

Abstract
Accumulation of β-amyloid (Aβ) and loss of synapses are hallmarks of Alzheimer's disease (AD). How synaptic activity relates to Aβ accumulation and loss of synapses is a current topic of major interest. Synaptic activation promotes Aβ secretion, and chronic reduction of synaptic activity reduced Aβ plaques in an AD transgenic mouse model. This suggested beneficial effects of reducing synaptic activity in AD. We now show that reduced synaptic activity causes detrimental effects on synapses and memory despite reducing plaques using two different models of chronic synaptic inhibition: deafferentation of the barrel cortex and administration of benzodiazepine. An interval of prolonged synaptic inhibition exacerbated loss of synaptophysin compared with synaptically more active brain in AD transgenic but not wild-type mice. Furthermore, an interval of benzodiazepine treatment, followed by a washout period, exacerbated memory impairment in AD transgenic mice. Exacerbation of synaptic and behavioral abnormalities occurred in the setting of reduced Aβ plaques but elevated intraneuronal Aβ immunoreactivity. These data support beneficial effects of synaptic activation on Aβ-related synaptic and behavioral impairment in AD.
Introduction
Alzheimer's disease (AD) is characterized by the aberrant accumulation of β-amyloid peptides (Aβ) and the progressive loss of synapses in the brain (Selkoe, 2002). Synapses are considered the earliest site of pathology, and synaptic loss is the best pathological correlate of cognitive impairment in subjects with AD (Terry et al., 1991; Gouras et al., 2010). The relation between synaptic activity and Aβ in AD pathogenesis is a topic of major interest. Synaptic activity has been shown to increase Aβ secretion in vitro and in vivo (Kamenetz et al., 2003; Cirrito et al., 2005). Since addition of extracellular Aβ alters synapses and impairs synaptic plasticity, chronic synaptic activity could therefore be detrimental. In support of this scenario, reduction of activity by induction of sleep reduced Aβ secretion and plaque burden in brains of AD mice (Kang et al., 2009). Moreover, epilepsy is associated with increased plaque pathology, and cortical hyper-excitability is seen in brains of AD mouse models (Mackenzie and Miller, 1994; Palop et al., 2006, 2007). On the other hand, synaptic activity is essential for synaptic plasticity and memory formation. Higher educational attainment and involvement in intellectually stimulating activities are associated with reduced risk of AD (Stern, 2006). In addition, environmental enrichment and behavioral training reduced amyloid deposition and improved memory in AD transgenic mice (Lazarov et al., 2005; Billings et al., 2007), although these behavioral manipulations are complex and are not necessarily straightforwardly related to synaptic activity. We previously provided mechanistic data demonstrating that synaptic activation reduced levels of intraneuronal Aβ and protected synapses in primary neuron models of AD (Tampellini et al., 2009). Intriguingly, these studies showed that synaptic activity promotes trafficking of the amyloid precursor protein (APP) to synapses, where Aβ is preferentially generated (Goldsbury et al., 2006; Cirrito et al., 2008; Tampellini et al., 2009). In addition, the Aβ-degrading protease neprilysin was critical for the reduction specifically of the intraneuronal pool of Aβ42 with synaptic activation. Although mechanistic biological studies showed quantitative beneficial effects of synaptic activity on synapses in AD transgenic neurons, in vivo studies to determine the role of synaptic activity on synapses and behavior are required to define the relation between synaptic activity and the pathophysiology of the disease. To investigate the role of synaptic activity on Aβ, synapses, and brain function, we chronically reduced activity in mice by two well established experimental methods used to investigate synaptic plasticity in the brain (Wong-Riley and Welt, 1980; Machín et al., 2006). We now demonstrate that inhibition of synaptic activity reduces plaque burden but increases immunoreactivity of Aβ within neurons and exacerbates loss of synaptophysin in AD transgenic mice. Moreover, despite reducing plaques, chronic treatment with diazepam worsened memory retention.
Materials and Methods
Chronic diazepam treatment.
Three-month-old Tg19959 mice and wild-type littermates (13 males and 19 females) were divided into 4 groups: wild-type + vehicle (control), wild-type + diazepam, Tg19959 + vehicle, Tg19959 + diazepam. Mice were treated with diazepam (4.5 mg/kg) or vehicle (saline containing 4% propylene glycol and 1% ethanol) by daily intraperitoneal injection for 6 d a week for 1 month. Mice were given 4 d of washout before testing in the Morris water maze (MWM).
Surgical procedure for unilateral removal of whiskers.
Two-month-old Tg19959 mice and wild-type littermates (5 males and 5 females) were anesthetized with an intraperitoneal injection of ketamine and xylazine. Surgery was performed as previously described (Tampellini et al., 2009). At 6 months of age, mice were killed with pentobarbital and perfused with 4% paraformaldehyde (PFA) or with 2% PFA and 3.75% acrolein for electron microscopy (EM) studies. Brains were removed and either cut by Vibratome (EM) or incubated at 4°C in increasing percentages of sucrose (10, 20 and 30%) before sectioning by cryostat (immunostaining and immunofluorescence).
Morris water maze.
Spatial learning and memory were assessed by the MWM as described previously (Dumont et al., 2009). In brief, the hidden platform was located in the middle of the northwest (NW) quadrant, 1 cm beneath water level. Latencies were recorded for 5 d with a video tracking system. Each day mice were placed next to and facing the wall of the basin in 4 different starting positions: north, east, south, and west, corresponding to 4 successive trials. Whenever the mouse failed to reach the platform within the maximally allowed time of 60 s, it was placed on the platform for 5 s. A probe trial was assessed 24 h after the acquisition period, removing the platform from the pool. The mice were released on the north side (exactly on the border between NW and NE quadrants) facing the wall. To ensure that any differences were not due to visual deficits, the visible platform version of the water maze was performed after the probe trial.
Electron microscopy.
Ultrastructural quantification of synapses was done using EM as described previously (Takahashi et al., 2002). Representative images were obtained from deafferented and contralateral barrels, n = 5 each. Synapses were identified by the following criteria: presence of a clearly defined synaptic density and evidence of presynaptic and postsynaptic compartments. Numbers of synapses were counted by an investigator unaware of the origin of the images, and the difference in synapse numbers was expressed as percentage of change from control.
Western blotting.
Western blots were performed as described previously (Dumont et al., 2009). Briefly, snap-frozen brain tissues were homogenized in 6% SDS. Equal protein amounts (25 μg) were electrophoresed through 4–12% Bis-Tris NuPage (synaptophysin) or 10–20% Tris-Tricine polyacrylamide (APP, CTFs, and Aβ) gels (Invitrogen). After transfer, membranes for Aβ were boiled in PBS for 5 min. Membranes were blocked in 5% nonfat dry milk and incubated with primary antibody overnight at 4°C. Horseradish peroxidase-conjugated secondary antibody binding was visualized with enhanced chemiluminescence. Antibodies used: rabbit polyclonal anti-APP 369, mouse monoclonal 6E10 (Covance), mouse monoclonal anti-synaptophysin (Millipore), and mouse monoclonal anti-tubulin (Sigma).
Immunostaining and quantitation.
Brains were cut at 40 μm thickness with a cryostat and stained for cytochrome oxidase (COX) (Wong-Riley and Welt, 1980) or immunolabeled for Aβ42 (Millipore) and/or synaptophysin (Millipore) in adjacent sections. The barrel cortices corresponding to the half snouts that did not undergo surgery were used as controls. Immunolabeling for light and fluorescence microscopy was performed as previously described (Li et al., 2004; Takahashi et al., 2010). Brain sections were viewed and images were acquired as described previously (Li et al., 2004; Tampellini et al., 2009; Takahashi et al., 2010). MetaMorph software 7.6 (Universal Imaging Co.) was used for quantitative analysis. Integrated fluorescence intensity per field was measured on thresholded images. Three images per stack were used for each section. Total fluorescence per thresholded picture was automatically quantified.
Statistical analysis.
Statistical comparisons were made using two-tailed t tests (paired for the deafferentation data, unpaired for the diazepam data) and ANOVA with significance placed at p < 0.05. Averages of measurements per section per mouse were considered as an individual measurement (n = 1). Data were expressed as mean ± SEM. Statistical analysis was performed using StatView (SAS Institute Inc.) and Excel (Microsoft).
Results
Chronic reduction of synaptic activity in vivo was accomplished using two different approaches: unilateral ablation of whiskers and chronic diazepam treatment. For the former, whisker bulbs were unilaterally and irreversibly removed in 2- to 3-month-old Tg19959 mice, which harbor the KM670/671NL and V717F familial AD mutations (Li et al., 2004). These mice develop plaques at 2–3 months of age. The effects on barrel cortices were examined at 6 months of age. Reduced COX staining confirmed reduced neuronal activity in the deafferented compared with the synaptically active control barrel cortices (Fig. 1A, top). Barrel cortices with reduced activity demonstrated a 29 ± 5% decrease in the number of Aβ plaques and a 32 ± 8% decrease in the area covered by plaques compared with the control side (Fig. 1A, bottom). At the same time, barrel cortices with reduced activity and reduced plaques showed a 64 ± 30% increase of Aβ42 immunoreactivity in neurons compared with controls (Fig. 1B).
Figure 1.
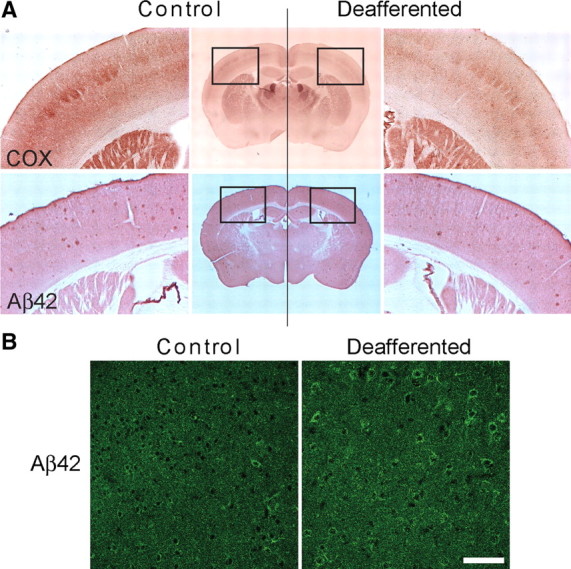
Chronic synaptic inhibition reduces amyloid plaques and increases intraneuronal Aβ42 immunoreactivity. A, Top, COX staining shows reduced activity in the deafferented compared with the control barrel cortex. Bottom: adjacent brain sections demonstrated reduced amyloid plaque burden in the deafferented barrel cortex compared with control. Quantification of plaque number and area covered by plaques revealed a 29 ± 5% and a 32 ± 8% decrease, respectively, in the deafferented compared with the control barrel cortex (n = 4, p < 0.05). B, Intraneuronal Aβ42 immunoreactivity was increased by 64 ± 30% in the deafferented compared with the control Tg19959 barrel cortex (n = 4; p < 0.05). Scale bar, 75 μm.
To determine the effect of chronic synaptic inhibition on the integrity of synapses, levels of the synaptic marker synaptophysin were examined in the barrel cortices by confocal microscopy. Progressive loss of the presynaptic marker synaptophysin is well established in AD transgenic mice (Mucke et al., 2000) and is considered the best brain correlate of cognitive decline in human AD (Terry et al., 1991). We have previously shown that Tg19959 mice have reduced synaptophysin levels compared with wild-type littermates (Dumont et al., 2009). Remarkably, and despite the reduction in plaques, confocal microscopy demonstrated that levels of synaptophysin in Tg19959 mice were further decreased by 80 ± 3% (Fig. 2A, left) on the deafferented compared with the synaptically active barrel cortex (control); there also was a 31 ± 14% reduction in levels of PSD-95 on the deafferented compared with control barrels (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In contrast, wild-type mouse brains with equivalent chronic synaptic inhibition did not show decreases in synaptophysin (Fig. 2A, right). To evaluate whether the decrease of synaptophysin in the deafferented barrel cortex of Tg19959 mice might reflect a loss of synapses, electron microscopy was used to quantify the number of synapses (Fig. 2B). There was a 31 ± 12% reduction in number of synapses in the deafferented barrels compared with control.
Figure 2.
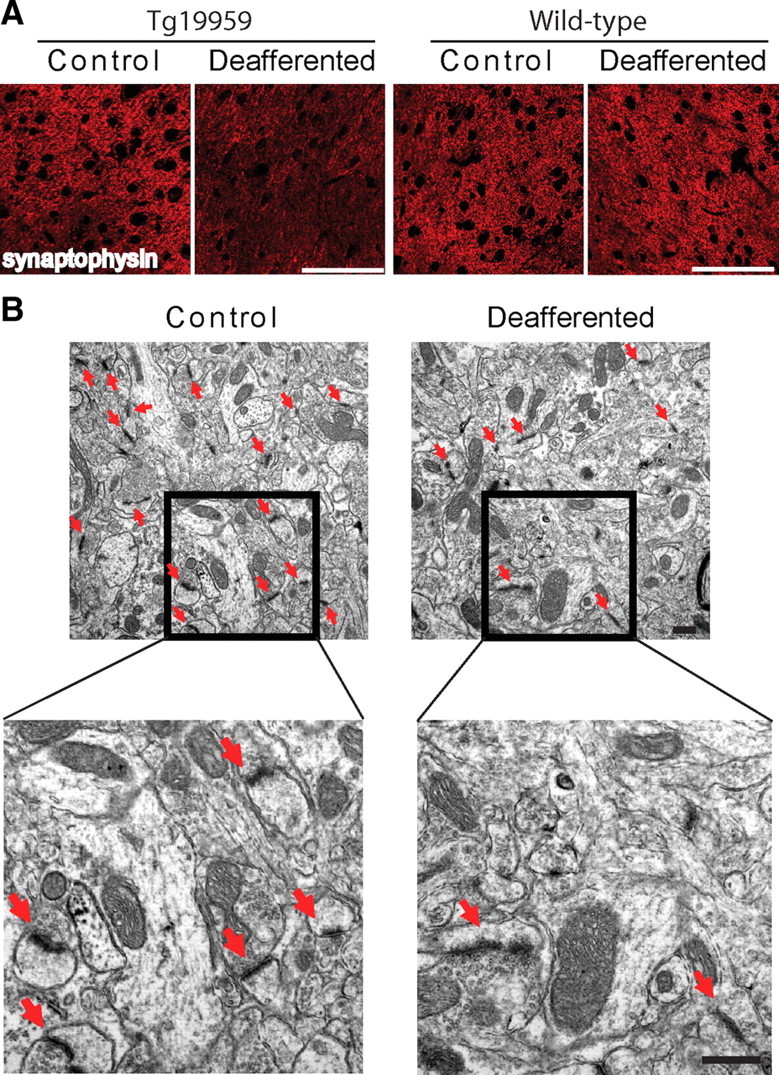
Chronic synaptic inhibition reduces levels of synaptophysin and number of synapses. A, Left, Fluorescent immunolabeling of synaptophysin in the barrel cortex of Tg19959 mice. The deafferented barrels demonstrated a marked 80 ± 3% reduction in synaptophysin compared with the control barrel cortices (n = 4; p < 0.01). Scale bar, 75 μm. Right, Fluorescent immunolabeling of synaptophysin in the barrel cortex of wild-type mice. No change in synaptophysin immunofluorescence was detectable between the deafferented and control sides (n = 3). Scale bar, 75 μm. B, EM images of Tg19959 barrel cortex: the number of synapses (red arrows) was reduced by 31 ± 12% in the deafferented compared with control barrels (n = 5/group; p < 0.05). Scale bar, 500 nm.
Next, diazepam treatment was used to investigate the effect of chronic synaptic inhibition on Aβ, synapses, and cognitive function. Tg19959 mice were treated with diazepam for 1 month beginning at 3 months of age, followed by a 4 d washout before behavioral testing. Diazepam treatment reduced plaque burden by 32 ± 12% and 20 ± 9% in cortex and hippocampus respectively, while confocal immunofluorescence of the hippocampus revealed a 41 ± 10% increase in intraneuronal Aβ42 immunoreactivity in CA1 neurons in diazepam-treated compared with vehicle-treated Tg19959 mice (Fig. 3A–C). Similar to the deafferentation results, confocal immunofluorescence demonstrated that the already decreased levels of synaptophysin in Tg19959 mice were further decreased by diazepam (Fig. 4A). In contrast, immunofluorescence demonstrated an increase in levels of synaptophysin in wild-type mice with synaptic inhibition by diazepam (supplemental Figure S2, available at www.jneurosci.org as supplemental material). Of note, chronic synaptic inhibition in wild-type barrel cortices was previously reported to lead to an increase in synapses (Machín et al., 2006). Our previous data linked intraneuronal Aβ with reduction in levels of synaptophysin in cultured AD transgenic neurons (Almeida et al., 2005). We now observe a link between Aβ42 accumulation and loss of synaptophysin in brain (supplemental Figure S3, available at www.jneurosci.org as supplemental material).
Figure 3.
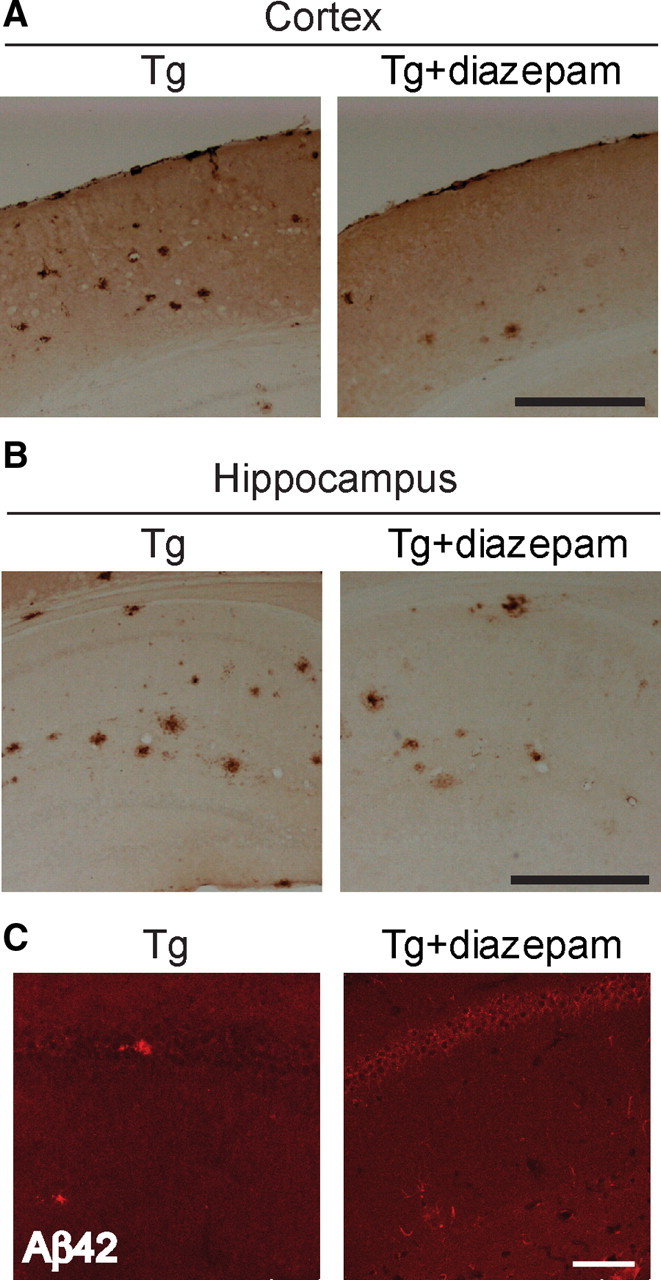
Chronic diazepam administration reduces amyloid plaques and increases intraneuronal Aβ42 immunofluorescence. A, Quantification of cortical plaque number demonstrated a 32 ± 12% decrease in diazepam-treated (Tg+diazepam) compared with untreated Tg19959 (Tg) mice (n = 5; p < 0.01). Scale bar, 500 μm. B, Quantification of hippocampal plaque number demonstrated a 20 ± 9% decrease in plaques in diazepam-treated compared with untreated Tg19959 mice (n = 5, p < 0.05). Scale bar, 500 μm. C, Confocal microscopy showing intraneuronal Aβ42 immunofluorescence in Tg19959 hippocampus. Diazepam-treated Tg19959 mice show 41 ± 10% higher levels of intraneuronal Aβ42 immunoreactivity compared with untreated Tg19959 mice (n = 5; p < 0.01). Scale bar, 75 μm.
Figure 4.
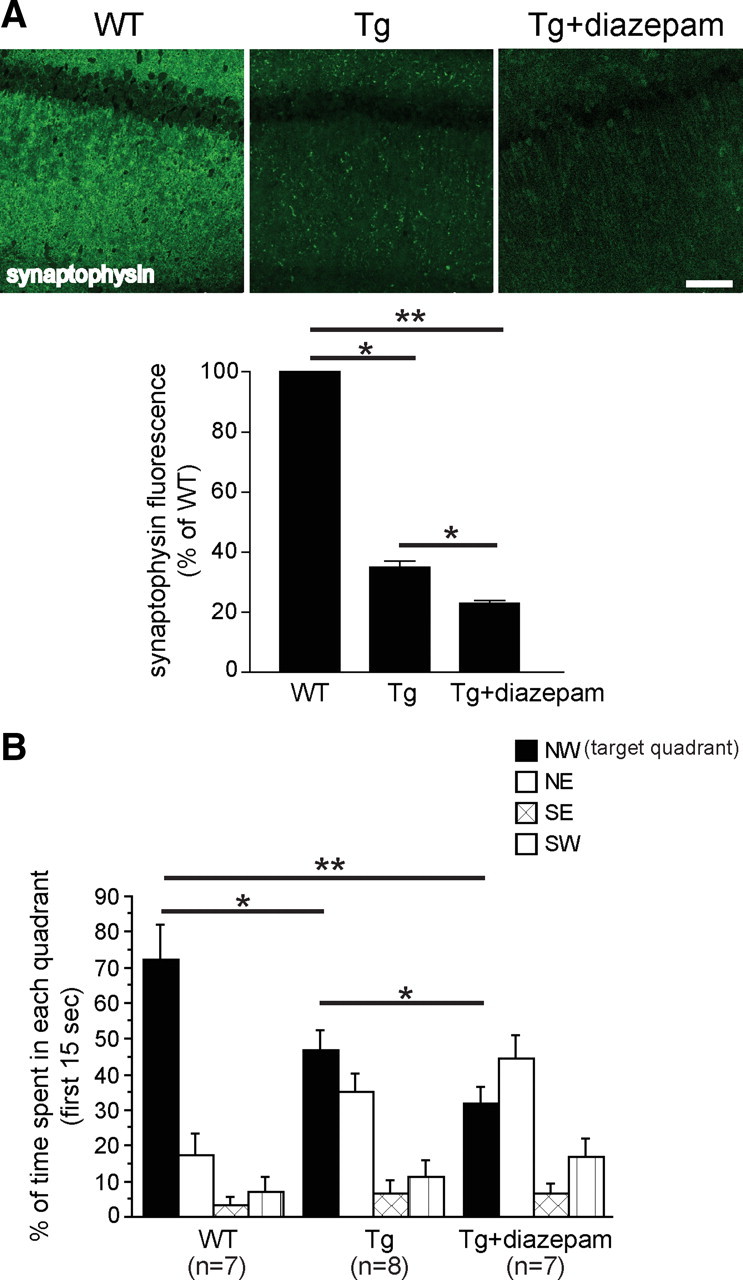
Chronic diazepam administration reduces levels of synaptophysin and worsens spatial memory in Tg19959 mice. A, Fluorescent immunolabeling of synaptophysin in the hippocampus of Tg19959 and wild-type mice. Compared with wild-type mice, both Tg19959 mice and Tg19959 mice treated with diazepam showed 77 ± 1% and 65 ± 2% decreases, respectively, in synaptophysin. Compared with untreated Tg19959 mice, levels of synaptophysin were also significantly reduced in Tg19959 mice treated with diazepam. n = 5; *p < 0.05; **p < 0.01). Scale bar, 75 μm. B, Percentage of time spent in each quadrant during the first 15 s of the probe trial. Both Tg19959 groups spent less time in the target quadrant compared with wild-type mice. Tg19959 mice treated with diazepam showed greater impairment in memory retention compared with untreated Tg19959 mice (*p < 0.05, **p < 0.01).
We also examined levels of full-length APP, Aβ, CTFs and synaptophysin in Tg19959 mouse brains by Western blotting (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Diazepam treatment did not significantly alter total brain levels of any of these in either wild-type or Tg19959 mice. This contrasts with the anatomically specific results seen by confocal immunofluorescence, above, likely because homogenization obscures cellular and anatomical structure.
We next tested whether the reduction in plaques from diazepam treatment was associated with behavioral improvement or decline in the Tg19959 mice. As expected, Tg19959 mice performed worse than wild-type littermates during the probe trial of the MWM (Fig. 4B; supplemental Figure S5A, available at www.jneurosci.org as supplemental material). Remarkably, despite plaque reduction, diazepam treatment further impaired spatial memory performance in Tg19959 mice in the first 15 s of the probe trial (Fig. 4B). Diazepam treatment did not affect performance in the acquisition phase of the MWM in Tg19959 and wild-type mice (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material), and did not significantly affect memory retention in wild-type mice (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material). In addition, there were no differences in noncognitive components of behavior, such as the visible platform test (supplemental Fig, S5D, available at www.jneurosci.org as supplemental material) and swim speed (data not shown), between the Tg19959 and wild-type mice or diazepam-treated compared with untreated mice.
Thus, we have obtained similar results with two different models of chronic synaptic inhibition, pharmacologic and surgical deafferentation. Although benzodiazepines might have effects other than their well established effect on GABA receptors, the concordance with the barrel cortex results further supports the primary role of synaptic inhibition in the results presented.
Discussion
In two different models of chronic synaptic inhibition, there was a reduction in plaque burden but an increase in Aβ42 immunoreactivity within neurons and an exacerbation in synaptophysin and synapse loss. Importantly, despite plaque reduction, chronic inhibition with diazepam exacerbated memory impairment in AD transgenic mice. Although mouse behavior may not necessarily reflect human cognition, the aborted active Aβ42 vaccine clinical trial suggested a similar dissociation between plaques and cognition, with plaque clearance despite continued cognitive decline (Holmes et al., 2008). As another example of dissociation between plaques and cognition, passive immunotherapy with an Aβ antibody improved behavior without plaque reduction in a mouse model of AD (Dodart et al., 2002). In the data reported here, the observed increase of Aβ42 within neurons with synaptic inhibition likely is the primary cause for the reduction of synaptic proteins and impaired memory given our previous mechanistic evidence for this in cultured primary neurons (Tampellini et al., 2009). Evidence increasingly supports that this intraneuronal pool of Aβ42 is critical in AD. Accumulation of intraneuronal Aβ immunoreactivity precedes the appearance of plaques (Gouras et al., 2000; Wirths et al., 2001; Oddo et al., 2003), and the emergence of intraneuronal Aβ correlated with the onset of synaptic, pathological, physiological and behavioral abnormalities in AD transgenic models (Runz et al., 2002; Takahashi et al., 2002; Oddo et al., 2003; Billings et al., 2005; Cruz et al., 2006; Knobloch et al., 2007; Lord et al., 2009; Gandy et al., 2010; Leon et al., 2010; Tomiyama et al., 2010). Of note, a recently described familial AD mutation in APP is associated with intraneuronal Aβ accumulation and oligomerization in the absence of plaques (Tomiyama et al., 2008, 2010). The current work shows the importance of normal synaptic activity in protecting against AD-related pathogenesis, but does not argue against a pathologic role also of synaptic hyperexcitability in AD (Buckner et al., 2005; Palop et al., 2007). Overall, these data have important therapeutic and diagnostic implications for AD, and support the idea that synaptic activity can be protective in AD pathogenesis.
Footnotes
This work was supported by an Alzheimer's Association New Investigator Award (D.T.) and Zenith Award (G.K.G.), and National Institutes of Health Grants AG020729 (M.T.L.) and AG027140, AG028174, and AG09464 (G.K.G.). We thank Dr. George Carlson, McLaughlin Research Institute (Great Falls, MT), for providing the Tg19959 mice. We thank Dr. Teresa Milner and Dr. Costantino Iadecola in our Division of Neurobiology for help with electron microscopy and advice on the barrel cortex experiments.
The authors declare that they have no conflicts of interest.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Billings LM, Green KN, McGaugh JL, LaFerla FM. Learning decreases A beta*56 and tau pathology and ameliorates behavioral decline in 3xTg-AD mice. J Neurosci. 2007;27:751–761. doi: 10.1523/JNEUROSCI.4800-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006;26:10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J. 2009;23:2459–2466. doi: 10.1096/fj.09-132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, Levey AI, Krafft GA, Levy E, Checler F, Glabe C, Bilker WB, Abel T, Schmeidler J, Ehrlich ME. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol. 2010;68:220–230. doi: 10.1002/ana.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsbury C, Mocanu MM, Thies E, Kaether C, Haass C, Keller P, Biernat J, Mandelkow E, Mandelkow EM. Inhibition of APP trafficking by tau protein does not increase the generation of amyloid-beta peptides. Traffic. 2006;7:873–888. doi: 10.1111/j.1600-0854.2006.00434.x. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010;119:523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007;28:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, DeWilde A, Vercauteren F, Atifeh R, Ducatenzeiler A, Klein W, Szyf M, Alhonen L, Cuello AC. A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis. 2010;20:113–126. doi: 10.3233/JAD-2010-1349. [DOI] [PubMed] [Google Scholar]
- Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- Lord A, Englund H, Söderberg L, Tucker S, Clausen F, Hillered L, Gordon M, Morgan D, Lannfelt L, Pettersson FE, Nilsson LN. Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer's disease transgenic mice. FEBS J. 2009;276:995–1006. doi: 10.1111/j.1742-4658.2008.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machín R, Pérez-Cejuela CG, Bjugn R, Avendaño C. Effects of long-term sensory deprivation on asymmetric synapses in the whisker barrel field of the adult rat. Brain Res. 2006;1107:104–110. doi: 10.1016/j.brainres.2006.05.096. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Miller LA. Senile plaques in temporal lobe epilepsy. Acta Neuropathol. 1994;87:504–510. doi: 10.1007/BF00294177. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci. 2002;22:1679–1689. doi: 10.1523/JNEUROSCI.22-05-01679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:S69–74. doi: 10.1097/00002093-200607001-00010. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer's disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging. 2010;31:1145–1152. doi: 10.1016/j.neurobiolaging.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid {beta} oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MT, Welt C. Histochemical changes in cytochrome oxidase of cortical barrels after vibrissal removal in neonatal and adult mice. Proc Natl Acad Sci U S A. 1980;77:2333–2337. doi: 10.1073/pnas.77.4.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]