

Abstract
Cadmium, a major metal constituent of tobacco smoke, elicits synergistic enhancement of cell transformation when combined with benzo[a]pyrene (BP) or other PAHs. The mechanism underlying this synergism is not clearly understood. We observed that (+/−)-anti-benzo[a]pyrene-7,8-diol-9,10-epoxide (BPDE), an ultimate carcinogen of BP, induces apoptosis in promotion sensitive mouse epidermal JB6 Cl41 cells at non-cytotoxic concentrations. BPDE also activates AP-1 several folds in AP-1 reporter JB6 cells. Cadmium at non-cytotoxic concentrations inhibits both AP-1 activation and apoptosis in response to BPDE. Since AP-1 is known to be involved in stress-induced apoptosis we investigated whether inhibition of AP-1 by cadmium has any role in the inhibition of BPDE-induced apoptosis. MAP kinases (particularly ERKs, p38 and JNKs) are known to have important role in DNA damage-induced AP-1 activation. We observed that ERK and JNK, but not p38 MAP kinase, are involved in BPDE-induced AP-1 activation. Effect of cadmium on MAP kinases and the effect of inhibition of above three MAP kinases on BPDE-induced AP-1 activation and apoptosis indicate that AP-1 is probably not involved in BPDE-induced apoptosis. Cadmium up-regulates BPDE-activated ERKs and ERK inhibition by U0126 relieves cadmium-mediated inhibition of BPDE-induced apoptosis. We suggest that cadmium inhibits BPDE-induced apoptosis not involving AP-1 but probably through a different mechanism by up-regulating ERK which is known to promote cell survival.
1. Introduction
Polynuclear aromatic hydrocarbons (PAHs) and the heavy metals like cadmium are regarded as pollutants of worldwide concern [1, 2]. It has been shown that cadmium produces a synergistic enhancement in the frequency of morphological transformation in mammalian cells in combination with benzo[a]pyrene (BP), a potent mutagenic/carcinogenic PAH [3]. The mechanism of the synergistic interaction of heavy metals with PAHs and other DNA-damaging agents is not well established. BP is metabolically activated by cytochrome P-450-dependent monooxygenase system preferentially to a electrophilic species (+)-anti-BP-7,8-diol-9,10-epoxide [(+)-anti-BPDE)] which binds to cellular DNA [4] and the DNA adduct formation is known to be the initial stage in BP-induced carcinogenesis [5]. An important component of cellular response to DNA damage is the induction of apoptosis. It was observed that BP and other PAHs induce apoptosis in vitro in hepatoma cells [6–8], murine cells (9) and others [10–12]. Interference with the DNA damage-induced apoptotic signaling may lead to tumor promotion.
It is known that the activation of the transcription factor activator protein 1 (AP-1) [13–16] and the induction of phosphorylation cascades that regulate MAP kinases [17, 18] play role in the regulation of cellular stress-induced apoptosis. Role of MAP kinases particularly ERKs and JNKs in regulation of AP-1 activity has been reported [19]. It is observed that AP-1 activation correlates cell death through activation of ERK/JNK [13, 14, 16]. Regarding association between BP-induced apoptosis and MAP kinase signaling pathways, different observations were reported indicating differential role of MAP kinases [20–22].
In this study, we report that BPDE induces apoptosis as well as AP-1 activation in promotion sensitive mouse epidermal JB6 cl41 cells and cadmium inhibits both the BPDE-induced apoptosis and AP-1 activation. Our results show that AP-1 activation is not involved in apoptosis induction by BPDE and inhibition of BPDE-induced apoptosis by cadmium is associated with up-regulation of ERK, which is known to be anti-apoptotic. The possible mechanism/s of the inhibition of apoptosis by cadmium is discussed.
2. Materials and Methods
2.1. Cells and reagents
Mouse epidermal JB6 Cl 41 (Cl 41) cells were obtained from American Type Culture Collection (ATCC, VA, USA). Cl 41 cells stably transfected with firefly luciferase reporter gene driven by a minimal AP-1-responsive region was a gift from Dr. Nancy Colburn, National Cancer Institute-Frederick Cancer Research Facility, Frederick, MD. (+/−)-anti-BPDE was purchased from the NCI Chemical Carcinogen Reference Standard Repository. Modified Eagle’s Medium (MEM) and fetal calf serum (FCS) were obtained from Invitrogen Life Technologies (CA, USA). Recombinant Elk-1 and ATF-2 fusion proteins, U0126 (MEK1/2 inhibitor) and primary antibodies were purchased from Cell Signaling, MA, USA; protein A/G PLUS-agarose was from Santa Cruz, CA, USA; SB202190 and SP600125 were from Calbiochem, CA, USA. All other chemicals were of analytical grade.
2.2. Cell culture
Cl 41 cells were cultured as monolayers at 37 °C in an atmosphere of 5% CO2 using modified Eagle’s medium (MEM) containing 5% fetal calf serum, 2 mM L-glutamine, 10 mM sodium pyruvate and penicillin/streptomycin (50μg/ml each). The cell cultures were checked on a routine basis for Mycoplasma contamination by the Gibco Mycotect.
2.3. AP-1 activation
AP-1 activation was determined in appropriately treated JB6 (Cl 41, P+) cells harboring AP-1-luciferase reporter plasmid. After selecting in presence of 400 μg/ml G418 cells in mid-log growth were treated with BPDE dissolved in DMSO (0.05–0.1% of the culture volume) for 90 minutes in serum-free medium and further incubated in 5% serum for 16 hours (unless mentioned otherwise) in the absence of BPDE. To examine the effect of cadmium, cells were pre-treated with cadmium chloride at non-cytotoxic concentration for four hours before BPDE treatment. Cells were harvested 16 hours after BPDE treatment. Luciferase activity in the respective cell extract was performed according to the manufacturer’s instructions using Luciferase Assay kit (Promega, CA). Briefly, the cells were lysed in lysis buffer (supplied in the kit), freeze-thawed and then centrifuged briefly at 14,000 rpm. Twenty μl of the supernatant was mixed with 100 μl of the luciferase assay reagent, and the light intensity was measured by a Luminometer with a 2 seconds measurement delay and 10 seconds measurement read. AP-1 activity in the respective cell extract was expressed as light units (LU) per mg protein.
2.4. DNA fragmentation and apoptosis assay
Appropriately treated cells were harvested 20 hours after BPDE treatment and DNA fragmentation was measured using Apoptotic DNA Ladder Kit of Roche Diagnostic (Indianapolis, IN). In brief DNA from each sample was isolated, purified, and separated on 1.2% agarose gel. The gel was stained with ethidium bromide and bands were visualized under UV light and analyzed using AlphaInotech gel imaging system (Alpha Innotech, CA). Apoptosis assay was performed by using a cell death enzyme-linked immunosorbent assay kit obtained from Roche Applied Science (Indianapolis, IN). The cells were cultured in a 96-well plate, and after appropriate treatments the cells were further incubated in 5% serum-containing MEM for 20 hours. Cells were washed with phosphate-buffered saline (PBS) and apoptosis was quantitated by measuring the levels of cytosolic histone-bound DNA fragments using the apoptosis ELISA kit provided by the manufacturer. Briefly, the cells were lysed with 200 μl of lysis buffer; 20 μl of the lysate was mixed with 80 μl of antibody solution in a well of the enzyme-linked immunosorbent assay plate and incubated at room temperature for 2 h. After washing the substrate was added and incubated at 37 °C for 10–20 min, and the optical density was measured using a microplate reader at a wavelength of 405 nm with a reference wavelength of 492 nm. The readings were used to represent the degree of apoptosis.
2.5. Phosphorylation of MAPKs and Kinase assay
To determine the phosphorylation of MAP kinases appropriately treated cells in 100 mm culture dishes were lysed 2 hours after BPDE treatment in SDS sample buffer. The extent of phosphorylation of ERKs, p38 MAPK and JNK in the respective cell extract was detected by Western immunoblotting using phospho-specific antibody.
Kinase assays of ERKs and p38 MAPK were carried out as described by others [23]. Cell extracts (300 μg protein) were immunoprecipitated by incubating with 3 μg of specific ERKs or p38 MAPK antibody with gentle rocking at 4°C overnight. The immunocomplexes were captured by Protein A/G plus agarose beads. The beads were washed 3 times with 500 μl ice-cold cell lysis buffer and then twice with 500 μl kinase buffer (25 mM Tris-HCl pH 7.5, 5 mM beta-glycerophosphate, 2 mM dithiothreitol, 0.1 mM Na3VO4, 10 mM MgCl2). The kinase reaction was carried out in the presence of 200 μM ATP in 20 μl kinase buffer at 30°C for 30 minutes using 2 μg of Elk-1 or ATF-2 as substrate for ERKs or p38 MAPK. The reaction was terminated by adding 20 μl 3X SDS sample buffer and the phosphorylated proteins were detected by immunoblotting using phospho-specific antibody of the enzyme substrate Elk-1 or ATF-2.
3. Results
3.1. Cadmium inhibits BPDE-induced apoptosis
We investigated whether BPDE induced DNA damage can elicit apoptosis and whether cadmium has any interfering effect on BPDE-induced apoptosis, which may explain synergistic enhancement of PAH-induced cell transformation by cadmium. We first monitored DNA fragmentation in JB6 cl41 cells in presence of BPDE or cadmium chloride plus BPDE. Cadmium chloride was used at non-cytotoxic concentration which is below 25 μM. Trypan blue exclusion assay showed 93% survival of JB6 cl41 cells at 25 μM cadmium chloride (data not shown). We observed that 1 μM BPDE treatment causes DNA fragmentation (figure 1) and pre-treatment of cells with 10 μM cadmium chloride significantly inhibits BPDE-induced DNA fragmentation (figure 1).
Figure 1.
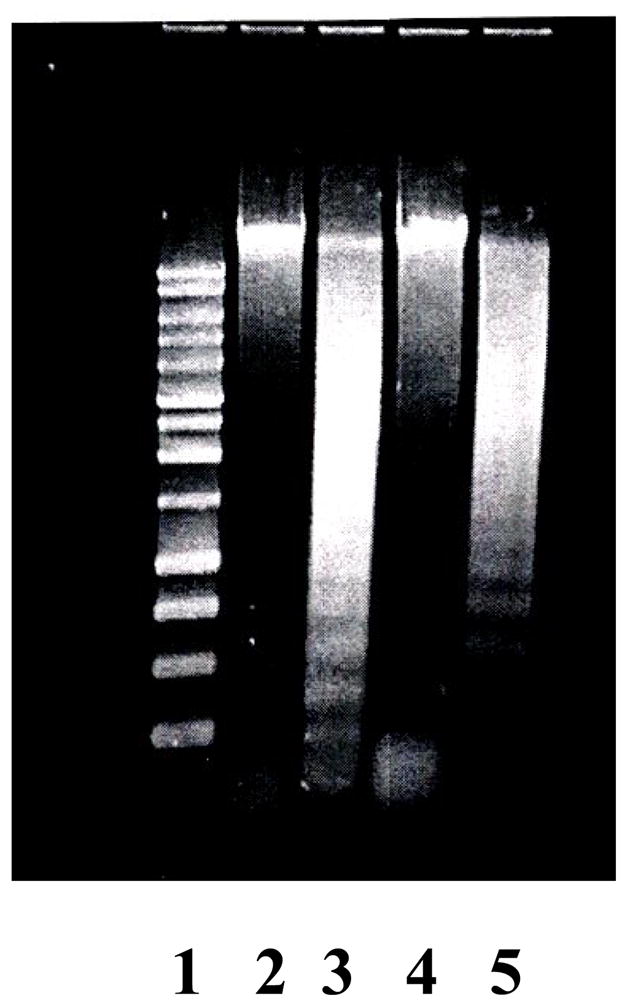
Effect of cadmium on DNA fragmentation induced by BPDE in JB6 Cl41 cells. Marker (Ln 1), no treatment (Ln 2), BPDE 1μM (Ln 3), CdCl2 10 μM (Ln 4) and CdCl2 10 μM plus BPDE 1 μM (Ln 5).
We also determined the extent of apoptosis using a cell death enzyme-linked immunosorbent assay kit obtained from Roche Applied Science (Indianapolis, IN). The cells were treated with different concentrations of BPDE (0.1 – 2.0 μM) and the extent of apoptosis was determined as described in Materials and methods. As evident in figure 2 BPDE caused dose-dependent induction of apoptosis and pre-treatment of cells with cadmium chloride caused inhibition of BPDE-induced apoptosis e.g. 19% and 43% inhibition by 5 μM and 10 μM cadmium chloride respectively. This finding indicates that inhibition of BPDE-induced apoptosis may represent one of the mechanisms by which cadmium potentiates BP-induced cell transformation.
Figure 2.

Inhibition of BPDE-induced apoptosis by cadmium. Extent of apoptosis was determined in JB6 Cl41 Cells, untreated (A); treated with 10 μM cadmium chloride only (F); treated with different concentrations of BPDE; 0.1 μM (B), 0.5 μM (C), 1.0 μM (D) and 2.0 μM (E). Effect of cadmium on the extent of apoptosis in BPDE (1 μM) treated cells was determined by pre-treatment of cells with cadmium chloride (5 μM, G; 10 μM, H) for 4 hours followed by BPDE treatment. Cell death enzyme-linked immunosorbent assay kit from Roche Applied Science was used to determine the extent of apoptosis. Experimental details are described in the text. Each bar graph represents mean ± S.D. of three different experiments.
3.2. Cadmium inhibits BPDE-induced AP-1 activation
The transcription factor AP-1 has been implicated in stress-induced apoptosis [15, 25, 26]. Since we observed that BPDE treatment of Cl41 cells induces apoptosis, we first examined whether AP-1 activation is an associated event with apoptosis induction by BPDE. BPDE treatment of cl 41 cells harboring AP-1-responsive luciferase gene showed a time- and dose-dependent activation of AP-1 (figures 3A, 3B). Up to 5 hours after 1 μM BPDE treatment, no significant increase in AP-1 activity was observed followed by a sharp increase in AP-1 activity up to 16 hours (12 fold activation) and then the rate was noticeably decreased with time (14 fold activation at 25 hours) (figure 3A). So BPDE indeed activates AP-1 in cl41 cells. We observed that pre-treatment of cells with cadmium chloride at non-cytotoxic doses caused a significant inhibition of BPDE-induced AP-1 activity, for example AP-1 activation in BPDE treated cells was decreased by 14% and 40% by 5 μM and 10 μM cadmium chloride respectively (Figure 3C). Treatment of cells by cadmium chloride alone did not have any effect on AP-1 activation. To our knowledge this is the first report that cadmium inhibits AP-1 activation in chemical carcinogen-damaged cells.
Figure 3.

Activation of AP-1 by BPDE and the effect of cadmium on BPDE-induced AP-1 activation. Determination of AP-1 activation corresponding to luciferase activity in the extracts of Cl41 cells harboring AP-1 -responsive luciferase reporter gene has been described elsewhere. (A) Cells were harvested at different time points after BPDE (1 μM) treatment, (B) cells were treated with different concentrations of BPDE for 90 minutes in serum-free medium followed by harvesting 16 hours after BPDE treatment and (C) cells were either untreated or treated with BPDE alone, CdCl2 alone or pre-treated with CdCl2 for 4 hours followed by BPDE (1 μM) for 90 minutes. After treatments cells were incubated in 5% serum-containing MEM and harvested 16 hours after BPDE treatment. The results are presented as relative AP-1 activation (fold increase) compared to untreated cells. Each bar indicates the mean ± SD of three parallel experiments.
3.3. Differential effect of cadmium on BPDE-induced activation of MAPKs
Role of MAP kinase family proteins in AP-1 activation and regulation of apoptosis is well documented [13, 17–19]. Since apoptosis induction and AP-1 activation are two associated phenomena in response to BPDE, and cadmium inhibits both of these events, we examined the role of MAPKs in this regard. BPDE-treated cells showed increased level of phosphorylation as well as activation of both the ERKs and p38 MAPK (Figures 4A and 4B). BPDE treatment also showed increased phosphorylation of JNK (Thr183/Tyr185) (Figure 4C) indicative of JNK activation. Pre-treatment of cells with highly selective inhibitor for each MAPK e.g. U0126 (MEK1/2 inhibitor), SB202190 (p38 MAPK inhibitor) and SP600125 (inhibitor of phosphorylation of JNK1/2 and of downstream c-Jun) inhibited BPDE-induced phosphorylation as well as activation of respective MAPK. Interestingly, cadmium chloride pre-treatment showed differential effects on aforementioned MAP kinases - ERK phosphorylation and activation were significantly increased, p38 phosphorylation and activation were inhibited marginally, JNK phosphorylation was unaltered (figures 4A, 4B and 4C). Inhibitory effect of cadmium on BPDE-induced AP-1 activation and the differential effects of cadmium on BPDE-induced activation of MAPKs necessitate further studies to understand the role of MAPKs in AP-1 activation and apoptosis induction by BPDE.
Figure 4.

Effect of cadmium on BPDE-induced activation of MAP kinases. Cl41 cells were treated with BPDE (1 μM) or pre-treated with cadmium chloride (10 μM) followed by BPDE (1 μM) and the phosphorylation/activation of MAP kinases were determined as described in the text. (A) Effect of cadmium chloride (10 μM) and U0126 (10 μM) on BPDE-induced phosphorylation and activation of ERKs, (B) Effect of cadmium chloride (10 μM) and SB202190 (2 μM) on BPDE-induced phosphorylation and activation of p38 MAPK, and (C) Effect of cadmium chloride (10 μM) and SP600125 (2 μM) on BPDE-induced phosphorylation of JNK.
3.4. Differential effects of MAPK inhibition on BPDE-induced AP-1 activation and apoptosis
We examined the effect of inhibition of MAP kinases on BPDE-induced AP-1 activation. It is observed that cells treated with U0126 (10 μM) and SP600125 (2 μM) at concentrations, which inhibited BPDE-induced phosphorylation and activation of these MAPKs as described in figure 4, also inhibited BPDE-induced AP-1 activation by 47% and 30% respectively (figure 5). In contrast, treatment of cells with p38 MAPK inhibitor SB202190 (2 μM) caused moderate potentiating effect on AP-1 activation (23%). These findings indicate that activation of ERKs and JNK, but not of p38 MAPK, may play a role in BPDE-induced AP-1 activation.
Figure 5.

Effects of MAPK inhibition on BPDE-induced AP-1 activation. AP-1 reporter cells were either treated with 1 μM BPDE for 90 minutes or with respective MAP kinase inhibitor (10 μM U0126 or 2 μM SB202190 or 2 μM SP600125) for 1 hour followed by 1 μM BPDE (90 minutes) and were harvested 16 hours after BPDE treatment. AP-1 activation in the respective cell extract was determined by luciferase reporter assay as described in figure 3 legend. The results are presented as relative AP-1 activation (fold increase) compared to untreated cells. Each bar indicates the mean ± SD of three parallel experiments.
We further examined the effect of MAPK inhibitors on BPDE-induced apoptosis in order to gain an insight into the role of MAPKs and AP-1 in this respect. We observed differential effects of the inhibition of MAPKs on apoptosis in BPDE treated cells (Figure 6). Surprisingly, U0126 (10 μM) significantly increased apoptosis induction by BPDE (72% increase). On the contrary JNK inhibitor SP600125 (2 μM) inhibited apoptosis induction marginally (25%), whereas p38 MAPK inhibitor SB202190 (2 μM) had practically no effect on BPDE-induced apoptosis. Our data of the effect of MAPK inhibitors on AP-1 activation and on apoptosis induction indicate lack of correspondence of BPDE-induced AP-1 activation with apoptosis induction. Data of the effect of cadmium on activation of MAPKs also indicate less likelihood of AP-1’s involvement in the inhibition of BPDE-induced apoptosis by cadmium.
Figure 6.

Effect of MAPK inhibitors on BPDE-induced apoptosis. JB6 Cl41 cells were either treated with 1 μM BPDE for 90 minutes or with respective MAP kinase inhibitor (10 μM U0126 or 2 μM SB202190 or 2 μM SP600125) for 1 hour followed by 1 μM BPDE (90 minutes) and were harvested 16 hours after BPDE treatment. Cell death enzyme-linked immunosorbent assay kit from Roche Applied Science was used to determine the extent of apoptosis. Experimental details are described in the text. Each bar graph represents mean ± S.D. of three different experiments.
3.5. ERK inhibition relieves the inhibition of BPDE-induced apoptosis by cadmium
To understand whether ERK up-regulation by cadmium has any role in inhibition of BPDE-induced apoptosis we further examined the effect of ERK inhibitor U0126 on cadmium-mediated inhibition of BPDE-induced apoptosis. Figure 7 shows that cadmium chloride (10 μM) inhibits BPDE induced apoptosis by 47% and pretreatment of cells with ERK inhibitor U0126 (10 μM) relieves cadmium-mediated inhibition of BPDE-induced apoptosis completely and even 15% more apoptosis compared to BPDE treatment was observed. Significant up-regulation of ERKs by cadmium (Figure 4A) indicates that the inhibition of BPDE-induced apoptosis by cadmium is mediated through a different mechanism probably involving up-regulation of ERKs and thereby promoting cell survival.
Figure 7.

Effect of ERK inhibitor on inhibition of BPDE-induced apoptosis by cadmium. JB6 Cl41 cells were either pretreated with 10 μM CdCl2 (4 hours) or with 10 μM CdCl2 plus 10 μM U0126 followed by 1 μM BPDE for 90 minutes. Cells were harvested 16 hours after BPDE treatment. Cell death enzyme-linked immunosorbent assay kit from Roche Applied Science was used to determine the extent of apoptosis. Experimental details are described in the text. Each bar graph represents mean ± S.D. of three different experiments.
4. Discussion
DNA damage induced by carcinogens may lead to permanent fixation of the damage as lethal mutation if the protective response elicited in response to the damage fails to remove the damage. Alternatively the extent of damage (adducted DNA) may be one of the determining factors in this respect. There may be a possibility that the co-carcinogenic effect of cadmium in presence of the carcinogen BPDE is due to its effect on BPDE-DNA adduct formation. BPDE, an ultimate carcinogenic metabolite of BP, is known to alkylate the N7 sites in guanines in DNA (30) as well as the exocyclic amino group of guanine in DNA (31). Since we used BPDE as DNA damaging agent, the effect of cadmium on metabolic activation of BP is not relevant here. Adduct formation with the exocyclic amino group of guanine in DNA is known to be mutagenic event (30). It is known that cadmium augments the DNA lesions (mutational fixation) by DNA damaging agents (32, 33). Regarding cadmium-PAH interaction with respect to DNA adduct formation it is observed that cadmium inhibits major groove alkylation of DNA by tobacco carcinogen metabolite BPDE, and it does not have any effect on the alkylation of minor groove which is known to be biologically relevant to mutation and carcinogenesis (34, 35). So, the effect of cadmium on BPDE-DNA binding may not have any role in the observed co-carcinogenic activity of cadmium (possibility of fixation of the damage as lethal mutation). Moreover inhibition of BPDE-mediated DNA alkylation by cadmium reflects less DNA adduct formation which can not justify the mechanism of co-carcinogenesis of cadmium in terms of DNA adduct formation. So, increased mutational event by cadmium (co-carcinogenic effect) must relate to its interference with the DNA damage-induced protective response leading to the fixation of the damage into mutation.
In this investigation, we examined the effect of cadmium on the cellular protective responses induced by BPDE in order to obtain an insight into the mechanism by which cadmium potentiates the mutagenicity and the cell transformation ability of BP, a potent mutagenic/carcinogenic PAH. An important component of cellular protective response to DNA damage by genotoxic stress is the induction of apoptosis. Our results show that BPDE induces apoptosis in promotion-sensitive mouse epidermal cells (JB6 cl 41) in a dose- and time-dependent manner. The promotion-sensitive JB6 cl 41 cells represent a suitable cellular model to study the tumor promoting activity of various compounds [24]. Interference with DNA damage-induced apoptotic response will be detrimental to normal cellular function and may lead to tumorogenesis. We observed that cadmium chloride at non-cytotoxic concentration significantly inhibits BPDE-induced apoptosis. We infer that inhibition of BPDE-induced apoptosis by cadmium may represent a possible mechanism for its co-carcinogenic function in presence of BPDE as DNA damaging agent.
Our results also show that induction of apoptosis by BPDE is associated with the activation of the transcription factor AP-1 which is also inhibited by cadmium. AP-1 is known to be involved in many stress-induced cellular functions [25, 26]. Amongst its diverse cellular functions, AP-1 is known to mediate apoptosis in response to cellular stress [13–16]. The aforementioned observation prompted us to examine the role of AP-1 activation in BPDE-induced apoptosis with respect to cadmium’s interference with AP-1 activation leading to inhibition of apoptosis. AP-1 activity is known to be regulated by several mechanisms, and it is also known that activation of the mitogen-activated protein kinase (MAPK) pathways has regulatory role in AP-1 activation [19]. We observed that BPDE treatment of cells activated all three MAPKs e.g. ERKs, p38 and JNK. Interestingly the effect of MAPK inhibition on BPDE-induced AP-1 activation showed differential effects e.g. inhibition of ERK or JNK down-regulated AP-1 activation, whereas p38 MAPK inhibition moderately up-regulated AP-1 activation in BPDE treated cells. These results imply that activation of ERKs/JNK by BPDE is involved in AP-1 activation, whereas p38 MAPK activation by BPDE is not directly related to AP-1 activation rather the possibility of the existence of negative relation between them. Activation of p38 MAPK by BPDE probably is involved in different signaling event, for example, our previous study showed that BPDE-induced p38 activation in JB6 cells is involved in the activation of nuclear factor kappaB transcription factor [27]. We also found that cadmium pre-treatment of cells had variable effects on BPDE-induced MAPK activation e.g. activation of ERKs was significantly increased, p38 MAPK activation was decreased and JNK phosphorylation did not show any change. Although ERKs and JNK are involved in BPDE-induced AP-1 activation, analyses of our results of the effect of cadmium on the activation of MAPKs and AP-1 suggest less likelihood of the role of any of the three MAPKs in the inhibition of BPDE-induced AP-1 activation by cadmium. MAPK-independent AP-1 activation through potentiation of JAB1 (Jun activation domain-binding protein 1), a co-activator of AP-1 has been reported [28, 29]. Our finding of MAPK-independent inhibition of BPDE-induced AP-1 activation by cadmium may need further studies to examine the effect of cadmium on JAB1 protein for the understanding of the underlying mechanism of AP-1 inhibition by cadmium.
Effect MAPK inhibitors on BPDE-induced apoptosis also showed differential effects e.g. ERK inhibition increased apoptosis significantly whereas JNK inhibition has marginal inhibitory effect on apoptosis, and p38 MAPK inhibition had no effect on apoptosis. Comparison of the effects of MAPK inhibition on apoptosis with AP-1 activation indicates that AP-1 is probably not involved in BPDE-induced apoptosis. We observed that cadmium up-regulates ERKs and inhibition of ERKs significantly increased BPDE-induced apoptosis. ERKs are also known to be anti-apoptotic. This observation probably suggests that cadmium inhibits BPDE-induced apoptosis through a different mechanism either by up-regulating ERKs and thereby promoting cell survival or by interfering with the signaling events involving activation of caspases without any involvement of AP-1. Further studies in these directions are needed to understand the mechanism of inhibition of BPDE-induced apoptosis by cadmium.
Acknowledgments
This work was partially supported by National Institute of Environmental Health Sciences (NIEHS) Grant R15ES12401 (to JJM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoffmann D, Hecht SS. Advances in tobacco carcinogenesis. In: Cooper CS, Grover PL, editors. Handbook of Experimental Pharmacology. 94/1. Heidelberg: Springer-Verlag; 1990. pp. 63–102. [Google Scholar]
- 2.Pinot F, Kreps SE, Bachelet M, Hainaut P, Bakonyi M, Polla BS. Cadmium in the environment: sources, mechanisms of biotoxicity, and biomarkers. Rev Environ Health. 2000;15:299–323. doi: 10.1515/reveh.2000.15.3.299. [DOI] [PubMed] [Google Scholar]
- 3.Rivedal E, Sanner T. Metal salts as promoters of in vitro morphological transformation of hamster embryo cells initiated by benzo[a]pyrene. Cancer Res. 1981;41:2950–2953. [PubMed] [Google Scholar]
- 4.Harvey RG. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge University Press; Cambridge: 1991. [Google Scholar]
- 5.Gelboin HV. Benzo[alpha]pyrene metabolism, activation and carcinogenesis: role and regulation of mixed-function oxidases and related enzymes. Physiol Rev. 1980;60:1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Nguyen N, Tamura K, Karin M, Tukey RH. The role of the Ah receptor and p38 in benzo[a]pyrene-7,8-dihydrodiol and benzo[a]pyrene-7,8 dihydrodiol-9,10-epoxide-induced apoptosis. J Biol Chem. 2003;278:19526–33. doi: 10.1074/jbc.M300780200. [DOI] [PubMed] [Google Scholar]
- 7.Lei W, Yu R, Mandlekar S, Kong AN. Induction of apoptosis and activation of interleukin 1beta-converting enzyme/Ced-3 protease (caspase-3) and c-Jun NH2-terminal kinase 1 by benzo(a)pyrene. Cancer research. 1998;58:2102–6. [PubMed] [Google Scholar]
- 8.Yoshii S, Tanaka M, Otsuki Y, Fujiyama T, Kataoka H, Arai H, Hanai H, Sugimura H. Involvement of alpha-PAK-interacting exchange factor in the PAK1-c-Jun NH(2)-terminal kinase 1 activation and apoptosis induced by benzo[a]pyrene. Mol Cell Biol. 2001;21:6796. doi: 10.1128/MCB.21.20.6796-6807.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burchiel SW, Davis DA, Ray SD, Barton SL. DMBA induces programmed cell death (apoptosis) in the A20.1 murine B cell lymphoma. Fundam Appl Toxicol. 1993;21:120–124. doi: 10.1006/faat.1993.1080. [DOI] [PubMed] [Google Scholar]
- 10.Solhaug A, Øvrebø S, Mollerup S, Låg M, Schwarze PE, Nesnow S, Holme JA. Role of cell signaling in B[a]P-induced apoptosis: characterization of unspecific effects of cell signaling inhibitors and apoptotic effects of B[a]P metabolites. Chem Biol Interact. 2005;151:101–119. doi: 10.1016/j.cbi.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Salas VM, Burchiel SW. Apoptosis in Daudi human B cells in response to benzo[a]pyrene and benzo[a]pyrene-7,8-dihydrodiol. Toxicol Appl Pharmacol. 1998;151:367–76. doi: 10.1006/taap.1998.8455. [DOI] [PubMed] [Google Scholar]
- 12.Roth W, Reed JC. Apoptosis and cancer: when BAX is TRAILing away. Nat Med. 2002;8:216–18. doi: 10.1038/nm0302-216. [DOI] [PubMed] [Google Scholar]
- 13.Taimor G, Rakow A, Piper HM. Transcription activator protein 1 (AP-1) mediates NO-induced apoptosis of adult cardiomyocytes. The FASEB Journal. 2001;15:2518–20. doi: 10.1096/fj.01-0353fje. [DOI] [PubMed] [Google Scholar]
- 14.Chen YN, Cheng CC, Chen JC, Tsauer W, Hsu SL. Norcantharidin-induced apoptosis is via the extracellular signal-regulated kinase and c-Jun-NH2-terminal kinase signaling pathways in human hepatoma HepG2 cells. British Journal of Pharmacology. 2003;140:461–70. doi: 10.1038/sj.bjp.0705461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobs-Helber SM, Wickrema A, Birrer MJ, Sawyer ST. AP1 Regulation of Proliferation and Initiation of Apoptosis in Erythropoietin-Dependent Erythroid Cells. Mol Cell Biol. 1998;18:3699–3707. doi: 10.1128/mcb.18.7.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu C, Shen G, Yuan X, Kim JH, Gopalkrishnan A, Keum YS, Nair S, Kong AN. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–45. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 17.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond Biol. 1996;351:127–134. doi: 10.1098/rstb.1996.0008. [DOI] [PubMed] [Google Scholar]
- 18.Cross TG, Scheel-Toellner D, Henriquez NV, Deacon E, Salmon M, Lord JM. Serine/threonine protein kinases and apoptosis. Experimental Cell Research. 2000;256:34–41. doi: 10.1006/excr.2000.4836. [DOI] [PubMed] [Google Scholar]
- 19.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–46. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 20.Solhaug A, Refsnes M, Holme JA. Role of cell signalling involved in induction of apoptosis by benzo[a]pyrene and cyclopenta[c,d]pyrene in Hepa1c1c7 cells. J Cell Biochem. 2004;93:1143–54. doi: 10.1002/jcb.20251. [DOI] [PubMed] [Google Scholar]
- 21.Chin BY, Choi ME, Burdick MD, Strieter RM, Risby TH, Choi AM. Induction of apoptosis by particulate matter: role of TNF-alpha and MAPK. Am J Physiol. 1998;27:L942–L949. doi: 10.1152/ajplung.1998.275.5.L942. [DOI] [PubMed] [Google Scholar]
- 22.Yoshii S, Tanaka M, Otsuki Y, Fujiyama T, Kataoka H, Arai H, Hanai H, Sugimura H. Involvement of alpha-PAK-interacting exchange factor in the PAK1-c-Jun NH(2)-terminal kinase 1 activation and apoptosis induced by benzo[a]pyrene. Mol Cell Biol. 2001;21:6796–6807. doi: 10.1128/MCB.21.20.6796-6807.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.She QB, Chen N, Dong Z. ERKs and p38 Kinase Phosphorylate p53 Protein at Serine 15 in Response to UV Radiation. J Biol Chem. 2000;275:20444–20449. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- 24.Suzukawa K, Weber TJ, Colburn NH. AP-1, NF[kappa]B, and ERK activation thresholds for promotion of neoplastic transformation in the mouse epidermal JB6 model. Environmental Health Perspectives. 2002;110:865–870. doi: 10.1289/ehp.02110865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong ZG, Birrer MJ, Watts RG, Matrisian LM, Colburn NH. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci USA. 1994;91:609–613. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang CS, Ma WY, Young MR, Colburn NH, Dong ZG. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc Natl Acad Sci USA. 1998;95:156–161. doi: 10.1073/pnas.95.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee JJ, Sikka HC. Attenuation of BPDE-induced p53 accumulation by TPA is associated with a decrease in stability and phosphorylation of p53 and down-regulation of NF-[[kappa]]B activation: Role of p38 MAP kinase. Carcinogenesis. 2006;27:631–638. doi: 10.1093/carcin/bgi247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C, Li Y, Zhao Y, Xing G, Tang F, Wang Q, Sun Y, Wei H, Yang X, Wu C, Chen J, Guan KL, Zhang C, Chen H, He F. Intracrine hepatopoietin potentiates AP-1 activity through JAB1 independent of MAPK pathway. FASEB J. 2002;16:90–92. doi: 10.1096/fj.01-0506fje. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi E, Denti S, Granata A, Bossi G, Geginat J, Villa A, Rogge L, Pardi R. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature. 2000;404:617–21. doi: 10.1038/35007098. [DOI] [PubMed] [Google Scholar]
- 30.Sage E, Haseltine WA. High ratio of alkali-sensitive lesions to total DNA modification induced by benzo(a)pyrene diol epoxide. J Biol Chem. 1984;259:11098. [PubMed] [Google Scholar]
- 31.Weinstein IB, Jeffrey AM, Jennette KW, Blobstein SH, Harvey RG, Harris C, Autrup H, Kasai H, Nakanishi K. Benzo(a)pyrene diol epoxides as intermediates in nucleic acid binding in vitro and in vivo. Science. 1976;193:592. doi: 10.1126/science.959820. [DOI] [PubMed] [Google Scholar]
- 32.Mandel R, Ryser HJ. Mutagenicity of cadmium in Salmonella typhimurium and its synergism with two nitrosamines. Mutation Res. 1984;138:9. doi: 10.1016/0165-1218(84)90079-x. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi K, Imaeda T, Kawazoe Y. Effect of metal ions on the adaptive response induced by N-methyl-N-nitrosourea in Escherichia coli. Biochem Biophys Res Commun. 1988;157:1124. doi: 10.1016/s0006-291x(88)80990-2. [DOI] [PubMed] [Google Scholar]
- 34.Prakash AS, Rao KS, Dameron CT. Cadmium Inhibits BPDE Alkylation of DNA in the Major Groove but Not in the Minor Groove. Biochemical and Biophysical Research Communications. 1998;244:198–203. doi: 10.1006/bbrc.1998.8241. [DOI] [PubMed] [Google Scholar]
- 35.Prakash AS, Tran HP, Peng C, Koyalamudi SR, Dameron CT. Kinetics of DNA alkylation, depurination and hydrolysis of anti diol epoxide of benzo(a)pyrene and the effect of cadmium on DNA alkylation. Chem-Biol-Interact. 2000;125:133–50. doi: 10.1016/s0009-2797(00)00145-9. [DOI] [PubMed] [Google Scholar]