 2008; Silva et al., 2009). Our analysis demonstrated the presence of Nanog protein in the nuclei of NSCs (Fig. 5E). To determine whether the same might be true of human NSCs, we analysed the human REN-VM NSC line, which has been shown to retain all the characteristics of an NSC; self-renewal and the ability to differentiate into glia and neurones (Donato et al., 2007). This cell line was derived from foetal human ventral mesencephalon brain tissue. Our analysis using immunocytochemistry demonstrated clear expression of Nanog in both the cytoplasm and nuclei of these cells (Fig. 5F).
Discussion
Pluripotency is a feature of only very restricted populations of cells in the early vertebrate embryo. These cells are marked out by the expression of a number of ‘pluripotency genes’ that, when over-expressed in somatic cells, can initiate a process through which some of those cells can become pluripotent, generating so-called iPS cells. Although none of the genes alone provides a mark unique to endogenous pluripotent cells, the combined expression of the full set of genes is regarded as a unique feature of such cells. Our data demonstrate that the cells of the CNS express several key genes typical of pluripotent stem cells, but Oct4 expression is rapidly lost as the CNS matures, coincident with methylation of its promoter and proximal enhancer regions.
The expression of pluripotency genes in neural stem cells
Expression of Sox2 and c-Myc in the developing CNS and NSCs is already well-described (Hirvonen et al., 1990; Graham et al., 2003; Eminli et al., 2008; Kerosuo et al., 2008; Kim et al., 2008) and Klf4 expression has recently been demonstrated in both mouse and human NSCs (Kim et al., 2008, 2009a,b).
Because of the problem of alternative transcripts and pseudogenes, there is little definitive evidence regarding the expression of Oct4 in NSCs. We have used RT-PCR to show that full-length Oct4 expression is maintained in the neural ectoderm beyond E8.5, when definitive NSCs are present, albeit at lower levels than in ES cells. Although our data suggested that Oct4 was predominantly localized to the cytoplasm, it was not excluded from the nucleus, so a functional role as a transcription factor remains possible. It may not be surprising that Oct4 protein is hard to detect despite significant levels of its transcript, as Oct4 protein has been shown to be processed by ubiquitination and rapidly turned over (Saxe et al., 2009; Wagner & Cooney, 2009).
Our observation that Nanog is expressed in the neuroepithelium and NSCs throughout development is contrary to the interpretation of most previous studies, but it does not in fact contradict their data. Previous reports have generally regarded levels of Nanog that are over 1000 times lower than that seen in ES cells, as negative. However, despite the fact Nanog RNA was present at a level 105-fold lower than in ES cells, this level was readily detectable in cells from several tissues. In addition, the presence of Nanog RNA in NSCs was reflected in the presence of nuclear Nanog protein (Fig. 4). Indeed, a recent study by Kim et al. (2009a) showed that endogenous Nanog expression in a foetal human NSC population was only approximately 14-fold lower than an ES cell line.
Overall it seems that early NSCs express all four ‘pluripotency’ genes but that Oct4 and Nanog are present at very low levels compared to ES cells and Oct4 is generally located in the cytoplasm.
Oct4 methylation
It is well-established that methylation is a common mechanism for the silencing of Oct4 expression. In the mouse embryo as a whole, Oct4 is unmethylated at the blastula stage and undergoes de novo methylation by E6.5, remaining modified at similar levels as development proceeds (Gidekel & Bergman, 2002). Our report is the first study to show that the Oct4 gene is progressively methylated during the in vivo maturation of NSCs in the neuroepithelium of the CNS, coincident with downregulation of its expression. Our data suggest that methylation plays a significant role in the decreasing expression Oct4 as the CNS matures.
It is notable, however, that not all CpGs analysed were methylated to the same extent. A core area of CpGs from −161 to −290 shows the most dramatic change from low methylation at E8.5 to high methylation after E8.5 (in particular CpGs −165, −189 and −227) and it is therefore possible that it is the methylation of these CpGs that is primarily responsible for the silencing of gene expression.
The functional significance of Oct4 expression in neural stem cells
As NSCs are clearly not pluripotent, what is the significance of the expression of the genes that we have described? Why is Oct4 still expressed in neural tissue at early embryonic stages and not immediately switched off when cells become neural precursors? One possibility is that a low level of Oct4 is necessary to regulate developmental events. Gerrard et al. (2005) analysed OCT4 expression as human ES cells were differentiated into neurones in vitro (Gerrard et al., 2005). They found that the level of OCT4 transcripts declined slowly compared to the decline of other hESC markers, and inhibition of OCT4 expression resulted in differentiation to extra-embryonic endoderm-like cells.
Although Oct4 is expressed in NSCs at early stages of development, this expression is low and the protein is predominantly in the cytoplasm. This brings into question the functional significance of this expression. Indeed, inactivation of Oct4, driven by Nestin-cre, had no discernible effect on brain development, suggesting that it is certainly not required for the basic features of NSCs once they are formed (Lengner et al., 2007). However, as Nestin is only robustly expressed from E8 in NSCs (Lothian & Lendahl, 1997; Kawaguchi et al., 2001), and it would then take time for the Nestin-cre used by Lengner et al. (2007) to be expressed and Oct4 mRNA and protein levels to decline, it seems likely that the loss of Oct4 would have occurred only a little before the time we have seen its loss endogenously. Its role in the very early stages of NSC formation remains to be tested.
The higher levels of Oct4 expression at the earlier stages of NSC development may explain observed differences in behaviour of the cells of the neural plate from E7.5 to E9.5. During this period, there is a change in the conditions under which cells of the neuroepithelium can be cultured in vitro. Prior to E8.5, cells from the neuroepithelium require LIF rather than EGF or FGF to be grown in culture, and these cells have been named ‘primitive NSCs’ (Hitoshi et al., 2004). These primitive NSCs not only exhibit features of neural stem cells, such as Sox1 and Nestin expression, but also have features in common with stem cells capable of forming other cell lineages. A role of Oct4 in this progression is suggested from the data of Akamatsu et al. (2009), who studied the development of NSCs derived from ES cells in culture. They found that Oct4 expression was increased in mice lacking the orphan nuclear receptor ‘germ cell nuclear factor’ gene (GCNF) and that primitive NSCs were inhibited from undergoing the transition to definitive NSCs. This block could be rescued by small interfering (si)RNA inhibition of Oct4 (Akamatsu et al., 2009). These data suggest that Oct4 may indeed be important in controlling the transition from primitive NSC to definitive NSC. A similar Oct4-positive population of cells was isolated by Takehara et al. from P0 mice, but this was achieved by growing cells in medium designed for ES cells, not for NSCs. Hence, it seems that a primitive population of cells may exist as late as birth, although it is not clear from their study if this primitive state was induced in cells by their unusual culture conditions.
Our data show that, even later in development, NSCs express three of the four ‘Yamanaka’ factors (Oct4 now being absent). This is likely to explain why NSCs are more readily reprogrammed to iPS cells than other somatic cell types (Eminli et al., 2008; Kim et al., 2009a,b;). Addition of Oct4 alone is sufficient to initiate reprogramming such that some of those cells convert to cells exhibiting full pluripotency, with increased expression of Nanog and other pluripotency genes as a consequence (Kim et al., 2009a,b;).
Oct4 and the origins of teratomas
A key assay of pluripotency is the ability of cells to form a teratoma (a tumour made up of components from all three embryonic germ layers) when transplanted into mice. Teratomas are in fact one of the most common forms of tumour seen in humans prenatally and in neonatal infants. Within the brain, teratomas make up about 50% of tumours diagnosed before or at birth, whereas they represent only a tiny proportion of brain tumours diagnosed after the first few months of life.
Although these tumours are classified as germ cell tumours, believed to arise from misplaced germ cell progenitors, we have hypothesized that they arise form endogenous progenitor cells in the developing brain. The data presented here provide a plausible explanation for their appearance so early in life and support this alternative hypothesis for their origins. As discussed above, simply over-expressing Oct4 in NSCs can be sufficient to convert them to a pluripotent state capable of forming a teratoma. We have shown that Oct4 expression is less robustly repressed in the NSCs of the early brain than at later stages and that this is coincident with methylation of its promoter. It therefore seems feasible that Oct4 expression would be more readily reactivated by demethylation at such early stages than at later stages. As intracranial teratomas normally present at birth and appear to develop at the expense of much of the brain's own normal development, they are likely to be initiated early during development consistent with the time when Oct4 methylation in NSCS is lowest.
Reactivation of Oct4 by demethylation would also be consistent with a role for global hypomethylation as an early event in cancer. Indeed, the earlier in life that a tumour forms, the less likely it is that its development is driven by a series of mutational events as seen in most adult cancers. Activation of key genes due to global epigenetic events is a strong alternative model for the mechanism by which these unusual tumours are initiated. Consistent with such a hypothesis, in contrast to virtually every other type of cancer examined, teratomas rarely exhibit any detectable cytogenetic abnormality.
However, high levels of Oct4 expression have not been described in these tumours. This is not surprising for two reasons. First, because such tumours go on to exhibit widespread differentiation into a range of different tissue types, any stem cell-like cells will represent a very small proportion of the tumour mass. Secondly, as in iPS cells, where over-expression of Oct4 is an initiating event for pluripotency after which its expression is down-regulated, any increase in Oct4 expression could be transient.
We would therefore propose a model in which rare disruption of global methylation in an early NSC could result in up-regulation of Oct4 expression (along with other aberrant gene expression) and so trigger conversion to pluripotency, resulting in teratoma formation. This would be restricted to the earliest stages of brain development, when repression of Oct4 by methylation is not yet complete, explaining the restriction of teratomas to the first few months of human life.
Summary
We have shown that Oct4 is initially expressed as both RNA and protein in NSCs during early embryogenesis, but the level of expression drops rapidly from E8.5 to become undetectable by E15.5. This loss of Oct4 expression is coincident with gradual methylation of its promoter and proximal enhancer region. Alongside the expression of other key genes associated with pluripotency this may explain the plasticity and other behavioural differences between NSCs at the different stages of CNS development and may explain why intracranial teratomas arise almost exclusively prior to birth.
Acknowledgments
V.A. was supported by The Samantha Dickson Brain Tumour Trust. J.N.J. was supported by Ali's Dream, Charlie's Challenge and the Children's Brain Tumour Research Centre. Thanks to Val Wilson for supplying the mouse ES cell line.
Author contribution
Shih-Han Lee: conception and design, collection and/or assembly of data, data analysis and interpretation, final approval of manuscript. Jennie N. Jeyapalan: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript. Vanessa Appleby: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript. Dzul Azri Mohamed Noor: collection and/or assembly of data. Virginie Sottile: conception and design, final approval of manuscript. Paul J. Scotting: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
References
- Akamatsu W, DeVeale B, Okano H, et al. Suppression of Oct4 by germ cell nuclear factor restricts pluripotency and promotes neural stem cell development in the early neural lineage. J Neurosci. 2009;29:2113–2124. doi: 10.1523/JNEUROSCI.4527-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen ND. Temporal and epigenetic regulation of neurodevelopmental plasticity. Philos Trans R Soc Lond B Biol Sci. 2008;363:23–38. doi: 10.1098/rstb.2006.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlasi Y, Mowla SJ, Ziaee SAM, et al. OCT4 spliced variants are differentially expressed in human pluripotent and nonpluripotent cells. Stem Cells. 2008;26:3068–3074. doi: 10.1634/stemcells.2008-0530. [DOI] [PubMed] [Google Scholar]
- Cauffman G, Liebaers I, Van Steirteghem A, et al. POU5F1 isoforms show different expression patterns in human embryonic stem cells and preimplantation embryos. Stem Cells. 2006;24:2685–2691. doi: 10.1634/stemcells.2005-0611. [DOI] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, et al. Functional expression cloning of nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Chin J-H, Shiwaku H, Goda O, et al. Neural stem cells express Oct-3/4. Biochem Biophys Res Commun. 2009;388:247–251. doi: 10.1016/j.bbrc.2009.07.165. [DOI] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, et al. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. Bmc Neurosci. 2007;8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eminli S, Utikal J, Arnold K, et al. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26:2467–2474. doi: 10.1634/stemcells.2008-0317. [DOI] [PubMed] [Google Scholar]
- Falk S, Sommer L. Stage- and area-specific control of stem cells in the developing nervous system. Curr Opin Genet Dev. 2009;19:454–460. doi: 10.1016/j.gde.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Feldman N, Gerson A, Fang J, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol. 2006;8:188–U55. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]
- Gerrard L, Zhao DB, Clark AJ, et al. Stably transfected human embryonic stem cell clones express OCT4-specific green fluorescent protein and maintain self-renewal and pluripotency. Stem Cells. 2005;23:124–133. doi: 10.1634/stemcells.2004-0102. [DOI] [PubMed] [Google Scholar]
- Gidekel S, Bergman Y. A unique developmental pattern of Oct-3/4 DNA methylation is controlled by a cis-demodification element. J Biol Chem. 2002;277:34521–34530. doi: 10.1074/jbc.M203338200. [DOI] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, et al. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- Hattori N, Nishino K, Ko Y, et al. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J Biol Chem. 2004;279:17063–17069. doi: 10.1074/jbc.M309002200. [DOI] [PubMed] [Google Scholar]
- Hirvonen H, Makela TP, Sandberg M, et al. Expression of the MYC protooncogenes in developing human fetal brain. Oncogene. 1990;5:1787–1797. [PubMed] [Google Scholar]
- Hitoshi S, Seaberg RM, Koscik C, et al. Primitive neural stem cells from the mammalian epiblast differentiate to definitive neural stem cells under the control of Notch signaling. Genes Dev. 2004;18:1806–1811. doi: 10.1101/gad.1208404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi A, Miyata T, Sawamoto K, et al. Nestin-EGFP transgenic mice: visualization of the self-renewal and multipotency of CNS stem cells. Mol Cell Neurosci. 2001;17:259–273. doi: 10.1006/mcne.2000.0925. [DOI] [PubMed] [Google Scholar]
- Kerosuo L, Piltti K, Fox H, et al. Myc increases self-renewal in neural progenitor cells through Miz-1. J Cell Sci. 2008;121:3941–3950. doi: 10.1242/jcs.024802. [DOI] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, et al. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Greber B, Arauzo-Bravo MJ, et al. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009a;461:649–643. doi: 10.1038/nature08436. [DOI] [PubMed] [Google Scholar]
- Kim JB, Sebastiano V, Wu GM, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009b;136:411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner CJ, Camargo FD, Hochedlinger K, et al. Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell. 2007;1:403–415. doi: 10.1016/j.stem.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-Y, Pu M-T, Hirasawa R, et al. Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in the methylation of Oct4 and Nanog. Mol Cell Biol. 2007;27:8748–8759. doi: 10.1128/MCB.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke S, Enczmann J, Waclawczyk S, et al. Oct4 and its pseudogenes confuse stem cell research. Cell Stem Cell. 2007;1:364–366. doi: 10.1016/j.stem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Liedtke S, Stephan M, Kogler G. Oct4 expression revisited: potential pitfalls for data misinterpretation in stem cell research. Biol Chem. 2008;389:845–850. doi: 10.1515/BC.2008.098. [DOI] [PubMed] [Google Scholar]
- Lothian C, Lendahl U. An evolutionarily conserved region in the second intron of the human nestin gene directs gene expression to CNS progenitor cells and to early neural crest cells. Eur J Neurosci. 1997;9:452–462. doi: 10.1111/j.1460-9568.1997.tb01622.x. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Alvarez-Buylla A. Neural stem cells in mammalian development. Curr Opin Cell Biol. 2006;18:704–709. doi: 10.1016/j.ceb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Mizuno N, Kosaka M. Novel variants of Oct-3/4 gene expressed in mouse somatic cells. J Biol Chem. 2008;283:30997–31004. doi: 10.1074/jbc.M802992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Tagawa K, Qi ML, et al. Oct-3/4 repression accelerates differentiation of neural progenitor cells in vitro and in vivo. Brain Res Mol Brain Res. 2004;132:18–30. doi: 10.1016/j.molbrainres.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Pain D, Chirn GW, Strassel C, et al. Multiple retropseudogenes from pluripotent cell-specific gene expression indicates a potential signature for novel gene identification. J Biol Chem. 2005;280:6265–6268. doi: 10.1074/jbc.C400587200. [DOI] [PubMed] [Google Scholar]
- Robertson M, Stenhouse F, Colby D, et al. Nanog retrotransposed genes with functionally conserved open reading frames. Mamm Genome. 2006;17:732–743. doi: 10.1007/s00335-005-0131-y. [DOI] [PubMed] [Google Scholar]
- Ruau D, Ensenat-Waser R, Dinger TC, et al. Pluripotency associated genes are reactivated by chromatin-modifying agents in neurosphere cells. Stem Cells. 2008;26:920–926. doi: 10.1634/stemcells.2007-0649. [DOI] [PubMed] [Google Scholar]
- Saxe JP, Tomilin A, Schöler HR, et al. Post-translational regulation of Oct4 transcriptional activity. PLoS ONE. 2009;4:e4467. doi: 10.1371/journal.pone.0004467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Desponts C, Do JT, et al. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Silva J, Nichols J, Theunissen TW, et al. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Kagalwala MN, Parker-Thornburg J, et al. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–U11. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm MP, Bone HK, Beck CG, et al. Regulation of nanog expression by phosphoinositide 3-kinase-dependent signaling in murine embryonic stem cells. J Biol Chem. 2007;282:6265–6273. doi: 10.1074/jbc.M610906200. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takeda J, Seino S, Bell GI. Human OCT3 gene family - cDNA sequences, alternative splicing, gene organization, chromosomal location, and expression at low levels in adult tissues. Nucleic Acids Res. 1992;20:4613–4620. doi: 10.1093/nar/20.17.4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehara T, Teramura T, Onodera Y, et al. Potential existence of stem cells with multiple differentiation abilities to three different germ lineages in mouse neurospheres. Stem Cells Dev. 2009;18:1433–1440. doi: 10.1089/scd.2008.0239. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Hitoshi S, Sirard C, et al. Direct neural fate specification from embryonic stem cells: a primitive mammalian neural stem cell stage acquired through a default mechanism. Neuron. 2001;30:65–78. doi: 10.1016/s0896-6273(01)00263-x. [DOI] [PubMed] [Google Scholar]
- Wagner RT, Cooney AJ. Less is more. Cell Res. 2009;19:527–528. doi: 10.1038/cr.2009.48. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]