

Abstract
A series of 3,6-disubstituted β-carbolines was synthesized and evaluated for their in vitro affinities at αxβ3γ2 GABAA/benzodiazepine receptor subtypes by radioligand binding assays in search of α1 subtype selective ligands to treat alcohol abuse. Analogues of β-carboline-3-carboxylate-t-butyl ester (βCCt, 1) were synthesized via a CDI-mediated process and the related 6-substituted β-carboline-3-carboxylates 6 including WYS8 (7) were synthesized via a Sonogashira or Stille coupling processes from 6-iodo βCCt (5). The bivalent ligands of βCCt (32 and 33) were also designed and prepared via a palladium-catalyzed homocoupling process to expand the structure-activity relationships (SAR) to larger ligands. Based on the pharmacophore/receptor model, a preliminary SAR study on 34 analogues illustrated that large substituents at position -6 of the β-carbolines were well tolerated. As expected, these groups are proposed to project into the extracellular domain (LDi region) of GABAA/Bz receptors (see 32 and 33). Moreover, substituents located at position -3 of the β-carboline nucleus exhibited a conserved stereo interaction in lipophilic pocket L1, while N(2) presumably underwent a hydrogen bonding interaction with H1. Three novel β-carboline ligands (βCCt, 3PBC and WYS8), which preferentially bound to α1 BzR subtypes permitted a comparison of the pharmacological efficacies with a range of classical BzR antagonists (flumazenil, ZK93426) from several different structural groups and indicated these β-carbolines were “near GABA neutral antagonists”. Based on the SAR, the most potent (in vitro) α1 selective ligand was the 6-substituted acetylenyl βCCt (WYS8, 7). Earlier both βCCt and 3PBC had been shown to reduce alcohol self-administration in alcohol preferring (P) and high alcohol drinking (HAD) rats but had little or no effect on sucrose self-administration.1–3 These data prompted the synthesis of the β-carbolines presented here.
Introduction
Alcoholism
Alcohol addiction and dependence remain significant public health concerns, impacting physical and mental well-being, family structure and occupational stability.4 While advances have been made in the development of novel therapies to treat alcoholism,5–8 alcohol-dependent individuals represent a heterogeneous group,9–11 and it is unlikely that a single pharmacological treatment will be effective for all alcoholics. Hence, a better understanding of the neuromechanisms which regulate alcohol seeking behaviors and the design of clinically safe and effective drugs that reduce alcohol addiction and dependence remain a high priority.7,12 While the precise neuromechanisms regulating alcohol-seeking behaviors remain unknown, there is now compelling evidence that the GABAA receptors within the striatopallidal and extended amygdala system are involved in the “acute” reinforcing actions of alcohol.13–18 Among the potential GABAA receptor isoforms within the VP regulating alcohol-seeking behaviors, GABA receptors containing the α1 receptor subtype (GABA α1) appear preeminent. Thus, Criswell and colleagues observed that acute alcohol administration selectively enhanced the effects of ionotophoretically applied GABA in the ventral pallidum (VP).19,20 However, no effects were seen in the septum, ventral tegmental area (VTA), and CA1 hippocampus. These data suggest the α1 Bz/Gaba(A)ergic receptor plays an important role in alcohol-motivated behaviors. Research on the neuroanatomical basis of alcohol reward has shown that the NACC, VTA, VP, central amygdala (CeA), and hippocampus are all involved in GABAergic regulation of ethanol (EtOH) reinforcement.2,21–24 Other investigators have identified a dense reciprocal projection from the VP to the NACC,25–27 and many of these have been found to be GABAergic neurons.28–30 The NACC is now well established as a substrate that regulates the reinforcing properties of abused drugs.13 Finally, immunohistochemical31,32 and in situ hybridization studies33–35 have demonstrated that the VP contains one of the highest concentrations of mRNA encoding the α1 subunit in the CNS. These findings, together with pharmacological studies suggesting the VP plays a role in reward-mediated behaviors of psychostimulants and opiates,28,36–39 suggest a possible role of the VP-α1 receptors in the euphoric properties of alcohol. Findings of previous studies concluded that inhibition of VP-α1 receptors by the α1 preferring antagonist 3-PBC produced marked reductions on alcohol-maintained responding.9,15,40,41 The α1-mediated suppression at the VP level by 3-PBC showed a high degree of neuroanatomical specificity. Specifically, the α1-mediated suppression was not observed with the more dorsal placements in the NACC or caudate putamen. The failure of 3-PBC to alter alcohol self-administration in the NACC/striatum is in agreement with previous research which has consistently reported a lack of expression of the α1 transcript in the NACC caudate.31–35
An understanding of the neuromechanisms that regulate alcohol drinking is key in the development of drugs to treat alcohol addiction and dependence in humans.2 In recent years, much evidence has accumulated in favor of the GABA system;22,23,42,43 however much remains unknown about the role of specific GABAA receptor subtypes in regulating ethanol reinforcement. This is due to both a lack of high-affinity and selective ligands capable of discriminating among the GABAA receptor subunits and the heterogeneity of various subunits within the known alcohol reward circuitry.31,34 Of the potential GABAA receptors involved in the reinforcing properties of alcohol, evidence suggests the α1 subtype within the VP may play an important role in regulating alcohol-seeking behaviors, as mentioned above. The VP contains one of the highest distributions of α1 subunits in the mesolimbic system.32–35 Finally, acute ethanol administration has been reported to selectively enhance the effects of iontophoretically applied GABA in the VP. These effects correlate highly with [3H] zolpidem binding (an α1-subtype selective agonist).19,20
The GABAA receptor is the major inhibitory neurotransmitter receptor of the central nervous system (CNS) and the site of action of a variety of pharmacologically and clinically important drugs, such as benzodiazepines, barbiturates, neuroactive steroids, anesthetics and convulsants.44 It is now clear that these receptors regulate the excitability of the brain, anxiety, muscle tone, circadian rhythms, sleep, vigilance, memory, and learning.44 There are several disease states thought to be associated with the improper functioning of this system, including anxiety, epilepsy,45 insomnia,46 depression and bipolar disorder,47,48 schizophrenia,49 as well as mild cognitive impairment and Alzheimer’s disease.50 A role of GABAA receptors in drug and alcohol abuse has also been reported.51–53 Briefly, GABAA receptors are composed of 5 subunits that form a central chloride channel and can belong to different subunit classes. A total of 19 subunits (6α, 3β, 3γ, 1ε, 1π, 1θ, 3ρ) of the GABAA receptor have been cloned and sequenced from the mammalian nervous system.54–59 All these polypeptides possess an approximate molecular mass of ~ 50 kD and are structurally related.
To evaluate the role of the α1 receptor in regulating alcohol reinforcement, 3-propoxy-β-carboline hydrochloride (3-PBC), a mixed benzodiazepine (BDZ) agonist-antagonist with binding selectivity at the α1 receptor was developed.2 Compared with the prototypical BDZ agonist zolpidem, 3-PBC exhibited a slightly higher binding selectivity for the α1 receptor.60,61 Preliminary behavioral studies in several species (e.g., rats, mice, and primates) show that 3-PBC is a BDZ antagonist, exhibiting competitive binding-site interactions with BDZ agonists at low to moderate doses (2.5–15 mg/kg).60,62,63 At higher doses (15–60 mg/kg), 3-PBC produces anxiolytic effects in the plus maze that are comparable with those of chlordiazepoxide in alcohol preferring (P) rats.62–65 Thus given the proposed subunit composition of the GABA receptors within the CeA,31,32,34,66 pharmacological compounds capable of exploiting the α1, α2, and α3 subunit-containing GABAA receptors represent optimal tools to evaluate the role of the GABAA receptors in alcohol reinforcement and better understand neurobehavior and ethanol responding.
α1 Subtype Selective Ligands
The β-carboline-3-carboxylate-t-butyl ester (βCCt) is a mixed benzodiazepine agonist-antagonist ligand with binding selectivity at α1 receptors;2,62,67 βCCt also exhibits some affinity (albeit lower) for both α2 and α3 receptors. Behavioural studies in several species (eg, rats, mice, primates) show that βCCt is a BDZ antagonist exhibiting competitive binding site interaction with BDZ agonists over a broad range of doses.60,62,68–72 Other studies show that βCCt produces anxiolytic effects in rodents62 and potentiates the anticonflict response induced by α1 subtype agonists in primates.70 Thus, βCCt displays a weak agonist or antagonist profile depending on the behavioral task, species, and dose employed.
In studies involving the α1 subtype, βCCT and 3-PBC were observed to selectively reduce alcohol-motivated behaviors in a variety of experiments.2,73 However, unlike the α5 selective inverse agonist RY-23, both the β-carboline antagonists βCCT and 3-PBC displayed mixed weak agonist-antagonist profiles in vivo in alcohol P and HAD rats. Therefore, in addition to being able to study the molecular basis of alcohol reinforcement, α1 Bz β-carboline ligands which display mixed agonist-antagonist pharmacology in alcohol P and HAD rats may be capable of reducing alcohol intake while eliminating or greatly reducing the anxiety associated with habitual alcohol, abstinence or detoxification. Thus, these types of ligands may be ideal clinical agents for the treatment of alcohol dependent individuals.2,73
Consequently, several series of structurally different compounds have been synthesized which possess some α1 subtype selectivity.67,74–77 The discovery of high affinity, saturable, and stereospecific ligands for the BzR has been coupled with the demonstration that β-carbolines exhibited an affinity for the BzR.78–85 Some of these agents act on the BzR to induce effects that are functionally opposite (inverse agonists/antagonists) to those of classical BDZs. Consequently, the affinities of a wide variety of β-carbolines have been reported on synaptosomal membranes from this laboratory,60,72,78,80,81,86–90 and the laboratories of others,91–96 and this prompted the study of the binding affinities of a series of β-carbolines67 at 5 recombinant GABAA/BzR subtypes (α1β3γ2, α2β3γ2, α3β3γ2, α5β3γ2 and α6β3γ2) expressed from recombinant human cell lines.91,97 In general, this series of β-carboline ligands exhibited some selectivity at α1 receptor subtypes including βCCt (1) and 3-PBC (2).2,3 These two ligands displayed a 20-fold and 10-fold selectivity, respectively, for the α1 subtype over the α2 and α3 receptors, as well as over 150-fold selectivity for the α1 site over the α5 subtype.2,3 βCCt (1) was more selective at the α1 subtype in vitro than the classical α1 selective agonists zolpidem (3) and CL 218872 (4) (Figure 1).60,98,99 A number of in vitro and in vivo studies employing α1 (e.g., zolpidem, CL 218872,68 βCCt, and 3-PBC3) selective ligands suggest the α1-containing GABAA/Bz receptors of the ventral pallidum (VP) play an important role in regulating alcohol’s neurobehavioral effects; particularly alcohol’s reinforcing properties as mentioned above.2,3,19,20,35
Figure 1.

In vitro binding affinities of a series of α1 selective ligands (Ki in nM).
Structure Activity Relationships
A predictive 3-D QSAR pharmacophore/receptor model for inverse agonist/antagonist β-carbolines was initially developed via Comparative Molecular Field Analysis (CoMFA) and later refined.100,101 Affinities of ligands from 15 different structural classes have been evaluated.61 Based on this CoMFA study of a series of β-carbolines, Huang et al. reported that β-carbolines bind to all diazepam sensitive (DS) sites of the BzR with some selectivity at the α1 containing receptor isoform and this was confirmed by in vitro binding affinity of these ligands.102 A lipophilic region (LDi) of the pharmacophore receptor model appears to be larger in the α1, α2 and α3-containing receptor isoforms and important for α1 subtype selectivity.61 More recently, during the design and synthesis of βCCt-related bivalent ligands,103–106 it was found that a series of 3,6-disubstituted β-carbolines (see Figure 1), including 6-iodo-βCCt (5) and 6-trimethylsilanyl-ethynyl-βCCt (6) (Figure 1) possessed α1 subtype selectivity.103
The rigidly linked linear bivalent ligands of βCCt at position “6” did bind to BzR receptors with some α1 subtype selectivity and may provide the desired α1 selectivity through specific occupation of the LDi region of the pharmacophore/receptor model.103 Although the two 3,6-disubstituted-β-carbolines 5 and 6 are less potent than βCCt (1), the potent binding affinities observed for 5 and 6 at the α1 subtype has stimulated the synthesis of the βCCt analogs: 3-substituted-β-carbolines as well as 3,6-disubstituted-β-carbolines.
On the other hand, these studies also indicated that the selectivity of GABAA/BzR site ligands could be described in relation to binding and pharmacological efficacy in vitro. This efficacy was based on the capacity of a ligand to modulate GABAergic function.54 BzR ligands act to modulate chloride flux over a continuum ranging from positive to negative modulation, with neutral antagonists acting theoretically, at a point on the continuum, with zero intrinsic efficacy (e.g. they bind to the receptor but exhibit no activity).74 Consequently, the pharmacological profiles of βCCt and 3-PBC at recombinant α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2 and α6β3γ2 receptor subtypes expressed in Xenopus oocytes were investigated.2,3,64
The results of this study illustrated that βCCt was a near “neutral” antagonist (i.e., little or no efficacy) at all receptor subtypes. In fact, the level of intrinsic efficacy of βCCt in oocytes was less at some receptor subtypes than the classical nonselective antagonist flumazenil (Ro 15-1788, for which intrinsic efficacy at all BZ-sensitive GABAA subtype was relatively low, but not zero). To date, no compound has been characterized that exhibits zero efficacy at all BzR subtypes, raising the possibility that a compound labeled as an “antagonist” may indeed exhibit functional activity given the right circumstances. For example, more recently, the efficacies of both βCCt and 3-PBC in the selective reduction of alcohol responding and production of anxiolytic effects were demonstrated in P and HAD rats following oral administration.107 When compared with naltrexone treatment, these reductions in alcohol responding were more selective and longer in duration.107 In summary, the antagonist βCCt exhibited either a neutral or low-efficacy agonist response at GABA receptors in oocytes. Although there has been some debate in the literature at present as to whether a ligand’s binding or efficacy selectivity was “the more salient factor” in determining a ligand’s capacity to function as an alcohol antagonist,3,67,107 the knowledge of the efficacy of an individual putative anti-alcohol reward ligand across all GABAA receptors was indeed critical to the knowledge of their mode of action in the CNS.
Based on the limited availability of data on the series of α1 “binding” and “efficacy” selective β-carbolines (βCCt, 3PBC) as anti-alcohol agents108 the present study was designed to expand the SAR and search for better α1 subtype selective ligands. These compounds may be promising modulators of alcohol-related co-morbid behaviors in alcohol dependence via the GABAA/BzR system. Although recent evidence suggests a salient role for GABAergic mechanisms in the regulation of excessive alcohol drinking and the negative affective states associated with abstinence, decreased GABAergic tone stemming from chronic alcohol use and withdrawal may serve to generate anxiety.109 Thus, compounds that enhance GABAergic tone may be effective and safe treatments for both excessive alcohol drinking and the negative affective states associated with abstinence and may represent novel pharmacotherapies to treat alcoholism.
In this regard, the chemistry and pharmacological evaluation of a series of structurally modified analogues of βCCt (1) as selective and potent α1 subtype-preferring ligands are described. The synthesis of the α1 selective compound 7 (WYS8) and the structure-activity relationships (SAR) of 3,6-disubstituted β-carbolines are also presented. The established pharmacophore/receptor model61,110 of BDZ binding sites was employed to design ligands with respect to the LDi region at position-6, as well as characterize the binding pocket L1 at position-3. Protein-ligand docking of the α1 subtype GABAA receptor protein and WYS8 illustrated the agreement between the pharmacophore/receptor model and BzR site prediction based on homology modeling.110–113
Chemistry
The synthesis of the ligands under study is outlined in Schemes 1, 2 and 3. The important precursor β-carboline-3-carboxylate-ethyl ester (βCCE, 8) and its corresponding acid (9) were the intermediates required for large-scale synthesis of βCCt (1), as well as an intermediate required for the synthesis of the new β-carbolines. As outlined in Scheme 1, D-L tryptophan 10 was converted into tetrahydro-β-carboline (11) via a Pictet-Spengler reaction on kilogram scale. Fischer esterification of 11, followed by oxidation with activated MnO2 provided the intermediate BCCE (8) on 200 gram scale. Hydrolysis of ester 8 to provide the acid 9, was then followed by esterification in t-butanol with CDI to provide BCCt (1). The synthesis of 3PBC·HCl (1) was more difficult to scale up due to the complex last step (14→2). It began with βCCE (8) from Scheme 1, which was heated with hydrazine to furnish hydrazide (13) in 82% yield. The hydrazide (13) was stirred with nitrous acid to provide an azide, which was unstable, and was converted into 3-amino-β-carboline (14) when stirred with acetic acid (Scheme 1) via a Curtius rearrangement. The last step, originally developed on a 100 mg scale, has now been scaled up to 4 gram levels to furnish 3PBC hydrochloride salt in reasonable yield for studies in primates.
Scheme 1.

Large scale synthesis of βCCt and synthesis of 3PBC.
Scheme 2.

CDI-mediated esterification of 3-substituted β-carbolines followed by the conversion into 3,6-disubstituted β-carbolines.
Scheme 3.

Preparation of 6-substituted acetylenyl βCCt (7, WYS8) and related bivalent ligands.
In Scheme 2, the β-carboline alkyl esters 16–20 as well as chiral βCCt analogs 21–24, and 25, 26 were prepared via the CDI-mediated process described above (see Scheme 1).113 Briefly, when β-carboline-3-carboxylic acid 9 was treated with 1,1-carbonydiimidazole (CDI) in dry DMF, the imidazole derivative 27 which resulted was subsequently transformed into the desired esters by treating it with the corresponding alcohols (individually) in the presence of DBU in a one-pot sequence. The key potential α1 chiral selective analogs CMD-30 R/S isomers (21 & 22) can be synthesized by the CDI method in 90% yield (individually) on 10 gram scale. The required starting chiral alcohols were obtained by asymmetric reduction of the corresponding trifluoromethyl ketones with (+)-DIP-chloride.114,115 or the (−)-DIP-enantiomer. The 6-substituted-iodo-β-carboline-3-carboxylates 28 were then prepared as intermediates to generate different functionality at position-6 through a palladium-mediated cross coupling process. For example, as illustrated in Scheme 2, βCCt (1) was treated with I2/CF3COOAg in chloroform to provide 6-iodo-βCCt 5 (see also 28a) in 80% yield and the 6-substituted targets 29–31 were obtained in 65%–83% yields via a Stille coupling process employing commercially available substituted tributyl-stannanes. The substitution by halogen occurred at position-6 as indicated by analysis of 5 (or 28a) by NMR spectroscopy especially with One Dimensional Nuclear Overhauser Effect (NOE) experiments.
Depicted in Scheme 3 are the synthetic routes for the βCCt related bivalent ligands 32 and 33.103 In order to efficiently effect a palladium mediated Sonogashira process at position-6 of β-carbolines (a reactive electron-rich indole heterocycle), protection/deprotection of the indole Na-H group in β-carbolines 34 and 35 was necessary. The Boc protected 6-ethynyl-β-carboline-3-carboxylic acid t-butyl ester (34) was prepared directly from 6-substituted acetylenyl βCCt (7), which was initially termed WYS8. The common intermediate iodo-βCCt 5 (see also 28a) was then converted into the 6-substituted trimethylsilylacetylenyl βCCt (6) via a Sonogashira coupling process.116,117 At this point, TBAF was employed to remove the trimethylsilyl group to provide the 6-substituted acetylenyl βCCt analog WYS8 (7) as well.
The ester 7, was then protected with a Boc group at the N(1) position to afford 34 under standard conditions. A Sonogashira process was then employed to couple 34 with Boc protected iodo-βCCt (35) to provide the rigid two carbon linked bivalent ligand 32 of βCCt. The Boc protecting group was removed thermally by heating in cumene at high dilution and bivalent ligand 32 was obtained. The bisacetylenic bridged ligand 33 was synthesized from the Boc protected 6-ethynyl-βCCt 34 via a homocoupling process,118 followed by the removal of the Boc group under thermal conditions in cumene at high dilution.
Results and Discussion
(1) βCCt bivalent ligands
The in vitro biological protocols employed in the present study follow the published procedure119,120 and are detailed in the Experimental Section. Although the α1β3γ2 BzR/GABAergic subtype is very similar in structure to the α2 and α3 subtypes, there are slight differences.61,121 One major difference is in region LDi, which appears larger in the α1 subtype than in either the α2, or α3 or α5 subtypes. This is located near position -6 of βCCt (1) and can be seen in the model of the Comparative Molecular Field Analysis (CoMFA) study for the α1 subtype (Figure 3).110,122 In particular, blue contours in the western region of the pharmacophore/receptor model imply positive lipophilic interactions in this area that corresponds to region LDi (a region in the pharmacophore adjacent to the extracellular domain of the receptor) of the unified pharmacophore/receptor model. In this region, bulky substituents are tolerated and occupation of this area with substituents appears to enhance affinity at α1 subtypes. This knowledge provided an opportunity to introduce a linker between two pharmacophoric β-carboline-3-carboxylate residues in order to design selective and rigid bivalent ligands. As described in the Introduction, initial efforts to find a novel series of α1-preferring ligands focused on design and synthesis of βCCt bivalent ligands. Although the α1 subtype selectivity was not amplified with the particular acetylenyl linked bivalent ligand 32, the ligand does bind preferentially at α1 subtypes (Table 1). It was proposed the two-carbon linker was not long enough and that crowding between the second βCCt unit and the receptor protein decreased the binding affinity at the α1 subtype, thereby negating some of the potential selectivity. However, these rigidly linked linear bivalent ligands 32 and 33 fit the GABAA/BzR pharmacophore/receptor model very well (Figure 4).110 The unit at C-6, presumably, protrudes into the extracellular domain of the BzR, as previously expected,102,103 and bound to BzR with some α1 subtype selectivity.102,103 To our knowledge these are the first two bivalent ligands in the β-carboline series, which bind to BzR. Further pharmacological evaluation in vivo of the βCCt bivalent ligand with the longer rigid linker should shed light on the above hypothesis and this would also provide some tools to determine the size and exact location of the LDi region.
Figure 3.

Orthogonal views of CoMFA contour maps for the affinity of 6-benzyl-substituted β-carbolines at the α1β3γ2 BzR.
Orthogonal view of CoMFA contour maps for the α1β3γ2 receptor subtype with 6-benzyl-substituted β-carbolines modeled by Huang.60 Green contours represent areas of positive steric interaction at a contribution level of 85%, which would result in reduced binding affinity. Blue contours represent areas of positive charge interaction at a level of 85%, which would increase the affinity of a ligand.
Table 1.
Affinities (Ki=nM) of 3,6-disubstituted β-carbolines at αxβ3γ2 (x=1–3,5,6) receptor subtypes.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ligands | R6 | R3 | α1 | α2 | α3 | α4 | α5 | α6 |
| 1(βCCt) | H | CO2tBu | 0.72 | 15 | 18.9 | 1000 | 111 | >5,000 |
| 8(βCCE) | H | CO2Et | 1.2 | 4.9 | 5.7 | ND | 26.8 | 2,700 |
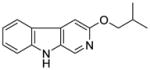
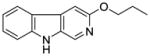
| 2(3-PBC) | H | OnPr | 5.3 | 52.3 | 68.8 | 1000 | 591 | >1,000 |
| 6(WYB14) | CO2tBu | 6.8 | 30 | 36 | 2000 | 108 | 1000 | |
| 6b(WY-B-25) | CO2CH2CF3 | 17 | 59 | 88 | 200 | 1444 | >3000 | |
| 6c(WY-B-99-1) | CO2Et | 4.4 | 4.5 | 5.58 | 2000 | 47 | 2000 | |
| 7(WYS8) | CO2tBu | 0.972 | 111 | 102 | 2000 | 1473 | 1980 | |
| 7b(WY-B-26-2) | CO2CH2CF3 | 4.5 | 44.6 | 42.7 | 2000 | 124 | 2000 | |
| 28a(iodo–βCCt) | I | CO2tBu | 14.4 | 44.9 | 123 | >4000 | 65.3 | >4000 |
| 28b(WY-B-20) | I | CO2CH2CF3 | 12 | 39 | 47 | 2000 | 122 | 3000 |
| 28c(iodo-βCCE) | I | CO2Et | 4.8 | 31 | 34 | 1000 | 286 | 1000 |
| 28d(WY-B-08) | I | CO2CH(CF3)2 | 78 | 301 | 131 | 3000 | 681 | 3000 |
| 29a(WYS13) |  |
CO2tBu | 2.4 | 13 | 27.5 | NA | 163 | 5000 |
| 29b(WYB27-1) |  |
CO2CH2CF3 | 26 | 143 | 117 | 3000 | 127 | 2000 |
| 30a(WYS12) |  |
CO2tBu | 37 | 166 | 314 | NA | 2861 | 5000 |
| 30b(WYB27-2) |  |
CO2CH2CF3 | 9.2 | 13 | 72 | 2000 | 449 | 2000 |
| 31a(WYS15) |  |
CO2tBu | 3.63 | 2.02 | 44.3 | NA | 76.5 | 5000 |
| 31b(WYB29-2) |  |
CO2CH2CF3 | 25 | 137 | 125 | 2000 | 299 | 2000 |
| CMA57 | F | COC3H7 | 3.7 | 27 | 40 | NA | 254 | >2500 |
| CM-A-82a | C(CH3)3 | CO2tBu | 2.78 | 8.93 | 24.5 | 1000 | 7.49 | 1000 |
| CM-A-87 | F | CO2tBu | 1.62 | 4.54 | 14.7 | 1000 | 4.61 | 1000 |
| 32(WYS2) | 30 | 124 | 100 | >300 | >300 | >4000 | ||
| 33(WYS6) | 120 | 1059 | 3942 | 5000 | 5000 | 5000 | ||
The affinity of compounds at GABAA/BzR recombinant subtypes was measured by competition for [3H]flunitrazepam binding to HEK cell membranes expressing human receptors of composition α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2 and α6β3γ2.119 Data represent the average of at least three determinations with a SEM of ±5%.
Figure 4.
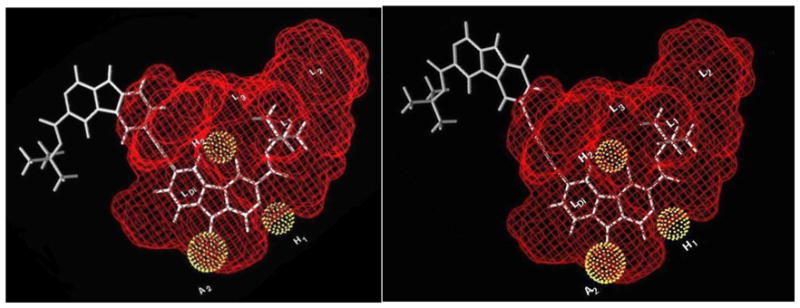
Alignment of bivalent ligands 32 and 33 within the included volume of the α1β3γ2 subtype.
(2) WYS8
A series of 6-substituted-β-carboline-3-carboxylates have been synthesized and bound in vitro to the α1β3γ2 BzR subtype preferentially as compared to other subtypes (see Tables 1–3).61,102 These ligands have also been modeled in the GABAA/BzR pharmacophore model, and the 6-substitutents align well in the LDi region.123 Occupation of this region should lead to enhanced selectivity of a ligand at the α1 containing isoform. Among the new 3,6-disubstituted-β-carbolines, 6-trimethylsilanylethnyl-βCCt 6 has been recently synthesized and found in vitro to prefer the α1 subtype. However, the most selective ligand for the α1 subtype was WYS8 (7). This α1 subtype selective ligand was 100 fold more selective over the other subtypes. This was the most α1 subtype selective ligand reported, to date, to these authors’ knowledge. This 6-substituted acetylenyl βCCt 7 was 214 fold more selective for α1 isoforms over α5 isoforms. Studies of SAR in Table 1 confirmed the occupation of region LDi of the receptor pharmacophore model did enhance α1 selectivity in comparison to the affinity of the non-selective ligand diazepam or the α5 selective ligand, RY080. As illustrated in the two dimensional Figure 5, full occupation of the LDi lip β-carbolines may account for the potency/selectivity of this class of ligands at the α1 subunit. Analysis of the in vitro binding data for this series of bulky 6-substituted β-carbolines (Table 1) has shown some selectivity for the α1 receptor subtype. In addition, it is important to note that binding affinity in this series of ligands of greater than 400nM usually results in zero efficacy at the subtype at pharmacologically relevant concentrations.
Table 3a.
Affinities (Ki=nM) of chiral 3-substituted β-carbolines at αxβ3γ2(x=1–3,5,6) receptor subtypes.
| Ligands | α1 | α2 | α3 | α4 | α5 | α6 | |
|---|---|---|---|---|---|---|---|
| 21 | 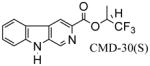 |
90 | 931 | 172 | >3000 | 1847 | >2000 |
| 22 | 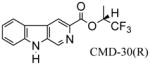 |
27.0 | 343.3 | 453 | >3000 | 1847 | >2000 |
| 23 |  |
17 | 59 | 88 | NA | 1444 | >3000 |
| 24 |  |
7.7 | 32.5 | 43 | NA | 69 | >2000 |
The affinity of compounds at GABAA/BzR recombinant subtypes was measured by competition for [3H]flunitrazepam binding to HEK cell membranes expressing human receptors of composition α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2 and α6β3γ2.119 Data represent the average of at least three determinations with a SEM of ±5%.
Figure 5.
Figure 5a. Overlap of diazepam and βCCt in the pharmacophore/receptor model.
The structure of WYS8 and diazepam in a simple representation of the pharmacophore model. WYS8 (7) (blue line) and diazepam (black line) fitted to the inclusive pharmacophore model for the BzR. Sites H1 and H2 represent hydrogen bond donor sites on the receptor protein complex, while A2 represents a hydrogen bond acceptor site necessary for potent inverse activity in vivo. L1, L2, L3 and LDi are four lipophilic regions in the binding pharmacophore. Descriptors S1, S2, and S3 are regions of negative steric repulsion.
Figure 5b. WYS8 docked in the BzR site of the α1 subtype GABAA receptor. The α1 and γ2 subunits are rendered in yellow and blue, respectively.

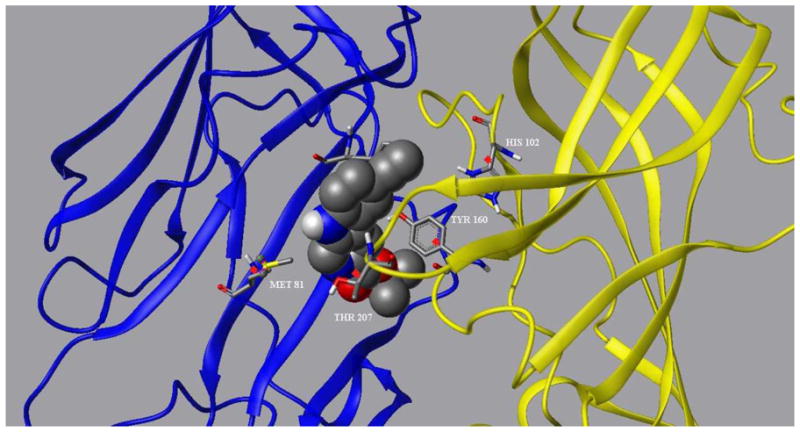
Homology models employed here of the GABAA receptor were as described previously111,124 except that a number of alternative models were considered for loop C, which was two residues shorter than the template and hence built from a loop database. The final model was selected based on assessment of model quality125 and consistency with published mutational data,126–130 particularly with the T207 side-chain appropriately positioned facing the benzodiazepine-binding pocket. Positioning of WYS8 in the BzR was executed using a genetic algorithm (FlexiDock®). Flexible docking provides a means of docking ligands into protein active sites.
(3) 3-Substituted β-carbolines
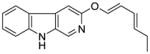
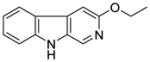
It was initially believed by Braestrup, Loew, and others that an ester moiety at position-3 of β-carbolines was required for a ligand to exhibit high affinity binding at Bz binding sites.84,95,131,132 However, high affinity binding of β-carbolines including the antagonist 3-propoxy β-carboline (3-PβC, Table 2b) demonstrated this was not the case.81,88,89 Examination of data from additional studies78,88,89 have suggested that at least two factors affected high affinity binding at BzR with respect to 3-alkoxy substituted β-carbolines,60,79,102,123 one of which was the lipophilicity of the substituent which interacted at L1. The L1 pocket tolerates linear groups up to 4 carbons in length. Comparison of the in vitro receptor binding affinity of the ligands depicted in Table 2b indicated the ethers 36 and 37 bind potently to α1 subtypes while 34 does not; binding affinity is lost, illustrating that the substituent at the 3-position is to large to allow the ligand to bind. Likewise, the 3-benzyloxy β-carboline 35 is also too bulky to fit the L1 pocket despite its lipophilic nature. The second factor was the ability of the substituent at position-3 to release electron density to the pyridine ring. This enhanced the basicity of the nitrogen atom at N(2) which resulted in a greater ligand-receptor interaction at H1. Analysis of the binding affinities of the novel trifluoroalkyl esters of β-carboline-3-carboxylic acid further supported this hypothesis (Table 2a). The trifluoroalkyl esters exhibited reduced binding affinity at all receptor subtypes when compared to their corresponding alkyl esters (20 vs. 19, 15 vs. βCCE). Since the trifluoromethyl was a strong electron-withdrawing group, when compared to the corresponding alkoxycarbonyl moiety, the 3-trifluoroalkoxycarbonyl substituent would decrease electron density to the pyridine (N2) ring reducing the basicity of the nitrogen atom. This would result in a weaker ligand-receptor interaction at H1. In addition, the trifluoroalkyl group was less lipophilic than the corresponding alkyl moiety, which may result in a weaker interaction at L1. Ramachandran and Hanzawa have reported that trifluoromethyl groups are nearly as large as isopropyl or t-butyl functions.114,133 It was possible, the trifluoromethyl substituted ligands are simply too large to exert high affinity binding; however, βCCT (1), WY-B-24 (25) and CM-A-77 (26) all bound with good potency to α1 BzR subtypes (see Tables 1 and 2), and these ester functions occupy a large molecular volume.
Table 2b.
Affinities (Ki=nM) of Ether-substituted β-carbolines at αxβ3γ2(x=1–3,5,6) receptor subtypes
| Ligands | α1 | α2 | α3 | α5 | α6 |
|---|---|---|---|---|---|
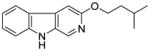 34 |
350.2 | 3000 | 3000 | 3000 | 10000 |
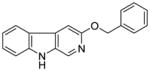 35 |
830 | 3000 | 3000 | 10000 | 10000 |
 36 |
36.9 | 194 | 245 | 1000 | 1000 |
 37 |
24.9 | 123.6 | 139.2 | 1000 | 10000 |
 38 |
245 | 818 | 859 | 10000 | 10000 |
 2 |
5.3 | 52.3 | 68.8 | 591 | 1000 |
 39 |
6.43 | 25.1 | 28.2 | 826 | 1000 |
The affinity of compounds at GABAA/BzR recombinant subtypes was measured by competition for [3H]flunitrazepam binding to HEK cell membranes expressing human receptors of composition α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2 and α6β3γ2.119 Data represent the average of at least three determinations with a SEM of ±5%.
Table 2a.
Affinities (Ki=nM) of 3-substituted β-carbolines at αxβ3γ2(x=1–3,5,6) receptor subtypes
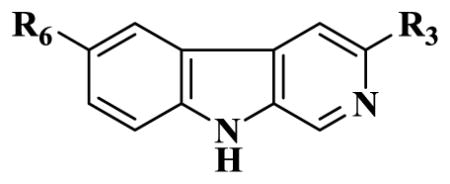 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ligands | R6 | R3 | α1 | α2 | α3 | α4 | α5 | α6 |
| BCCE | H | CO2Et | 1.2 | 4.9 | 5.7 | 1000 | 26.8 | 2700 |
| 15 | H | CO2CH2CF3 | 3.0 | 24.5 | 41.7 | >500 | 125.7 | >2000 |
| 16(WYB09-1) | H | CO2CH(CF3)2 | 3.99 | 8 | 32 | 1000 | 461 | 2000 |
| 17(WYB23-1) | H | CO2CH2CCl3 | 10 | 33 | 43 | 1000 | 189 | 2000 |
| 18(WYB17) | H | CO2CH(CH3)CCl3 | 2000 | 2000 | 2000 | 3000 | 2000 | 5000 |
| 19(CMA64) | H | CO2CH(CH3)C2H5 | 18 | 60 | 116 | NA | 216 | >2000 |
| 20(CMA69) | H | CO2CH(CF3)C2H5 | 1000 | 1000 | 1000 | NA | 1000 | >2000 |
| 25(WY-B-24) | 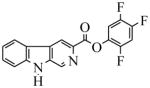 |
22.0 | 177 | 44.8 | 3000 | 422 | 3000 | |
| 26(CM-A-77) | 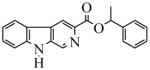 |
33.5 | 1000 | 1000 | 1000 | 1000 | 3000 | |
The affinity of compounds at GABAA/BzR recombinant subtypes was measured by competition for [3H]flunitrazepam binding to HEK cell membranes expressing human receptors of composition α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, α5β3γ2 and α6β3γ2.119 Data represent the average of at least three determinations with a SEM of ±5%.
(4) Chiral 3-substituted β-carbolines
Examination of the binding data for the enantiomeric pair of β-carboline sec-butyl esters 23 and 24 (Table 3) indicated that the (R)-enantiomer 24 bound tighter to the receptor subtypes than the (S) isomer 23. Although both enantiomers exhibited approximately a 4-fold selectivity for the α1β3γ2 subtype, the (R) isomer remained more potent in vitro at all 5 BzR subtypes. Because the receptor subtype selectivity remained about the same for the (R) and (S) isomers, this indicated the stereoenvironment in lipophilic pocket L1 was highly conserved across the entire series of BzR subtypes in agreement with earlier work on the binding affinities of the enantiomers of the framework-constrained 4,5-substituted pyrroloimidazobenzodiazepines.134 It is possible that lipophilc pocket L1 is simply a large area in the pharmacophore/receptor model with only small steric differences between receptor subtypes. More work will be required to determine if this is the case. A similar result was observed in the case of (R) and (S) isomers of CMD-30. The (R)-enantiomer CMD-30 R (22) bound slightly tighter to the receptor subtype than the (S) isomer (21) with almost 70 fold more selectivity for the α1 subtype over the α5 isoform. In addition, previously it was reported that a hydrogen bond between the N (9) H atom of a β-carboline and the secondary site A2 in the receptor pharmacophore was required for potent inverse agonist activity in vivo.78,79 Therefore, a series of ligands with the Boc protection at position-9 such as 40 and 41 were evaluated and were not α1 subtype selective ligands. In fact, they did not bind to BzR at all in agreement with previous work.123
(5) Efficacy of α1 Preferring Ligands in oocytes at GABAA Receptor Channels
The physiological efficacy of βCCt, as compared to other Bz antagonists, was investigated across all diazepam sensitive (DS) receptor subunits at recombinant α1, α2, α3, and α5 receptor subunits in the Xenopus oocytes assay and is depicted in Figure 2 by Harvey et al.2,64 In comparison to other BzR antagonists such as flumazenil and ZK 93426, as mentioned, βCCt exhibited either a neutral or low-efficacy agonist response at GABA α1 (96±7%), α2 (99±10%), α3 (108±6%), and α4 (107±5%) receptors. However, a low-efficacy partial inverse agonist response was observed at the α5 receptor (88±7% of the GABA response). Flumazenil exhibited an efficacy profile that was qualitatively similar to βCCt at the α1 (99±5%), α3 (118±7%), and α5 (96±6%) subtypes. At the α2 receptor, flumazenil produced a low-efficacy agonist response (115±4%), while βCCt was GABA neutral (99±10%). Flumazenil also produced a qualitatively similar response to βCCt at the α4 receptor, albeit the magnitude of GABA potentiation by flumazenil far exceeded that of βCCt (132±6 vs. 108±6%, respectively). However, it is important to note, with regard to α4/α6β3γ2 subtypes, the agonist effect was observed at 10 μM, far above that required for agonist efficacy at the DS subtypes. In contrast, ZK 93426 produced a clear agonist profile, potentiating GABAergic activity by 137±8–148±11% across the α1–α4 subtypes, but was GABA neutral at the α5 receptor (96±6%). These findings suggested that βCCt had no appreciable intrinsic efficacy. The rationale for referring to this agent as a “mixed agonist-antagonist” was based on the fact that, despite the ability to potentiate GABA at certain receptor subtypes, it was “GABA neutral” at select doses. In addition, at select doses, βCCt and 3-PβC were capable of competitive antagonism of classical benzodiazepine agonists,60,64,72 therefore, the development of subtype-selective antagonists for GABAA receptors, such as βCCt, which targeted the GABAA α1 receptor as a weak agonist-like antagonist,74 should facilitate efforts to understand the antialcohol action of β-carbolines in nonhuman primates and humans as well.
Figure 2.
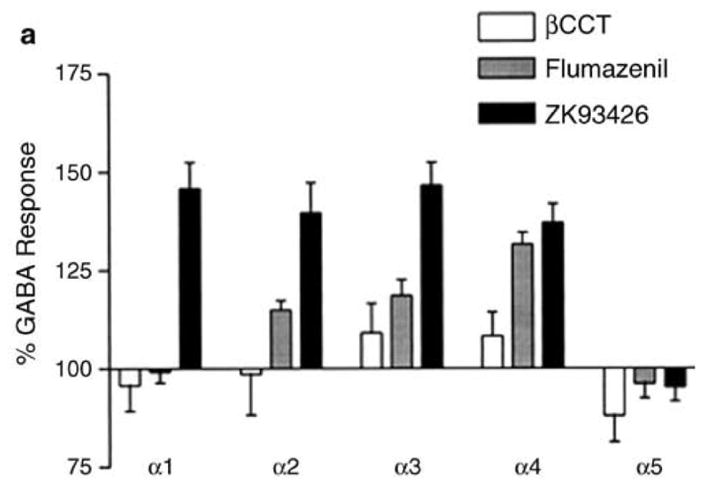
Efficacy of βCCt in modulating GABA at recombinant GABAα1-α5 receptors2 in Xenopus oocytes: comparison with other BzR antagonists.
Modulation of GABAA α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, and α5β3γ2 receptor subunit combinations expressed in Xenopus oocytes by βCCt (open bars), flumazenil (shaded bars), and ZK 93426 (black bars). A saturating concentration (1–10 μM) was coapplied over voltage-clamped oocytes along with an EC50 of GABA.
In the NIMH supported PDSP screen neither βCCT, 3PBC, nor WYS8 exhibited significant interactions at other receptors (see http://pdsp.med.unc.edu for details).
Conclusion
Ethanol allosterically modulates the GABA receptor complex to open the chloride channel and hyperpolarize cells. At the pharmacological level, the effects of ethanol can be antagonized with GABA antagonists.109 Unfortunately, the paucity of high affinity subtype selective ligands capable of discriminating among the various GABAA receptor subtypes has, thus far, precluded study of the precise role GABAA subunits play in mediating EtOH-maintained responding.
A series of β-carboline ligands described here has exhibited some selectivity at the α1 receptor subtype which included β-carboline-3-carboxylate-t-butyl ester (βCCt, 1) and 3-propoxy-β-carboline hydrochloride (3-PBC, 2). These ligands displayed a 20-fold and 10-fold selectivity, respectively, for the α1 subtype over the α2 and α3 receptors, as well as over 150-fold selectivity for the α1 site over the α5 subtype.2,3 βCCt (1) was more selective at the α1 subtype in vitro than the classical α1 selective agonists zolpidem (3) and CL 218872 (4).60,98,99 βCCt and 3-PBC are capable of competitive antagonism of classical benzodiazepine agonists,3,60,72 therefore, the development of subtype-selective antagonists for GABAA receptors which targets the GABAA α1 receptor as a weak agonist-like antagonist,74 should facilitate efforts to understand the antialcohol actions of β-carbolines in nonhuman and human primates alike. Compared with Naltrexone, the reductions in alcohol responding were more selective and longer in duration.107 The goal of the present study was to identify novel α1 GABAA subtype-preferring ligands that may serve as prototypes for further evaluation of clinical efficacy. These types of compounds may provide treatments for excessive alcohol drinking and the negative affective states associated with abstinence. Ligands that enhance GABAergic tone may be effective and safe treatments for both excessive alcohol drinking and the negative affective states associated with abstinence. This may represent novel, new pharmacotherapies to treat alcoholism.
Studies of the structure-activity relationships confirmed that occupation of region LDi of the receptor pharmacophore model did enhance α1 selectivity in comparison to the affinity of the non-selective ligands or the α5 selective ligands. Full occupation of the LDi lipophilic region by β-carbolines may account for the potency/selectivity of this class of ligands at the α1 subunit. Based on the SAR, the most potent α1 selective ligand was 6-substituted acetylenyl βCCt (WYS8, 7). It was suggested the attenuation of EtOH-motivated responding effected by WYS8 (7) was mediated via the α1 selective antagonism of the GABAA/.BzR receptor.2 In regard to ester functions at C(3), although both (R) and (S) enantiomers exhibited approximately a 4-fold selectivity for the α1β3γ2 subtype, the (R) isomer remained more potent in vitro at all 5 BzR sites. Two factors affected high affinity binding at BzR with respect to β-carbolines, one of which was the lipophilicity of the substituent which interacted at L1. The second factor was the ability of the substituent at position-3 to release electron density to the pyridine ring.
The most selective ligand for α1 subtypes, to date, to these authors’ knowledge was WYS8 (7). This 6-substituted acetylenyl βCCt (7) was 214 fold more selective for α1 isoforms over α5 isoforms. WYS8 can clearly be differentiated from nonselective BDZs by its selective binding affinity at the α1 receptor subunit and reduced capacity to potentiate GABA in Xenopus oocytes.
Innate elevations of the α1 and α2 subunits of the HAD rat may contribute to the capacity of novel β-carboline ligands to function as both anxiolytic agents and alcohol antagonists in this genetic rat line.107 These differences may explain the capacity of these novel β-carboline ligands to block alcohol drinking and still exhibit anxiolytic actions in the P and HAD alcoholic rats. WYS8 may be a suitable ligand to evaluate as a preclinical agent to reduce alcohol dependence. Its reduced efficacy at the α1 – 2 subunits in potentiating GABA may render it a safe BDZ receptor ligand devoid of synergistic interactions with alcohol.
Experimental Section
Biological & Pharmacological Testing
Methods for in vitro receptor binding and efficacy in oocytes follow previous work.129, 130 Competition binding assays were performed in a total volume of 0.5 mL at 4 °C for 1 hour using [3H] flunitrazepam as the radiolabel. For these binding assays, 20–50 μg of membrane protein harvested with hypotonic buffer (50 mM Tris-acetate pH 7.4 at 4 degree) was incubated with the radiolabel as previously described.119 Nonspecific binding was defined as radioactivity bound in the presence of 100 μM diazepam and represented less than 20% of total binding. Membranes were harvested with a Brandel cell harvester followed by three ice-cold washes onto polyethyleneimine-pretreated (0.3%) Whatman GF/C filters. Filters were dried overnight and then soaked in Ecoscint A liquid scintillation cocktail (National Diagnostics; Atlanta, GA). Bound radioactivity was quantified by liquid scintillation counting. Membrane protein concentrations were determined using an assay kit from Bio-Rad (Hercules, CA) with bovine serum albumin as the standard.
The results are summarized in Tables 1–3.
The electrophysiological analyses of all selective compounds were performed with whole cell variation of the patch-clamp-technique, in HEK cells employing GABA concentrations around the subtype-specific EC203,120 to depict the quantitative efficacy difference [i.e., GABA modulation] and qualitative subunit modulation [i.e., subunit type] of these ligands relative to diazepam.
Melting points were taken on a Thomas-Hoover melting point apparatus or an Electrothermal Model IA8100 digital melting point apparatus and are reported uncorrected. Proton NMR spectra were recorded on a Bruker 250- or 300-MHz multiple-probe instrument. Infrared spectra were recorded on a Nicolet DX FTIR BX V5.07 spectrometer or a Mattson Polaris IR-10400 instrument. Low-resolution mass spectral data (EI/CI) were obtained on a Hewlett-Packard 5985B GC-mass spectrometer, while high resolution mass spectral data were taken on a VG autospectrometer (Double Focusing High Resolution GC/Mass Spectrometer, UK). Microanalyses were performed on a CE Elantech EA1110 elemental analyzer. Analytical TLC plates employed were E. Merck Brinkman UV active silica gel (Kieselgel 60 F254) on plastic, and silica gel 60b for flash chromatography was purchased from E. M. Laboratories. All chemicals were purchased from Aldrich Chemical Co. unless otherwise stated. All solvents were dried according to the published procedures.
1,2,3,4-Tetrahydro-9H-pyrido [3,4-b] indole-3-carboxylic acid (11)
D, L-tryptophan (1000 g, 4.9 mol) was added to a solution of aq sodium hydroxide (12 L, 0.4 N) after which the mixture was stirred until it dissolved. Formaldehyde (560 mL of a 37% aq solution, 6.9 mol) was added and the solution was allowed to stir for three days at 37 °C. Glacial acetic acid (400 mL) was added which resulted in the precipitation of a solid as a fine suspension. The mixture was allowed to stir for two days, after which additional solid formed. The solid was filtered from the medium, washed with water (4 × 1000 mL), and dried to give 11 (953g, 90.0%). 11: mp 295 °C (lit mp 293 °C)135 (lit mp 286 °C);136 (IR (KBr) 3600-2300, 1630 cm−1; MS (CI, CH4), m/z (relative intensity) 217 (M+ + 1, 50), 216(62), 169(59), 144(100). This material was employed directly in the next step.
Ethyl 1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole-3-carboxylate (12)
The 1,2,3,4-tetra-hydro-9H-pyrido[3,4-b]indole-3-carboxylic acid 11 (500 g, 2.3 mol) was dissolved in anhydrous ethanol (9 L) in a 12L (3 neck) flask, and conc sulfuric acid (98%, 245 mL, 4.6 mol) was carefully added to the solution until most of the solid dissolved. The reaction mixture was heated to reflux under nitrogen until the starting material was no longer detected by TLC on silica gel (48 h), and the solution became homogeneous. The reaction solution was cooled and the solvent removed under reduced pressure. The residue was dissolved in H2O (6.4 L) and the pH of the solution adjusted to 8 with cold aq NH4OH (conc.) after which a precipitate formed. This mixture was then extracted with CHCl3 (6 × 2.5 L). The combined organic layers were dried (Na2SO4) and the solvent was removed under reduced pressure to yield a light tan solid which was dried in a vacuum oven at 100 °C to provide 12 (465 g, 83%). 11: mp 150 °C (lit. mp 149–150 °C);90,137 1H (300 MHz, CDCl3) δ 1.33 (t, J = 7.32 Hz, 3H), 2.45 (s(br), 1H), 2.88 (dd, J = 9.70 Hz, J = 15.37 Hz, 1H), 3.13 (dd, J = 4.76 Hz, J = 15.5 Hz, 1H), 3.77 (dd, J = 4.76 Hz, J = 9.70 Hz, 1H), 4.07 (s(br), 2H), 4.26 (q, J = 7.14 Hz, 2H), 7.18-7.08 (m, 2H), 7.29 (d, J = 7.87 Hz, 1H), 7.48 (d, J = 7.68 Hz, 1H), 8.17 (s(br), 1H); MS (CI CH4) m/e 144 (97.3), 245 (M+1, 87), 244 (100), 183 (6), 171 (33), 144 (83). This material was employed directly in the next step.
Ethyl 9H-pyrido[3,4-b]indole-3-carboxylate (8)
Into a round bottom flask (12 L) equipped with a reflux condenser and an overhead stir was added 1,2,3,4-tetrahydro-β-carboline-3-carboxylic acid ethyl ester 12 (200g, 0.86 mol) and dry benzene (8 L). The solution was allowed to heat to reflux at which time activated MnO2 (200 g) was added to the flask. Additional quantities of activated MnO2 were added until analysis by TLC (silica gel/ethyl acetate) indicated the absence of starting material. The hot solution was filtered through a bed of celite to remove the MnO2 and the filter cake was washed with hot benzene. The benzene layers were allowed to cool. A precipitate formed and was collected by vacuum filtration, which provided (100–120g, 50%–60%) of pure β-carboline-3-carboxylic acid ethyl ester 8 (βCCE). The benzene which remained in the filtrate was removed under reduced pressure to provide 25–35 g of additional βCCE, but as crude material. The crude material could be purified by recrystalization from ethanol. 8: mp 225–227 °C (lit. 224–229 °C);137,138 1H (300 MHz, DMSO-d6) δ 1.36 (t, J = 6.95 Hz, 3H), 4.37 (q, J = 6.95 Hz, 2H), 7.37-7.24 (m, 1H), 7.68-7.57 (m, 2H), 8.38 (d, J = 7.87 Hz, 1H), 8.90 (s, 1H), 8.97 (s, 1H), 10.7 (br, 1H); MS (CI, CH4) m/e 241 (M+ + 1, 47), 195 (22), 168 (100), 140 (9). This material was employed directly in the next step.
β-Carboline-3-carboxylic acid (9)
β-Carboline-3-carboxylic acid ethyl ester 8 (30.0 g, 0.126 mol) was suspended in aq NaOH (10%, 1.5L) and heated to reflux until all the material had gone into solution (1 h). The heating was continued for an additional 3 h. The reaction mixture was cooled to rt and acidified by addition of ice cold aq conc HCl to pH 4. The precipitate which resulted was stirred overnight. The solid was collected by vacuum filtration and washed with H2O (2 × 150 mL). The product was dried at 80 °C under vacuum for 24 h to provide 9 (26.1 g, 99%). 9: mp 220–221 °C (lit. mp 220 °C);67 IR (KBr) 3260, 2970, 1710 cm−1; 1H NMR (CDCl3) δ 7.31 (t, J = 7.32 Hz, 1H), 7.69-7.57 (m, 2H), 8.38 (d, J = 7.87 Hz, 1H), 8.90 (s, 1H), 8.96 (s, 1H), 12.10 (s, 1H); MS (CI, CH4), m/e (M+ + 1, 269). Anal. Calcd. for C16H16N2O2 (0.55 H2O): C, 69.07; H, 6.19; N, 10.07. Found: C, 68.81; H, 5.77; N, 10.00.
β-Carboline-3-carboxylic acid t-butyl ester (1)
To a solution of carbonyl diimidazole (28.2 g, 0.177 mol) in anhydrous DMF (1.2 L) was added dry β-carboline-3-carboxylic acid 9 (25 g, 0.118 mol) under argon. The reaction mixture was initially a pale yellow-colored suspension, but after stirring for 30 min, a purple or red-colored solution resulted. The reaction mixture was stirred for an additional 2h at rt and carbon dioxide was released during the reaction. Analysis by TLC (silica gel) indicated the absence of starting material on the baseline. To this reaction mixture was added dry DBU (18 g, 0.118 mol) and dry freshly distilled t-butyl alcohol (437 g/560 mL, 50 eq). The mixture was heated at 85°C for 18h until analysis by TLC indicated the disappearance of the imidazole intermediate. The solvent was then removed under reduced pressure. The residue was partitioned between CH2Cl2 (1.2 L) and H2O (800 mL). The organic layer was separated and the H2O layer was extracted with CH2Cl2 (2 × 500 mL). The combined organic layer was washed with an aq solution of 10% K2CO3, water, brine and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, EtOAc/hexane = 1:1) to provide βCCt (20 g, 65%) as a white solid. βCCt can be recrystallized from EtOAc to provide white crystals 1: mp 301–303°C (lit. mp 298–300);80 IR (KBr) 3500-3400, 3200-3000, 1610, 1560, 1370, 1340 cm−1; 1H (300 MHz, DMSO-d6) δ 1.74 (s, 9H), 7.37 (t, J = 7.48 Hz, 1H), 7.63 (t, J = 7.66 Hz, 1H), 7.73 (d, J = 8.52 Hz, 1H), 8.24 (d, J = 7.89 Hz, 1H), 8.85 (s, 1H), 9.12 (s, 1H), 8.97 (s, 1H), 10.19 (br, 1H); The spectral data for 1 were identical to those reported in the literature.88
2,2,2-Trifluoroethyl β-carboline-3-carboxylate 15 was prepared from β-carboline-3-carbo-xylic acid 9 and 2,2,2-trifluoroethyl alcohol following the procedure employed for the preparation of 1. 15: mp 264–266 °C; IR (NaCl) 3275, 1735 cm−1; 1 H NMR (300 MHz, CDCl3) 4.87 (m, 2H), 7.42 (m, 1H), 7.65 (m, 2H), 8.24 (d, J = 7.9 Hz, 1H), 8.93 (s, 1H), 9.09 (s, 1H), 9.10 (s, br, 1H);; 13C NMR (75.5 MHz, CDCl3) δ 60.4, 60.8, 112.8, 118.9, 120.7, 121.2, 122.7, 127.8, 129.2, 134.5, 135.2, 138.1, 141.3, 164.5; MS (EI) m/e (relative intensity) 294(M+, 30), 195(7), 168(100), 167(42). Anal. Calcd. for C14H9F3N2O2: C, 57.15; H, 3.08; N, 9.52. Found: C, 57.22; H, 3.14; N, 9.23.
9H-β-Carboline-3-carboxylic acid 2,2,2-trifluoro-1-trifluoromethyl-ethyl ester 16 was prepared following the procedure employed for the preparation of 1. 16: 1H NMR (300 MHz, DMSO-d6) δ 7.05–7.21 (m, 1H), 7.33–7.44 (t, 1H), 7.61–7.72 (m, 2H), 8.48 (d, J = 7.89 Hz, 1H), 9.09 (s, 2H), 12.31 (s, 1H); 13C NMR (75.5 MHz, DMSO-d6) δ 65.9, 66.8, 112.9, 120.1, 120.9, 122.8, 127.8, 129.4, 133.3, 134.9, 138.4, 141.3, 162.9. This material was pure by TLC (silica gel).
9H-β-Carboline-3-carboxylic acid 2,2,2-trichloro-ethyl ester 17 was prepared following the procedure employed for the preparation of 1. 17: 1H NMR (300 MHz, CDCl3) δ 5.17 (s, 2H), 7.38–7.44 (m, 1H), 7.63–7.74 (m, 2H), 8.27 (d, J = 7.95 Hz, 1H), 8.95 (s, 1H), 9.21 (s, 1H), 9.66 (s, 1H). This material was pure by TLC (silica gel).
9H-β-Carboline-3-carboxylic acid 2,2,2-trifluoro-1-methyl-ethyl ester 18 was prepared following the procedure employed for the preparation of 1. 18: m.p. 247 249 °C; IR (NaCl) 2359, 1729, 1345, 1251, 1092, 729, 450 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.82 (d, J = 6.1 Hz, 3H), 5.96 (q, 1H), 7.42 (t, 1H), 7.67 (m, 2H), 8.26 (d, J = 7.6 Hz, 1H), 8.8 (s, 1H), 8.9 (s, 1H), 9.5 (br, s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 16.5, 79.1, 112.3, 118.5,121.2, 121.5, 121.9, 128.8, 129.2, 133.9, 137.4. This material was pure by TLC (silica gel).
(S)-1,1,1-Trifluoroisopropyl β-carboline-3-carboxylate (21)
To a solution of carbonyl diimidazole (0.168 g, 1.03 mmol) in anhydrous DMF (5 mL), β-carboline-3-carboxylic acid 9 (0.10 g, 0.47 mmol) was added. The reaction mixture which resulted was stirred for 2 h at rt until analysis by TLC (silica gel) indicated the absence of starting material on the baseline. The solution which resulted was then cooled to −6 °C and this was followed by addition of (S)-1,1,1-trifluoropropan-2-ol (2.3 eq) which was contaminated with some EtOH. The dry DBU (100 mg, 0.68 mmol) in dry DMF (0.5 mL) was slowly syringed into the reaction mixture at −6 °C. The mixture was stirred at 0 °C for 8 h until analysis by TLC (silica gel) indicated the diaspperance of the imidazole intermediate. The reaction mixture was then poured into ice water (30 mL) and extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were washed with H2O (5 × 40 mL), brine and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, EtOAc/hexanes = 2:1) to provide 21 (0.113 g, 78%) as a white solid. 21: mp 239–241 °C; (c = 0.81, in CHCl3); IR (NaCl) 3266, 1725, 1502 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.63 (d, J = 6.6 Hz, 3H), 5.75 (m, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.64 (t, J = 7.6 Hz, 1H), 7.72 (d, J = 8.1 Hz, 1H), 8.25 (d, J = 7.9 Hz, 1H), 8.91 (s, 1H), 9.18 (s, 1H), 10.04 (s, br, 1H); MS (EI) m/e (relative intensity) 308 (M+, 17), 168 (100), 140 (21). Exact mass calcd. for C15H11F3N2O2: 308.0773. Found: 308.0773. Anal. Calcd. for C15H11F3N2O2: C, 58.45;H, 3.60; N, 9.09. Found: C, 58.15; H, 3.63; N, 8.88.
(S)-1,1,1-Trifluorobutan-2-ol was prepared following the literature procedure.114 To an oven-dried, 25 mL round-bottom flask was transferred (−)-DIP-Chloride (10.68 g, 33 mmol) in a glove box. Then 1,1,1-trifluorobutan-2-one (4.03 g, 32 mmol) was added at rt under argon. The reaction mixture was stirred at rt for 10 h. Ethyl ether was added and the reaction solution was cooled to 0 °C followed by addition of acetaldehyde (1.6 g, 1.1 eq). The reaction mixture was allowed to warm to rt and stirring was continued for 24 h while the second equivalent of α-pinene was liberated. An aq solution of sodium hydroxide (2.5M, 30 mL) was added and the solution which resulted was extracted with ether (3 × 30 mL). The ether layer was dried (Na2SO4) and fractionally distilled through a Pyrex distilling column packed with glass beads. The desired alcohol was collected along with EtOH (20%). (S)-1,1,1-Trifluorobutan-2-ol: 1H NMR (300 MHz, CDCl3) 1.04 (dt, J = 0.6 Hz and J = 7.4 Hz, 3H), 1.58 (m, 1H), 1.72 (m, 1H), 3.82 (m, 1H). This material was used in a later step without further purification.
(R)-1,1,1-Trifluoroisopropyl β-carboline-3-carboxylate 22 was prepared from the acid 9 and (R)-1,1,1-trifluoropropan-2-ol following the procedure employed for preparation of (S)-1,1,1-trifluoro-propan-2-ol. 22: mp 239–241 °C; (c = 0.88, in CHCl3); The spectral data for 22 were identical to those for 21; however, the optical rotation was in the opposite direction.
(S)-β-Carboline-3-carboxylic acid sec-butyl ester (23)
To a solution of carbonyl diimidazole (1.53 g, 9.4 mmol) in anhydrous DMF (50 mL) was added β-carboline-3-carboxylic acid 9 (1.0 g, 4.7 mmol). The reaction mixture was stirred for 2 h at rt and carbon dioxide was released during the reaction. Analysis by TLC (silica gel) indicated the absence of starting material on the baseline. To this reaction mixture was added dry DBU (0.72 g, 4.7 mmol) and dry (S)-butyl alcohol (1.13 g, 15.2 mmol). The mixture which resulted was heated at 55 °C for 8 h until analysis by TLC (silica gel) indicated the disappearance of the imidazole intermediate. The solvent was then removed under reduced pressure. The residue was partitioned between CH2Cl2 (100 mL) and H2O (100 mL). The organic layer was separated and the H2O layer was extracted with CH2Cl2 (2 × 80 mL). The combined organic layer was washed with H2O (3 × 100 mL), brine and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, EtOAc/hexane = 2:1) to provide 23 (0.96 g, 76%) as a white solid. 23: mp 212–213 °C; [α]25 = 35.6° (CHCl3, c = 1.43); IR (KBr) 3222, 1706, 1622, 1494 cm−1; 1 H NMR (300 MHz, CDCl3) δ 0.99 (t, J = 7.5 Hz, 3H), 1.40 (d, J = 6.3 Hz, 3H), 1.79 (m, 2H), 5.27 (m, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), 8.21 (d, J = 7.9 Hz, 1H), 8.44 (s, 1H), 9.10 (s, 1H), 11.47 (s, br, 1H); MS (CI, CH4) m/e (relative intensity) 269 (M+ + 1, 100), 241(15), 213(41), 195(14). Anal. Calcd. for C16H16N2O2: C, 71.62; H, 6.01; N, 10.44. Found: C, 71.33; H, 6.09; N, 10.26.
(R)-β-Carboline-3-carboxylic acid sec-butyl ester 24 was prepared in 75% yield following the procedure for preparation of 1. 24: [α]25 = -35.2° (CHCl3, c = 1.25) The spectral data for 22 were identical to those for 23, except the optical rotation was opposite in direction.
6-Iodo-9H-β-carboline-3-carboxylic acid t-butyl ester (5)
Into a round bottom flask (250 mL) was added CF3CO2Ag (1.03 g, 4.67 mmol), β-carboline-3-carboxylic acid t-butyl ester 9 (1.02 g, 3.83 mmol), and CHCl3 (100 mL). This was followed by addition of iodine (1.15 g, 4.66 mmol). The reaction mixture was allowed to stir at rt for 6 h after which another portion of CF3CO2Ag (500 mg, 2.26 mmol) was added and stirring continued for another 10 h at reflux. Analysis by TLC (silica gel) indicated that most of the βCCt had disappeared. The reaction mixture was filtered through a bed of celite to remove the solid salts and the filter cake was washed with EtOH (3 × 50 mL). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, EtOAc/hexane = 4:1) to provide 5 (908 mg, 65%) as a white solid. 5: mp 347–348 °C (dec.); IR (NaCl) 3223, 1710, 1485, 1323, 1245, 1161, 1104, 1020 cm−1; 1 H NMR (300 MHz, CDCl3) δ 1.68 (s, 9H), 7.63 (d, J =8.6 Hz, 1H), 7.85 (dd, J = 1.5 Hz and J = 8.6 Hz, 1H), 8.12 (s, 1H), 8.53 (d, J = 1.4 Hz, 1H), 9.31 (s, 1H), 11.47 (s, br, 1H); 13C NMR (75.7 MHz, CDCl3) δ 28.3, 80.8, 83.6, 115.1, 118.1, 123.9, 126.7, 131.2, 134.1, 136.8, 137.5, 138.7, 140.4, 164.9; MS(EI800) m/e (relative intensity) 395 (M+ + 1, 18), 367 (20), 339 (100), 268 (33). Anal. Calcd. for C16H15IN2O2: C, 48.75; H, 3.84; N, 7.11. Found: C, 49.01; H, 3.91; N, 6.95.
6-Trimethylsilanylethynyl-9H-β-carboline-3-carboxylic acid t-butyl ester (6)
Into a round bottom flask (50 mL) which contained a solution of degassed THF/Et3N (10 mL/2 mL), was added 6-iodo-9H-β-carboline-3-carboxylic acid t-butyl ester 5 (400 mg, 1.02 mmol), bis[triphenylphosphine] palladium dichloride (35 mg, 5 mol%) and copper(I) iodide (7 mg, 5 mol%) Note: Practically, on a small scale, CuI could be used up to 10–15 mol% because of its lower molecular weight compared to the palladium catalyst; on a bigger scale, both Pd(PPh3)2Cl2 and CuI can be used as low as 0.5–1 mol%. The reaction mixture was then degassed 2 times with an oil pump at −78 °C, and then the trimethylsilyl acetylene (300 mg, 3.06 mmol) was added into the mixture and it was degassed one more time at −78 °C. The reaction mixture was gradually allowed to warm to rt and stirred at rt for an additional 0.5–1h until all the starting material had disappeared (TLC analysis indicated that the original red spot changed color to purple, since both s.m. and product had very similar Rf values). The solvent was removed under reduced pressure at this point and the residue was purified by flash chromatography (silica gel, EtOAc/hexane = 4:1) to provide 6 (340 mg, 92%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 1.71 (s, 9H), 1.75 (s, 9H), 7.67 (m, 2H), 8.26 (d, J = 7.6 Hz, 1H), 8.8 (s, 1H), 8.9 (s, 1H), 9.5 (br, 1H); 13C NMR (75.5 MHz, CDCl3) δ 0.021, 28.3, 81.9, 92.6, 105.7, 113.2, 114.9, 117.7, 121.3, 125.5, 128.3, 132.4, 133.8, 138.2, 138.4, 141.5, 165.4; EIMS 364(M+, 38), 293(12), 264(100), 249(48), 124(60). This material was employed directly in the next step.
6-Ethynl-9H-β-carboline-3-carboxylic acid t-butyl ester (7)-WYS8
To a solution of 6-trimethyl-silanyl-β-carboline-3-carboxylic acid t-butyl ester 6 (850 mg, 2.32 mmol) in THF (10 mL) was added 1.2 eq of TBAF (2.8 mL of 1M TBAF/THF solution) at 0 °C and then the solution was allowed to warm to rt. After consumption of the starting material as indicated by TLC, H2O (10 mL) was added and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was concentrated under reduced pressure and the residue was chromatographed on a short silica gel column (EtOAc/hexane = 4:1) to give 7 (620 mg, 92%) as a white solid. 7: 1H NMR (300 MHz, CDCl3) δ 1.75 (s, 9H), 3.54 (s, 1H), 7.62–7.85 (m, 2H), 8.41 (s, 1H), 8.83 (s, 1H), 9.33 (s, 1H), 11.1 (s, 1H); EIMS 292 (M+, 25), 236(12), 192 (100), 164(30). This material was pure by TLC (silica gel) and used directly in a later step.
6-Thiophen-2-yl-9H-β-carboline-3-carboxylic acid t-butyl ester (30a). Representative procedure for preparation of 6-subsitituted β-carbolines
A solution of 6-Iodo-β-carboline-3-carboxylic acid t-butyl ester 5 (265 mg, 0.67 mmol) in dry toluene (15 mL) was degassed under vacuum and purged with dry N2 through the solution 3 times. The mixture was then heated to 140 °C under nitrogen after which tetrakis(triphenylphosphine) palladium (0) (77 mg, 0.067 mmol, 10mol%) and 2-(tributylstannyl) thiophene (718 mg, 2.01 mmol) were added in one portion. The mixture was heated to reflux under nitrogen. After 12 h, the mixture was allowed to cool to rt and the precipitate which resulted was removed by vacuum filtration. The filtrate was concentrated under reduced pressure and the residue was treated with a saturated aq solution of NaHCO3 (30 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined extracts were washed with brine and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, EtOAc/hexane = 5:1) to provide a white solid 30a (195 mg, 83%). 30a: 1H NMR (300 MHz, CDCl3) δ 2.07 (s, 9H), 7.15 (t, 1H), 7.31 (d, J = 3Hz, 1H), 7.40 (d, J = 3Hz, 1H), 7.89 (q, 2H), 8.44 (s, 1H), 8.89 (s, 1H), 9.32 (s, 1H), 11.2 (s, 1H). This material was pure by TLC (silica gel).
6-Furan-2-yl-9H-β-carboline-3-carboxylic acid t-butyl ester 29a was prepared following the procedure for preparation of 30a. 29a: 1H NMR (300 MHz, CDCl3) δ 1.63(s, 9H), 6.63 (m, 1H), 6.97 (d, J = 6Hz, 1H), 7.70 (d, J = 9Hz, 1H), 7.77 (s, 1H), 7.96 (d, J = 9Hz, 1H), 8.73 (s, 1H), 8.90 (s, 1H), 8.96 (s, 1H), 12.1 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 28.4, 80.7, 102.3, 106.3, 112.4, 113.2, 117.4, 117.8, 127.1, 127.8, 133.6, 134.1, 138, 140.6, 142.6, 154.1, 165.0, 166.0. This material was pure by TLC (silica gel).
6-Furan-2-yl-9H-β-carboline-3-carboxylic acid 2,2,2-trifluoro-ethyl ester 29b was prepared following the procedure for preparation of 30a. 29b: 1H NMR (250 MHz, CDCl3) δ 4.82–4.92 (m, 2H), 6.54 (s, 1H), 6.75 (s 1H), 7.38 (s, 1H), 7.65 (d, J = 10Hz, 1H), 7.96 (d, J = 10Hz, 1H), 8.5 (s, 1H), 8.93 (s, 1H), 9.13 (s, 1H), 9.59 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 60.9, 105.3, 112.5, 112.8, 113.3, 117.6, 119.2, 122.3, 123.8, 125.3, 127.9, 134.7, 135.3, 138.6, 140.6, 142.7, 154.1, 164.4. This material was pure by TLC (silica gel).
6-Iodo-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester (35)
To a solution of 6-iodo-β-carboline-3-carboxylic acid t-butyl ester 5 (2 g, 5.06 mmol) in anhydrous CH2Cl2 (30mL), (Boc)2O (1.32 g, 6.07 mmol) and DMAP(123 mg, 1.01 mmol) were added. The reaction mixture was allowed to stir at rt for half an hour until analysis by TLC (silica gel) indicated that the starting material had been converted into the Boc protected indole 35. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, EtOAc/hexane = 5:95) to provide 35 (2.3 g, 92%) as a white solid. 35: mp 317–320 °C; IR (NaCl) 2975, 2917, 1728, 1457, 1343, 1238, 1154, 1119, 1031, 811cm−1; 1H NMR (300 MHz, CDCl3) δ 1.71 (s, 9H), 1.77 (s, 9H), 7.91 (d, J = 8.972 Hz, 1H), 8.22 (d, J = 8.972Hz, 1H), 8.42 (s, 1H), 8.61 (s, 1H), 9.61 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 28.1, 28.2, 82.1, 85.8, 87.2, 115.9, 118.5, 125.7, 129.9, 130.6, 135.7, 138.9, 143.0 149.6 164.1; MS (EI) m/e (relative intensity) 494 (M+, 42), 438 (71), 338 (100), 294 (70), 268 (8), 212 (8), 168 (35). Anal. calcd. for C21H23IN2O4(0.1 C6H14): C, 51.60; H, 4.85; N, 5.57; Found: C, 51.84; H, 4.88; N: 5.45.
6-Trimethylsilanylethynyl-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester (36)
Dichlorobis(triphenylphosphine)palladium(II) (140 mg; 2 mol %), and CuI (40 mg; 2 mol %) were added to a solution of 6-iodo-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester 35 (4.9 g; 10 mmol) in anhydrous THF (30 mL) and triethylamine (10 mL). The mixture was degassed, and back-filled three times with argon. Then trimethylsilyl acetylene (1.08 g; 11 mmol) was added with stirring under argon. After the mixture was allowed to stir for 1 h, the solvents were removed under vacuum and the residue was chromatographed on a short column (silica gel, hexane/CH2Cl2 = 7:3) to give 36 (4.36 g, 94%) as a white solid. 36: mp 334–336 °C (dec); IR (NaCl) 2974, 2137, 1732, 1559, 1476, 1469, 1368, 1343, 1309, 1247, 1156, 1109, 872, 842, 760 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.31 (s, 9H), 1.71 (s, 9H), 1.78 (s, 9H), 7.77 (d, J = 8.97 Hz, 2H), 8.25 (s, 1H), 8.41 (d, J = 8.61 Hz, 2H), 8.65 (s, 1H), 9.62 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 0.11, 28.1, 28.2, 82.1, 85.8, 94.4, 104.4, 116, 116.5, 118.7, 123.5, 124.7, 131.6, 133.7, 136.2, 138.1, 139.2, 142.9, 149.7, 164.1; MS (EI) m/e (relative intensity) 465 (M+, 30), 409 (100), 365 (21), 308 (80), 262 (25), 249 (40). This material was pure by TLC (silica gel) and used in the next step.
6-Ethynyl-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester 34
To a solution of 6-trimethylsilanylethynyl-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester 36 (2.14g, 4.6 mmol) in THF (20 mL), 1.2 eq of TBAF (5.52 ml of 1M TBAF/THF solution) at 0 °C was added and then the solution was allowed to warm to rt. After consumption of the starting material as indicated by TLC (silica gel), H2O (10 mL) was added and the mixture was extracted with CH2Cl2 (20 mL, 3X). The combined organic layer was concentrated under reduced pressure and the residue was chromatographed on a short column (silica gel, hexane/CH2Cl2 = 4:1) to give 34 (1.66 g, 92%) as a white solid. 34: mp 226–229 °C; IR (NaCl) 3303, 2978, 2346, 2232, 1734, 1622, 1560, 1463 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.54 (s, 9H), 1.72 (s, 9H), 1.79 (s, 9H), 3.17 (s, 1H), 7.79 (d, J = 8.76 Hz, 2H), 8.27 (s, 1H), 8.44 (d, J = 8.94 Hz, 2H), 8.67 (s, 1H), 9.64 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 27.3, 28.1, 28.2, 67.8, 82.1, 83.0, 85.1, 85.5, 116.0, 116.7, 123.5, 124.9, 133.7, 136.2, 138.1, 139.4, 143.0, 164.2; MS (EI) m/e (relative intensity) 393 (M+, 32), 338 (13), 321 (26), 293 (100), 167 (28), 139 (24). Anal. Calcd. for C23H24N2O4 (0.05 CH2Cl2): C, 69.78; H, 6.12; N, 7.06; Found: C: 69.70; H: 6.10; N: 6.81.
1,2-Bis(9H-β-carboline-3-carboxylic acid tert-butyl ester) ethyne (32)
Dichlorobis (tri-phenylphosphine)palladium(II) (60 mg, 2 mol%) and copper iodide (16 mg, 2 mol%) were added to a mixture of 6-ethynyl-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester 34 (1.6g, 4.1mmol) and 6-iodo-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester 35 (2.1g, 4.25mmol) in THF/TEA(30 mL; 4:1). The reaction mixture which resulted was degassed, and back-filled three times with argon. The reaction mixture was then allowed to stir at rt for about 1h until analysis by TLC (silica gel) indicated the starting materials were absent. The solution was concentrated under reduced pressure and the residue was chromatographed on a silica gel column with CH2Cl2 as the eluent to give 1,2-bis(β-carboline-3,9-dicarboxylic acid di-tert-butyl ester) ethyne (2.97 g, 95%) as a white solid: mp 305–307°C; IR (NaCl) 2972, 2929, 1737, 1559, 1466, 1338, 1156, 1102, 823, 624cm−1; 1H NMR (300 MHz, CDCl3) δ 1.72 (s, 9H), 1.79 (s, 9H), 7.86 (d, J = 8.97 Hz, 1H), 8.32 (s, 1H), 8.47 (d, J = 8.79Hz, 1H), 8.68 (s, 1H), 9.63 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 28.2, 82.1, 85.8, 89.0, 116.0, 116.7, 118.6, 123.6, 124.3, 131.5, 133.3, 136.1, 138.1, 139.1, 143.0, 149.7, 164.2; MS (FAB) 759((M + H)+, 13). Anal. calcd. for C44H46N4O8 (H2O): C, 68.03; H, 6.22; N, 7.21; Found: C: 68.10; H: 6.22; N: 7.21.
The 1,2-bis(β-carboline-3,9-dicarboxylic acid di-tert-butyl ester) ethyne 32 (800mg, 1.05 mmol) was added to a distilled solution of cumene (40 mL), which had been degassed. The reaction vessel was evacuated and refilled with nitrogen three times. The temperature was then brought to reflux for about 30 min until a yellow precipitate had formed. The mixture which resulted was filtered and washed with hexane to give pure dimer 32 (545 mg, 93%). 32: m.p. >350 °C (dec.); IR (KBr) 3227, 1716, 1327, 1162, 738, 450 cm−1; 1H NMR (300 MHz, DMSO) δ 1.62 (s, 9H), 7.70–7.80 (m, 2H), 8.7 (s, 1H), 8.94 (s, 1H), 8.99 (s, 1H), 12.25 (s,1H); 13C NMR (300MHz, CDCl3) δ 28.3, 80.8, 113.3, 114.6, 117.9, 125.8, 127.5, 131.9, 134.2, 138.0, 140.8, 164.9; MS (FAB) 559 ((M +H)+, 41). This material was pure by TLC (silica gel).
1,4-bis(9H-β-carboline-3-carboxylic acid tert-butyl ester) buta-1,3-diyne (33)
In a round bottom flask (200 mL), PdCl2(PPh3)2 (58 mg, 2 mol%), CuI (16mg, 2 mol%), and N, N-diisopropyl-ethylamine (534 mg, 4.92 mmol) were added and the mixture stirred under argon. The flask was evacuated (degassed) and refilled with argon. The THF (40 mL) and 6-ethynyl-β-carboline-3,9-dicarboxylic acid di-tert-butyl ester 34 (1.6 g, 4.1 mmol) were then added (under argon) to the above mixture. To this flask methyl bromoacetate (410 mg, 2.5 mmol) was added, and the reaction mixture was stirred at rt for 6–8 h. The progress of this reaction was monitored by TLC on silica gel. After the reaction was complete, 8–10 g of silica gel was added and the solvent was removed under vacuum. The solid residue (a plug) was then placed on a column and subjected to column chromatography (CH2Cl2) to give 1,4-bis(β-carboline-3,9-dicarboxylic acid di-tert-butyl ester) buta-1,3-diyne (2.08 g, 65%): 1H NMR (300 MHz, CDCl3) δ 1.74 (s, 9H), 1.81 (s, 9H), 7.82 (d, J = 8.79 Hz, 1H), 8.34 (s, 1H), 8.48 (d, J = 8.79 Hz, 1H), 8.69 (s, 1H), 9.65 (s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 28.2, 74.0, 81.2, 82.2, 86.0, 116.1, 116.9, 117.2, 123.7, 125.5, 131.3, 134.0, 136.2, 138.1, 139.6, 143.2, 149.6, 164.1; MS (FAB) 783((M + H)+, 100).
The 1,4-Bis-(β-carboline-3,9-dicarboxylic acid di-tert-butyl ester) buta-1,3-diyne (411 mg, 0.5 mmol) was added to a distilled solution of cumene (20 mL), which had been degassed. The reaction mixture was evacuated and refilled with nitrogen three times. The temperature was then brought to reflux for about 30 min until a yellow precipitate had formed. The mixture was filtered and the solids washed with hexane to give pure dimer 33. 33: mp >350 °C (dec.) IR(KBr) 3424, 1708, 1627, 1466, 1369, 1302, 1251, 1154, 1107, 1025, 846, 645 cm−1; 1H NMR (300 MHz, DMSO) δ 1.63(s, 9H), 7.60–7.69(m, 2H), 8.40(s, 1H), 8.96(s, 1H), 9.04(s, 1H), 12.5(s, 1H); 13C NMR (75.5 MHz, CDCl3) δ 28.3, 78.8, 82.5, 112.9, 118.1, 121.0, 122.1, 129.5, 131.5, 134.8, 141.1, 163.0, 131.9, 134.2, 138.0, 140.8, 164.9; MS (FAB) 583(M+, 100). This material was pure by TLC (silica gel).
The synthesis of the ligands in Table 2b had been reported previously in reference 89.
Table 3b.
Affinities (Ki=nM) of Boc-protected 3-substituted β-carbolines at αxβ3γ2(x=1–3,5,6) receptor subtypes.
| Ligands | α1 | α2 | α3 | α4 | α5 | α6 | |
|---|---|---|---|---|---|---|---|
| 40 |  |
450 | 5000 | ND | ND | 5000 | 5000 |
| 41 |  |
ND | ND | ND | ND | 1847 | ND |
ND = not determined yet (see previous tables for details of receptor binding). Data represent the average of at least three determinations with a SEM of ±5%.
Acknowledgments
This work was supported in part by NIMH 46851 (J.M.C.). We acknowledge support of this work by the Research Growth Initiative of the University of Wisconsin Milwaukee and the Lynde and Harry Bradley Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cook JM, Van Linn ML, Yin W. Preparation of aza-beta-carbolines and methods of using same for treatment of chemical addiction, anhedonia, and anxiety. 2009:92. CODEN: PIXXD2. (UWM Research Foundation, Inc., USA). WO 2009-US45014 WO 2009143445 A1 CAN 151:571093 AN 2009:1468879. [Google Scholar]
- 2.Harvey SC, Foster KL, McKay PF, Carroll MR, Seyoum R, Woods JE, Grey C, Jones CM, McCane S, Cummings R, Mason D, Ma CR, Cook JM, June HL. The GABA(A) receptor alpha(1) subtype in the ventral pallidum regulates alcohol-seeking behaviors. J Neurosci. 2002;22:3765–3775. doi: 10.1523/JNEUROSCI.22-09-03765.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.June HL, Foster KL, McKay PF, Seyoum R, Woods JE, Harvey SC, Eiler WJA, Grey C, Carroll MR, McCane S, Jones CM, Yin WY, Mason D, Cummings R, Garcia M, Ma CR, Sarma P, Cook JM, Skolnick P. The reinforcing properties of alcohol are mediated by GABA(A1) receptors in the ventral pallidum. Neuropsychopharmacol. 2003;28:2124–2137. doi: 10.1038/sj.npp.1300239. [DOI] [PubMed] [Google Scholar]
- 4.Kessler RC, Frank RG. The impact of psychiatric disorders on work loss days. Psychiatric Medicine. 1997;27:861–873. doi: 10.1017/s0033291797004807. [DOI] [PubMed] [Google Scholar]
- 5.O’Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence: A controlled study. Arch Gen Psychiatry. 1992;49:881–889. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- 6.Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone and the treatment of alcohol abuse. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 7.Kranzler HR. Pharmacotherapy of alcoholism: Gaps in knowledge and opportunities for research. Alcohol. 2000;35:537–547. doi: 10.1093/alcalc/35.6.537. [DOI] [PubMed] [Google Scholar]
- 8.Spanagel R, Zieglgansberger W. Anti-craving compounds for ethanol: New pharmacological tool to study addictive processes. Trends Pharmacol Sci. 1997:18. [PubMed] [Google Scholar]
- 9.Cloninger CR. Neurogenetic adaptive mechanisms in alcoholism. Science. 1987;236:410–416. doi: 10.1126/science.2882604. [DOI] [PubMed] [Google Scholar]
- 10.Li TK. Pharmacogenics of responses to alcohol and genes that influence alcohol drinking. J Stud Alcohol. 2000;61:5–12. doi: 10.15288/jsa.2000.61.5. [DOI] [PubMed] [Google Scholar]
- 11.Li T-K, Crabb DW, Lumeng L. Neuropharmacology of Ethanol. Birkhauser; Boston: 1991. pp. 107–124. [Google Scholar]
- 12.Johnson BA, Ait-Daoud N. Neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Psychopharmacology. 2000;149:327–344. doi: 10.1007/s002130000371. [DOI] [PubMed] [Google Scholar]
- 13.Koob GF, Roberts AJ, Schulteis Gea. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- 14.June HL, Cason CR, Cheatham G, Liu RY, Gan T, Cook JM. GABA(A)-benzodiazepine receptors in the striatum are involved in the sedation produced by a moderate, but not an intoxicating ethanol dose in out-bred Wistar rats. Brain Res. 1998;794:103–118. doi: 10.1016/s0006-8993(98)00222-4. [DOI] [PubMed] [Google Scholar]
- 15.McBride WJ, Li T. Animal models of alcoholism: Neurobiology of high alcohol-drinking behavior in rodents. Critical Reviews in Neurobiology. 1998;12:339–369. doi: 10.1615/critrevneurobiol.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- 16.Allain H, Belliard S, Decertaines J, Bentueferrer D, Bureau M, Lacroix P. Potential biological targets for anti-Alzheimer drugs. Dementia. 1993;4:347–352. doi: 10.1159/000107344. [DOI] [PubMed] [Google Scholar]
- 17.Heimer L, Alheid GF. The basal forebrain: anatomy and function. Plenum Press; New York: 1991. pp. 1–42. [DOI] [PubMed] [Google Scholar]
- 18.Heimer L, Zahm DS, Churchill P, Kalivas W. Specificity in the projection patterns of accumbal core and shell in the rat. Neurosci. 1991;41:89–125. doi: 10.1016/0306-4522(91)90202-y. [DOI] [PubMed] [Google Scholar]
- 19.Criswell HE, Simson PE, Duncan GE, Mc Cown TJ, Herbert JS, Morrow L. Molecular basis for regionally specific action of ethanol on γ-aminobutyric acid (A) receptors: Generalization to other ligand-gated Ion channels. J Pharmacol Exper Ther. 1993;267:522–527. [PubMed] [Google Scholar]
- 20.Criswell HE, Simson PE, Knapp DJ, Devaud LL, Mc Cown TJ, Duncan GE. Effect of zolpidem on γ-aminobutyric acid (GABA)-induced inhibition predicts the interaction of ethanol with GABA on individual neurons in several rat brain regions. J Pharmacol Exper Ther. 1995;273:525–536. [PubMed] [Google Scholar]
- 21.June HL, Zuccarelli D, Torres L, Craig KS, DeLong J, Allen A, Braun MR, Cason CR, Murphy JM. High-affinity benzodiazepine antagonists reduce responding maintained by ethanol presentation in ethanol-preferring rats. J Pharmacol Exp Ther. 1998;284:1006–1014. [PubMed] [Google Scholar]
- 22.June HL, Harvey SC, Foster KL, McKay PF, Cummings R, Garcia M, Mason D, Grey C, McCane S, Williams LS, Johnson TB, He XH, Rock S, Cook JM. GABA(A) receptors containing alpha 5 subunits in the CA1 and CA3 hippocampal fields regulate ethanol-motivated behaviors: An extended ethanol reward circuitry. J Neurosci. 2001;21:2166–2177. doi: 10.1523/JNEUROSCI.21-06-02166.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyytia P, Koob GF. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol. 1995;283:151–159. doi: 10.1016/0014-2999(95)00314-b. [DOI] [PubMed] [Google Scholar]
- 24.Nowak KL, McBride WJ, Lumeng L, Li TK, Murphy JM. Blocking GABAA receptors in the anterior ventral tegmental area attenuates ethanol intake of the alcohol-preferring P rat. Psychopharmacol. 1998;139:108–116. doi: 10.1007/s002130050695. [DOI] [PubMed] [Google Scholar]
- 25.Nauta HJ, Smith GP, Faull RLM, Domesick VB. Efferent connections and nigral afferents of the nucleus accumbens septi in the rat. Neurosci. 1978:3. doi: 10.1016/0306-4522(78)90041-6. [DOI] [PubMed] [Google Scholar]
- 26.Zahm DS, Heimer L. Ventral striatopallidal parts of the basal ganglia in the rat: I. Neurochemical compartmentation as reflected by the distributions of neurotensin and substance P immunoreactivity. J Comp Neurology. 1988;272:516–535. doi: 10.1002/cne.902720406. [DOI] [PubMed] [Google Scholar]
- 27.Groenwegen HJ, Berende HW. Organization of the output of the ventral striatopallidal system in the rat: Ventral pallidal efferents. Neurosci. 1993;57:113–142. doi: 10.1016/0306-4522(93)90115-v. [DOI] [PubMed] [Google Scholar]
- 28.Churchill L, Kalivas PW. A topographical organized GABA projection from the ventral pallidum to the nucleus accumbens in the rat. J Comp Neurology. 1994;345:579–595. doi: 10.1002/cne.903450408. [DOI] [PubMed] [Google Scholar]
- 29.Kuo H, Chang HT. Ventral-pallidostriatal pathway in the rat brain: A light electron microscopic study. J Comp Neurol. 1992;321:626–636. doi: 10.1002/cne.903210409. [DOI] [PubMed] [Google Scholar]
- 30.Morgenson GJ, Nielson M. Evidence that an accumbens to subpallidal GABAergic projection contributes to locomotor activity. Brain Res Bulletin. 1983;11:309–314. doi: 10.1016/0361-9230(83)90166-1. [DOI] [PubMed] [Google Scholar]
- 31.Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult-rat brain: differential regional and cellular-distribution of 7 major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- 32.Turner JD, Bodewitz G, Thompson CL, Stephenson FA. Anxiolytic beta carbolines: from molecular biology to the clinic. Springer-Verlag; New York: 1993. pp. 29–49. [Google Scholar]
- 33.Churchill L, Bourdelais A, Austin MC, Lolait SJ, Mahan LC, O’Carroll AM, Kalivas PW. GABAA receptors containing alpha 1 and beta 2 subunits are mainly localized on neurons in the ventral pallidum. Synapse. 1991;8:75–85. doi: 10.1002/syn.890080202. [DOI] [PubMed] [Google Scholar]
- 34.Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13-Gaba-A receptor subunit messenger-RNAs in the rat-brain. 1. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncan GE, Breese GR, Criswell HE, McCown TJ, Herbert JS, Devaud LL, Morrow AL. Distribution of [3H] zolpidem binding sites in relation to messenger RNA encoding the alpha 1, beta 2 and gamma 2 subunits of GABAA receptors in rat brain. Neurosci. 1995;64:1113–1128. doi: 10.1016/0306-4522(94)00433-6. [DOI] [PubMed] [Google Scholar]
- 36.Napier TC, Chrobak JJ. Evaluation of ventral pallidal dopamine receptor activation in behaving rats. Neuroreport. 1992;3:609–611. doi: 10.1097/00001756-199207000-00016. [DOI] [PubMed] [Google Scholar]
- 37.Hubner CB, Koob GF. GABA(A) receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol. 1995;283:151–159. doi: 10.1016/0014-2999(95)00314-b. [DOI] [PubMed] [Google Scholar]
- 38.Gong W, Justice JB. Dissociation of locomotor and conditioned place preference responses following manipulation of GABA-A and AMPA receptors in ventral pallidum. Prog Neuropsychohpharmacol Bio Psych. 1997;21:839–852. doi: 10.1016/s0278-5846(97)00084-5. [DOI] [PubMed] [Google Scholar]
- 39.Gong W, Neill D. Place preference conditioning and locomotor activation induced by local injection of psychostimulants into ventral pallidum. Brain Res. 1996;707:64–74. doi: 10.1016/0006-8993(95)01222-2. [DOI] [PubMed] [Google Scholar]
- 40.Cicero TJ. Biochemistry and Pharmacology of Ethanol. Vol. 2. Plenum Press; New York: 1979. pp. 533–560. [Google Scholar]
- 41.Lumeng L, Murphy JM, McBride WJ, Li T-K. Genetic Influences on alcohol preference in animals. Oxford University Press; New York: 1995. pp. 165–201. [Google Scholar]
- 42.June HL, Eggers MW, Warren-Reese C, DeLong J, Ricks-Cord A, Durr LF, Cason CR. The effects of the novel benzodiazepine receptor inverse agonist Ru 34000 on ethanol-maintained behaviors. Eur J Pharmacol. 1998;350:151–158. doi: 10.1016/s0014-2999(98)00260-x. [DOI] [PubMed] [Google Scholar]
- 43.June HL, Torres L, Cason CR, Hwang BH, Braun MR, Murphy JM. The novel benzodiazepine inverse agonist RO19–4603 antagonizes ethanol motivated behaviors: neuropharmacological studies. Brain Res. 1998;784:256–275. doi: 10.1016/s0006-8993(97)01380-2. [DOI] [PubMed] [Google Scholar]
- 44.Sieghart W, Ernst M. Heterogeneity of GABA(A) receptors: Revived interest in the development of subtype-selective drugs. Curr Med Chem: Cent Nerv Syst Agents. 2005;5:217–242. [Google Scholar]
- 45.Bowser DN, Wagner DA, Czajkowski C, Cromer BA, Parker MW, Wallace RH, Harkin LA, Mulley JC, Marini C, Berkovic SF, Williams DA, Jones MV, Petrou S. Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [g2(R43Q)] found in human epilepsy. Proc Natl Acad Sci US A. 2002;99:15170–15175. doi: 10.1073/pnas.212320199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bateson AN. The benzodiazepine site of the GABAA receptor: An old target with new potential? Sleep Medicine. 2004;5:S9–S15. doi: 10.1016/s1389-9457(04)90002-0. [DOI] [PubMed] [Google Scholar]
- 47.Otani K, Ujike H, Tanaka Y, Morita Y, Katsu T, Nomura A, Uchida N, Hamamura T, Fujiwara Y, Kuroda S. The GABA type A receptor alpha 5 subunit gene is associated with bipolar I disorder. Neurosci Lett. 2005;381:108–113. doi: 10.1016/j.neulet.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 48.Dean B, Scarr E, McLeod M. Changes in hippocampal GABAA receptor subunit composition in bipolar I disorder. Brain Res Mol Brain Res. 2005;138:145–155. doi: 10.1016/j.molbrainres.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Guidotti A, Auta J, Davis JM, Dong EB, Grayson DR, Veldic M, Zhang XQ, Costa E. GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacol. 2005;180:191–205. doi: 10.1007/s00213-005-2212-8. [DOI] [PubMed] [Google Scholar]
- 50.Maubach K. GABAA receptor subtype selective cognition enhancers. Drug Targets-CNS & Neuro Disorders. 2003;2:233–239. doi: 10.2174/1568007033482779. [DOI] [PubMed] [Google Scholar]
- 51.Barrett AC, Negus SS, Mello NK, Caine SB. Effect of GABA agonists and GABA-A receptor modulators on cocaine- and food-maintained responding and cocaine discrimination in rats. J Pharmacol Exp Ther. 2005;315:858–871. doi: 10.1124/jpet.105.086033. [DOI] [PubMed] [Google Scholar]
- 52.Anthenelli R, Schuckit MA. Genetic studies in alcoholism. Inter J Addict. 1990/1991;25:81–94. doi: 10.3109/10826089009067006. [DOI] [PubMed] [Google Scholar]
- 53.Schuckit MA. Reactions to alcohol in sons of alcoholics and controls. Alcohol Clin Exp Res. 1988;12:465–470. doi: 10.1111/j.1530-0277.1988.tb00228.x. [DOI] [PubMed] [Google Scholar]
- 54.Barnard E, Skolnick P, Olsen R, Möhler H, Sieghart W, Biggio G, Braestrup C, Bateson A, Langer S. International union of pharmacology. XV. Subtypes of γ-aminobutyric acid(A) receptors: classification on the basis of subunit structure and function. J Pharmacol Exp Ther(Pharmacol Rev) 1998;50:291–313. [PubMed] [Google Scholar]
- 55.Simon J, Wakimoto H, Fujita N, Lalande M, Barnard EA. Analysis of the set of GABA(A) receptor genes in the human genome. J Biol Chem. 2004;279:41422–41435. doi: 10.1074/jbc.M401354200. [DOI] [PubMed] [Google Scholar]
- 56.Sieghart W, Sperk G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- 57.Mossier B, Togel M, Fuchs K, Sieghart W. Immunoaffinity purification of gamma-aminobutyric Acid(A) (GABA(A)) receptors containing gamma(1)-subunits - evidence for the presence of a single type of gamma-subunit in GABA(A) receptors. J Biol Chem. 1994;269:25777–25782. [PubMed] [Google Scholar]
- 58.Togel M, Mossier B, Fuchs K, Sieghart W. Gamma-aminobutyric acid(A) receptors displaying association of gamma(3)-subunits with beta(2/3) and different alpha-subunits exhibit unique pharmacological properties. J Biol Chem. 1994;269:12993–12998. [PubMed] [Google Scholar]
- 59.Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: Immunocytochemical distribution of 13 subunits in the adult rat brain. Neurosci. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- 60.Cox ED, Diaz-Arauzo H, Huang Q, Reddy MS, Harris B, Mckernan RM, Skolnick P, Cook JM. Synthesis and evaluation of analogues of the partial agonist 6-(propyloxy)-4-(methoxymethyl)-beta-carboline-3-carboxylic acid ethyl ester (6-PBC) and the full agonist 6-(benzyloxy)-4-(methoxymethyl)-beta-carboline-3-carboxylic acid ethyl ester (Zk 93423) at wild type and recombinant GABA(A) receptors. J Med Chem. 1998;41:2537–2552. doi: 10.1021/jm970460b. [DOI] [PubMed] [Google Scholar]
- 61.Huang H, He X, Ma CR, Liu RY, Yu S, Dayer CA, Wenger GR, McKernan R, Cook JM. Pharmacophore/receptor models for GABAA/BzR subtypes (α1β3γ2, α5β3γ2, and α6β3γ2) via a comprehensive ligand-mapping approach. J Med Chem. 2000;43:71–95. doi: 10.1021/jm990341r. [DOI] [PubMed] [Google Scholar]
- 62.Carroll M, Woods JE, II, Seyoum RA, June HL. The role of the GABA(A) alpha1 subunit in mediating the sedative and anxiolytic properties of benzodiazepines. Alcohol Clin Exp Res. 2001:25. [Google Scholar]
- 63.Rowlett JK. 2010 unpublished results. [Google Scholar]
- 64.June HL, Cook JM, Ma C. Methods for reducing alcohol cravings in chronic alcoholics. US Pat Appl Publ. 2003:60. CODEN: USXXCO. (USA). US 2002–329100 US 20030176456 AN 2003:737374 CAN 139:241570. [Google Scholar]
- 65.Foster KL, McKay PF, Seyoum R, Milbourne D, Yin W, Sarma PVVS, Cook JM, June HL. GABAA and opioid receptors of the central nucleus of the amygdala selectively regulate ethanol-maintained behaviors. Neuropsychopharmacol. 2004;29:269–284. doi: 10.1038/sj.npp.1300306. [DOI] [PubMed] [Google Scholar]
- 66.Araki T, Tohyama M. Region-specific expression of GABA(A) receptor alpha 3 and alpha 4 subunits mRNAs in the rat brain. Mol Brain Res. 1992;12:295–314. doi: 10.1016/0169-328x(92)90132-u. [DOI] [PubMed] [Google Scholar]
- 67.Cox ED, Hagen TJ, Mckernan RM, Cook JM. Bz(1) receptor subtype specific ligands. Synthesis and biological properties of BCCt, a Bz(1) receptor subtype specific antagonist. Med Chem Res. 1995;5:710–718. [Google Scholar]
- 68.Griebel G, Perrault G, Letang V, Grainger P, Avenet P, Schoemaker H. New evidence that the pharmacological effects of benzodiazepine receptor ligands can be associated with activities at different BZ (α) receptor subtypes. Psychopharmacol. 1999;146:205–213. doi: 10.1007/s002130051108. [DOI] [PubMed] [Google Scholar]
- 69.Carroll ME, Carmona G, May S. Modifying drug-reinforced behavior by altering the economic conditions of the drug and the drug reinforcer. J Exp Anal Behav. 1991;18:361–376. doi: 10.1901/jeab.1991.56-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paronis CA, Cox ED, Cook JM, Bergman J. Different types of GABA(A) receptors may mediate the anticonflict and response rate-decreasing effects of zaleplon, zolpidem, and midazolam in squirrel monkeys. Psychopharmacol. 2001;156:461–468. doi: 10.1007/s002130100754. [DOI] [PubMed] [Google Scholar]
- 71.Rowlett JK, Tornatzky W, Cook JM, Ma CR, Miczek KA. Zolpidem, triazolam, and diazepam decrease distress vocalizations in mouse pups: Differential antagonism by flumazenil and β–carboline-3-carboxylate-t-butyl ester (βCCt) J Pharmacol Exp Ther. 2001;297:247–253. [PubMed] [Google Scholar]
- 72.Shannon HE, Guzman F, Cook JM. β-Carboline-3-carboxylate-t-butyl ester: a selective BZ1 benzodiazepine receptor antagonist. Life Sci. 1984;35:2227–2236. doi: 10.1016/0024-3205(84)90464-8. [DOI] [PubMed] [Google Scholar]
- 73.Rowlett JK, Spealman RD, Lelas S, Cook JM, Yin WY. Discriminative stimulus effects of zolpidem in squirrel monkeys: role of GABA(A)/(alpha 1) receptors. Psychopharmacol. 2003;165:209–215. doi: 10.1007/s00213-002-1275-z. [DOI] [PubMed] [Google Scholar]
- 74.Rowlett JK, Cook JM, Duke AN, Platt DM. Selective antagonism of GABA(A) receptor subtypes: An in vivo approach to exploring the therapeutic and side effect of benzodiazepine-type drugs. CNS Spectrums. 2005;10:40–48. doi: 10.1017/s1092852900009895. [DOI] [PubMed] [Google Scholar]
- 75.Liu R, Zhang P, Mckernan RM, Wafford KA, Cook JM. Synthesis of novel imidazobenzodiazepines selective for the α5β2γ2 (Bz5) GABAA/benzodiazepine receptor subtype. Med Chem Res. 1995;5:700–709. [Google Scholar]
- 76.Liu R, Hu RJ, Zhang P, Skolnick P, Cook JM. Synthesis and pharmacological properties of novel 8-substituted imidazobenzodiazepines: High-affinity, selective probes for α5-containing GABAA receptors. J Med Chem. 1996;39:1928–1934. doi: 10.1021/jm950887n. [DOI] [PubMed] [Google Scholar]
- 77.Li XY, Cao H, Zhang CC, Furtmueller R, Fuchs K, Huck S, Sieghart W, Deschamps J, Cook JM. Synthesis, in vitro affinity, and efficacy of a bis 8-ethynyl-4H-imidazo[1,5a]-[1,4]benzodiazepine analogue, the first bivalent alpha 5 subtype selective BzR/GABA(A) antagonist. J Med Chem. 2003;46:5567–5570. doi: 10.1021/jm034164c. [DOI] [PubMed] [Google Scholar]
- 78.Allen MS, Tan YC, Trudell ML, Narayanan K, Schindler L, Martin MJ, Schultz CA, Hagen TJ, Koehler KF, Codding P, Skolnick P, Cook J. Synthetic and computer-assisted analyses of the pharmacophore for the benzodiazepine receptor inverse agonist site. J Med Chem. 1990;33:2343–2357. doi: 10.1021/jm00171a007. [DOI] [PubMed] [Google Scholar]
- 79.Allen MS, Laloggia AJ, Dorn LJ, Martin MJ, Constantino G, Hagen TJ, Koehler KF, Skolnick P, Cook J. Predictive binding of β-carboline inverse agonists and antagonists via the CoMFA/GOLPF approach. J Med Chem. 1992;35:4001–4010. doi: 10.1021/jm00100a004. [DOI] [PubMed] [Google Scholar]
- 80.Cain M, Weber RW, Guzman F, Cook JM, Barker SA, Rice KC, Crawley JN, Paul SM, Skolnick P. β-Carbolines: Synthesis and neurochemical and pharmacological actions on brain benzodiazepine receptors. J Med Chem. 1982;25:1081–1091. doi: 10.1021/jm00351a015. [DOI] [PubMed] [Google Scholar]
- 81.Hagen TJ, Skolnick P, Cook JM. Synthesis of 6-substitutedβ-carbolines that behave as benzodiazepine receptor antagonists or inverse agonists. J Med Chem. 1987;30:750–753. doi: 10.1021/jm00387a033. [DOI] [PubMed] [Google Scholar]
- 82.Cook JM, Diaz-Arauzo H, Allen MS. Inverse agonists, probes to study the structure, topology and function of the benzodiazepine receptor. NIDA Res Monogr. 1990:133–139. [PubMed] [Google Scholar]
- 83.Schweri M, Cain M, Cook JM, Paul S, Skolnick P. Blockade of 3-carbomethoxy-β-carboline induced seizures by diazepam and the benzodiazepine antagonists, Ro 15–1788 and CGS 8216. Pharmaco Biochem Behav. 1982;17:457–460. doi: 10.1016/0091-3057(82)90304-5. [DOI] [PubMed] [Google Scholar]
- 84.Fryer RI, Cook C, Gilman NW, Walser A. Conformational shifts at the benzodiazepine receptor related to the binding of agonists, antagonists and inverse agonists. Life Sci. 1986;39:1947–1957. doi: 10.1016/0024-3205(86)90318-8. [DOI] [PubMed] [Google Scholar]
- 85.Trullas R, Ginter H, Jackson B, Skolnick P, Allen MS, Hagen TJ, Cook JM. 3-Ethoxy-β-carboline: a high affinity benzodiazepine receptor ligand with partial inverse agonist properties. Life Sci. 1988;43:1193–1197. doi: 10.1016/0024-3205(88)90208-1. [DOI] [PubMed] [Google Scholar]
- 86.Ninan PT, Insel TM, Cohen RM, Cook JM, Skolnick P, Paul SM. Benzodiazepine receptor-mediated experimental “anxiety” in primates. Science. 1982;218:1332–1334. doi: 10.1126/science.6293059. [DOI] [PubMed] [Google Scholar]
- 87.Mendelson WB, Cain M, Cook JM, Paul SM, Skolnick P. A benzodiazepine receptor antagonist decreases sleep and reverses the hypnotic actions of flurazepam. Science. 1983;219:414–416. doi: 10.1126/science.6294835. [DOI] [PubMed] [Google Scholar]
- 88.Hagen TJ, Guzman F, Schultz C, Cook JM, Shannon HE. Synthesis of 3,6-disubstituted β-carbolines which possess either benzodiazepine antagonist or agonist activity. Heterocycles. 1986;24:2845–2855. [Google Scholar]
- 89.Allen MS, Hagen TJ, Trudell ML, Codding PW, Skolnick P, Cook JM. Synthesis of novel 3-substituted β-carbolines as benzodiazepine receptor ligands: probing the benzodiazepine receptor pharmacophore. J Med Chem. 1988;31:1854–1861. doi: 10.1021/jm00117a029. [DOI] [PubMed] [Google Scholar]
- 90.Diaz-Arauzo H, Evoniuk GE, Skolnick P, Cook JM. The agonist pharmacophore of the benzodiazepine receptor. Synthesis of a selective anticonvulsant/anxiolytic. J Med Chem. 1991;34:1754–1756. doi: 10.1021/jm00109a035. [DOI] [PubMed] [Google Scholar]
- 91.Wafford KA, Bain CJ, Whiting PJ, Kemp JA. Functional comparison of the role of γ subunits in recombinant human γ-aminobutyric acidA/benzodiazepine receptors. Mol Pharmacol. 1993;44:437–442. [PubMed] [Google Scholar]
- 92.Braestrup C, Nielsen M. GABA reduces binding of 3H-methyl β-carboline-3-carboxylate to brain benzodiazepine receptors. Nature. 1981;294:472–474. doi: 10.1038/294472a0. [DOI] [PubMed] [Google Scholar]
- 93.Venault P, Chapouthier G, de-Carvalho LP, Simiand J, Morre M, Dodd RH, Rossier J. Benzodiazepine impairs and β-carboline enhances performance in learning and memory tasks. Nature. 1986;321:864–866. doi: 10.1038/321864a0. [DOI] [PubMed] [Google Scholar]
- 94.Lippke KP, Schunack WG, Wenning W, Müller WE. β-Carbolines as benzodiazepine receptor ligands. 1. Synthesis and benzodiazepine receptor interaction of esters of β-carboline-3-carboxylic acid. J Med Chem. 1983;26:499–503. doi: 10.1021/jm00358a008. [DOI] [PubMed] [Google Scholar]
- 95.Corda MG, Blaker WD, Mendelson WB, Guidotti A, Costa E. β-Carbolines enhance shock-induced suppression of drinking in rats. Proc Natl Acad Sci USA. 1983;80:2072–2076. doi: 10.1073/pnas.80.7.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Havoundjian H, Reed GF, Paul SM, Skolnick P. Protection against the lethal effects of pentobarbital in mice by a benzodiazepine receptor inverse agonist, 6,7-dimethoxy-4-ethyl-3-carbomethoxy-β-carboline. J Clin Invest. 1987;79:473–477. doi: 10.1172/JCI112836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hadingham KL, Wingrove P, Le-Bourdelles B, Palmer KJ, Ragan CI, Whiting PJ. Cloning of cDNA sequences encoding human α2 and α3 γ-aminobutyric acidA receptor subunits and characterization of the benzodiazepine pharmacology of recombinant α1-,α2-,α3-, and α5-containing human γ-aminobutyric acidA receptors. Mol Pharmacol. 1993;43:970–975. [PubMed] [Google Scholar]
- 98.Sanger DJ, Benavides J, Perrault G, Morel E, Cohen C, Joly D, Zivkovic B. Recent developments in the behavioral pharmacology of benzodiazepine-(omega) receptors - Evidence for the functional-significance of receptor subtypes. Neurosci Biobehav Rev. 1994;18:355–372. doi: 10.1016/0149-7634(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 99.McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, Ragan CI, Dawson GR, Whiting PJ. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 100.Braestrup C, Nielsen M, Olsen R. Urinary and brain β-carboline-3-carboxylates as potent inhibitors of brain benzodiazepine receptors. Proc Natl Acad Sci USA. 1980;77:2288–2292. doi: 10.1073/pnas.77.4.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nielsen M, Braestrup C. Ethyl β-carboline-3-carboxylate shows differential benzodiazepine receptor interaction. Nature. 1980;286:606–607. doi: 10.1038/286606a0. [DOI] [PubMed] [Google Scholar]
- 102.Huang Q, Cox ED, Gan T, Ma CR, Bennett DW, Mckernan RM, Cook JM. Studies of molecular pharmacophore/receptor models for GABAA/benzodiazepine receptor subtypes: Binding affinities of substituted β–carbolines at recombinant α1β3γ2 subtypes and quantitative structure-activity relationship studies via a Comparative Molecular Field Analysis. Drug Des Discov. 1999;16:55–76. [PubMed] [Google Scholar]
- 103.Yin W, Sarma PVVS, Ma J, Han D, Chen JL, Cook JM. Synthesis of bivalent ligands of β-carboline-3-carboxylates via a palladium-catalyzed homocoupling process. Tetrahedron Lett. 2005;46:6363–6368. [Google Scholar]
- 104.Portoghese PS. From models to molecules: Opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 105.Portoghese PS, Lin CE, Farouzgrant F, Takemori AE. Structure-activity relationship of N17′-substituted norbinaltorphimine congeners - role of the N17′ basic group in the interaction with a putative address subsite on the kappa-opioid receptor. J Med Chem. 1994;37:1495–1500. doi: 10.1021/jm00036a015. [DOI] [PubMed] [Google Scholar]
- 106.Halazy S. G-protein coupled receptors bivalent ligands and drug design. Expert Opinion on Therapeutic Patents. 1999;9:431–446. [Google Scholar]
- 107.June HL, Sr, Foster KL, Eiler WJAI, Goergen J, Cook JB, Johnson N, Mensah-Zoe B, Simmons JO, June HL, Jr, Yin W, Cook JM, Homanics GE. Dopamine and benzodiazepine-dependent mechanisms regulate the EtOH-enhanced locomotor stimulation in the GABA(A) α1 subunit null mutant mice. Neuropsychopharmacol. 2007;32:137–152. doi: 10.1038/sj.npp.1301097. [DOI] [PubMed] [Google Scholar]
- 108.Bell RL, AZ, Lumeng L, Murphy JM, McBride WJ. The alcohol-preferring P rat and animal models of excessive alcohol drinking. Addict Biol. 2006;11:270–288. doi: 10.1111/j.1369-1600.2005.00029.x. [DOI] [PubMed] [Google Scholar]
- 109.Koob GF. A Role for GABA Mechanisms in the Motivational Effects of Alcohol. Biochem Pharmacol. 2004;68:1515–1525. doi: 10.1016/j.bcp.2004.07.031. (review and references therein) [DOI] [PubMed] [Google Scholar]
- 110.Clayton T, Chen JL, Ernst M, Richter L, Cromer BA, Morton HN, Kaczorowski CC, Helmstetter FJ, Furtmuller R, Ecker G, Parker MW, Sieghart W, Cook JM. Analysis of the benzodiazepine binding site on γ-aminobutyric acid (A) receptors: Correlation of experimental data with pharmacophore and comparative models. Curr Med Chem. 2007;14:2755–2775. doi: 10.2174/092986707782360097. [DOI] [PubMed] [Google Scholar]
- 111.Cromer B, Morton C, Parker MW. Anxiety of GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–287. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- 112.Trudell JR. Unique assignment of inter-subunit association in GABA(A) α1β3 γ2 receptors determined by molecular modeling. Biochim Biophys Acta. 2002;1565:91–96. doi: 10.1016/s0005-2736(02)00512-6. [DOI] [PubMed] [Google Scholar]
- 113.Ma C. Ph D Thesis. University of Wisconsin; Milwaukee: 2000. [Google Scholar]
- 114.Ramachandran PV, Teodorovic AV, Brown HC. Chiral synthesis via organoboranes. 38. Selective reductions. 48. Asymmetric reduction of trifluoro-methyl ketones by B-chlorodiisopinocampheylborane in high enantiomeric purity. Tetrahedron. 1993;49:1725–1738. [Google Scholar]
- 115.Ramachandran PV, Gong B, Brown HC. Chiral synthesis via organo-boranes. 40. Selective reductions. 55. A simple one-pot synthesis of the enantio-mers of trifluoromethyloxirane. A general synthesis in high optical purities of α-trifluoromethyl secondary alcohols via the ring-cleavage reactions of the epoxide. J Org Chem. 1995;60:41–46. [Google Scholar]
- 116.Sonogashira K, Tohda Y, Hagihara N. Convenient synthesis of acetylenes. Catalytic substitutions of acetylenic hydrogen with bromo alkenes, iodo arenes, and bromopyridines. Tetrahedron Lett. 1975;50:4467–4470. [Google Scholar]
- 117.Sonogashira K. Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. Organometal Chem. 2002;653:46–49. [Google Scholar]
- 118.Lei A, Srivastava M, Zhang X. Transmetalation of palladium enolate and its application in palladium-catalyzed homocoupling of alkynes: a room-temperature, highly efficient route to make diynes. J Org Chem. 2002;67:1969–1971. doi: 10.1021/jo011098i. [DOI] [PubMed] [Google Scholar]
- 119.Choudhary MS, Craigo S, Roth BL. Identification of receptor domains that modify ligand binding to 5-hydroxytryptamine2 and 5-hydroxytryptamine1c serotonin receptors. Mol Pharmacol. 1992;42:627–633. [PubMed] [Google Scholar]
- 120.Lüddens H, Korpi ER, Seeburg PH. GABAA/benzodiazepine receptor heterogeneity: neurophysiological implications. Neuropharmacol. 1995;34:245–254. doi: 10.1016/0028-3908(94)00158-o. Review. [DOI] [PubMed] [Google Scholar]
- 121.Huang Q, Zhang WJ, Liu RY, McKernan RM, Cook JM. Benzo-fused benzodiazepines employed as topological probes for the study of benzodiazepine receptor subtypes. Med Chem Res. 1996;6:384–391. [Google Scholar]
- 122.Huang Q, Liu RY, Zhang PW, He XH, McKernan R, Gan T, Bennett DW, Cook JM. Predictive models for GABA(A)/benzodiazepine receptor subtypes: Studies of quantitative structure-activity relationships for imidazobenzodiazepines at five recombinant GABA(A)/benzodiazepine receptor subtypes [α × β 3 γ2 (x = 1–3, 5, and 6)] via Comparative Molecular Field Analysis. J Med Chem. 1998;41:4130–4142. doi: 10.1021/jm980317y. [DOI] [PubMed] [Google Scholar]
- 123.He XH, Huang Q, Ma CR, Yu S, McKernan R, Cook J. Pharmacophore/receptor models for GABAA/BzR α2β3γ2, α3β3γ3, and α4β3γ2 recombinant subtypes. Included volume analysis and comparison to α1β3γ2, α5β3γ2 and α6β3γ2 subtypes. Drug Des Discov. 2000;17:131–171. [PubMed] [Google Scholar]
- 124.O’mara M, Cromer B, Parker M, Chung SH. Homology model of the GABA(A) receptor examined using Brownian dynamics. Biophys J. 2005;88:3286–3299. doi: 10.1529/biophysj.104.051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 126.Buhr A, Baur R, Sigel E. Subtle changes in residue 77 of the gamma subunit of α1β2γ 2 GABA(A) receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J Biol Chem. 1997;272:11799–11804. doi: 10.1074/jbc.272.18.11799. [DOI] [PubMed] [Google Scholar]
- 127.Buhr A, Schaerer MT, Baur R, Sigel E. Residues at positions 206 and 209 of the α1 subunit of gamma-aminobutyric acid(A) receptors influence affinities for benzodiazepine binding site ligands. Mol Pharmacol. 1997;52:676–682. doi: 10.1124/mol.52.4.676. [DOI] [PubMed] [Google Scholar]
- 128.Buhr A, Sigel E. A point mutation in the gamma(2) subunit of gamma-aminobutyric acid type A receptors results in altered benzodiazepine binding site specificity. Proc Natl Acad Sci US A. 1997;94:8824–8829. doi: 10.1073/pnas.94.16.8824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mihic SJ, Whiting P, Klein RL, Wafford K, Harris RA. A single amino acid of the human γ-aminobutyric acid type A receptor g2 subunit determines benzodiazepine efficacy. J Biol Chem. 1994;269:32768–32773. [PubMed] [Google Scholar]
- 130.Sigel E, Schaerer MT, Buhr A, Baur R. The benzodiazepine binding pocket of recombinant α1β2γ2 γ-aminobutyric acidA receptors: Relative orientation of ligands and amino acid side chains. Mol Pharmacol. 1998;54:1097–1105. doi: 10.1124/mol.54.6.1097. [DOI] [PubMed] [Google Scholar]
- 131.Braestrup C, Honore T, Nielsen MC, Peterson EN, Jensen LH. Ligands for benzodiazepine receptors with positive and negative efficacy. Biochem Pharmacol. 1984;33:859–862. doi: 10.1016/0006-2952(84)90438-6. [DOI] [PubMed] [Google Scholar]
- 132.Lawson J, Uyeno ET, Nienow J, Loew GH, Toll L. Structure-activity studies of β-carboline analogs. Life Sci. 1984;34:2007–2013. doi: 10.1016/0024-3205(84)90365-5. [DOI] [PubMed] [Google Scholar]
- 133.Hanzawa Y, Kawagoe K, Ito M, Kobayashi Y. Kinetic resolution of (E)-[(fluoroalkyl)vinyl]carbinol derivatives by asymmetric epoxidation with titanium-tartrate catalysts. Chem Pharm Bull. 1987;35:1633–1636. [Google Scholar]
- 134.Liu R, Zhang P, Gan T, Mckernan RM, Cook JM. Evidence for the conservation of conformational topography at five major GABA(A)/benzodia-zepine receptor subsites. Potent affinities of the (S)-enantiomers of framework-constrained 4,5-substituted pyrroloimidazobenzodiazepines. Med Chem Res. 1997;7:25–35. [Google Scholar]
- 135.Saiga Y, Iijima I, Ishida A, Miyagishima T, Shigezane K, Oh-Ishi T, Matsumoto M, Matsuoka Y. Synthesis of 1,2,3,4-tetrahydro-b-carboline derivatives as hepatoprotective agents. II. Alkyl 1,2,3,4-tetrahydro-b-carboline-2-carbodithioates. Chem Pharm Bull. 1987;35:3262–3269. doi: 10.1248/cpb.35.3262. [DOI] [PubMed] [Google Scholar]
- 136.Eftink MR, Jia J, Hu D, Ghiron CA. Fluorescence studies with tryptophan analogs: excited state interactions involving the side chain amino group. J Phy Chem. 1995;99:5713–5723. [Google Scholar]
- 137.Plate R, Nivard RJF, Ottenheijm HCJ, Kardos J, Simonyi M. Synthesis and pharmacological activity of C(1)- and N(2)-substituted β-carboline derivatives. Heterocycles. 1986;24:3105–3114. [Google Scholar]
- 138.Moody CJ, Ward JG. [2,3] Fused indoles. Synthesis of β-carbolines and azepino[4,5-b]indoles from 3-(2-alkylindol-3-yl)-2-azidoacrylates. J Chem Soc Perkin Trans. 1984;112:2895–2901. [Google Scholar]