

Abstract
Bone is one of the few tissues in the body with the capacity to regenerate and repair itself. In most cases, fractures are completely repaired in a relatively short period of time; however, in a small percentage of cases, healing never occurs and non-union is the result. Fracture repair and bone regeneration require the localized re-activation of signaling cascades that are crucial for skeletal development. The Wnt/β-catenin signaling pathway is one such developmental pathway whose role in bone formation and regeneration has been recently appreciated. During the last decade, much has learned about how Wnt pathways regulate bone mass. Small molecules and biologics aimed at this pathway are now being tested as potential new anabolic agents. Here we review recent data demonstrating that Wnt pathways are active during fracture repair and that increasing the activities of Wnt pathway components accelerates bone regeneration.
Keywords: β-catenin, Dkk1, PTH, Sclerostin, SOST, Sfrp1, osteoblasts
Introduction
Bone is one of the few tissues in the body with the capacity to regenerate and repair itself throughout the lifespan of the organism. Fracture healing occurs by two general mechanisms (direct or indirect repair) that mimic early developmental processes. Direct or primary repair takes place when there is contact between adjacent bone cortices. It requires the recruitment of osteoprogentor cells, osteoclasts and undifferentiated mesenchymal stem cells to the fracture site. This process of bone formation across the fracture gap is similar to intramembranous bone formation that forms the flat bones of the skull and clavicles. Direct repair usually occurs when the injury is treated surgically [1]. The second mechanism, indirect or secondary healing, is similar to endochondral bone formation, which is a developmental process that produces the long bones. Secondary fracture healing involves the formation of a soft callus, which is cartilaginous template that undergoes calcification into a hard callus and eventually is replaced by new woven bone. Woven bone is slowly remodeled into lamellar bone, the final stage of a process requiring several months time before the afflicted bone is able to support normal load bearing.
Most fractures heal following temporary immobilization and/or surgical fixation. However, a small percentage (3-10%) of fractures fail to heal properly [2]. Therapies aimed at inducing bone formation at the break points could not only increase the chances of a successful bone union, but also decrease the time required for normal fracture healing. To date, the only anabolic drugs approved in the United States to stimulate bone formation are bone morphogenic protein (BMP)-2, BMP-7 (OP-1) and parathyroid hormone (PTH). Although effective, these biologics have limitations and can only be given locally (e.g. BMPs) or for short periods of time because of safety concerns (e.g PTH). Genetic studies in mice and humans have revealed numerous signaling pathways that are potential new therapeutic targets for regenerating new bone. Particular interest has been focused on the Wnt signaling pathway, mainly due to the essential role that β-catenin-dependent Wnt signaling (commonly referred to as the canonical pathway) plays in initiating and maintaining bone mass (reviewed in [3]). As a result of the relatively newfound appreciation for Wnts in bone biology, several small molecules and biologics that enhance canonical Wnt signaling have been tested in pre-clinical models and some are entering clinical trials. This review will focus on components of canonical Wnt signaling likely to be targeted by future treatment regimens to augment fracture healing. An overview of the Wnt pathway will be provided and followed by discussions of specific canonical Wnt-signaling molecules that are favorable targets for facilitating fracture repair.
Overview of Wnt-β-Catenin Signaling
Originally discovered in Drosophila during the examination of the wingless phenotype, Wnt (Wingless-type MMTV integration site) proteins are now known to play crucial roles in mammalian embryogenesis, organ development and regeneration, cell migration and proliferation, and carcinogenesis [4]. Wnts are secreted glycoproteins that bind several classes of plasma membrane bound receptors, including Frizzled (Fzd), Ror2 and Ryk. Depending on the receptor complex that is ligated, Wnts stimulate one or more intracellular signaling cascades (Figure 1) [5]. The best understood Wnt pathway involves the stabilization of β-catenin. This pathway is initiated when Wnts bind to a Fzd receptor. The complex is further stabilized by the recruitment of a low-density lipoprotein receptor-related protein (LRP5 or LRP6) co-receptor. Through a series of complex molecular events (reviewed in [6]), β-catenin phosphorylation by two kinases, glycogen synthase kinase (GSK)3β and casein kinase (CK)-1, is inhibited and proteosome-mediated degradation is thwarted. Increased cytoplasmic levels of β-catenin eventually allow for the translocation of β-catenin into the nucleus, where it can associate with the transcription factors, T cell factor (Tcf) 7 and lymphoid enhancing factor (Lef1).
Figure 1. Wnts initiate multiple signaling pathways.
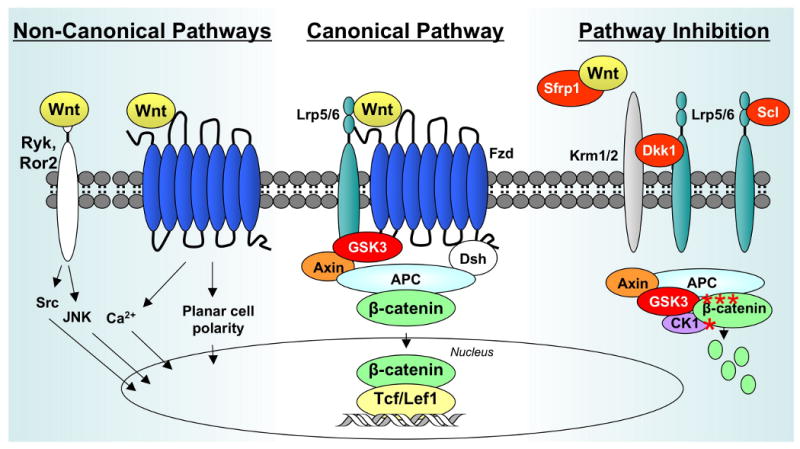
The “canonical pathway” (center) is stimulated when Wnts bind to Fzd receptors and Lrp5/6 co-receptors. A cytoplasmic complex composed of GSK3β, Axin, and APC is inhibited, allowing cytoplasmic levels of β-catenin levels to rise. Some β-catenin translocates to the nucleus where it associates with Tcf/Lef transcription factors to regulate gene expression. During non-canonical Wnt signaling (left), Wnts bind to a Fzd receptor, Ryk or Ror2, and activate downstream signaling events that do not involve GSK3β or β-catenin. Wnt signaling pathways are inhibited by several mechanisms (right). Secreted frizzled-related proteins (Sfrps) antagonize canonical Wnt signaling by binding the ligands and preventing their association with Fzd receptors. Dkk1 suppresses Wnt signaling by forming a ternary complex with Lrp5/6 and Krm1/2. Sclerostin (Scl) also binds to Lrp5/6, but not Krm1/2, to antagonize canonical Wnt signaling. In the absence of Wnt/Lrp signaling, GSK3β phosphorylates (asterisks) β-catenin, which marks it for ubiquitination and proteosomal degradation.
Like all developmental pathways, Wnts are tightly regulated by paracrine factors and autocrine feedback loops. The most fruitful therapeutic strategy for targeting the Wnt-β-catenin pathway in bone regeneration settings has focused on agents that neutralize or inhibit the negative regulators of Wnt signaling, particularly GSK3β, Dickkopfs (Dkk), Secreted frizzled related proteins (Sfrp), and Sclerostin (Scl) (Figure 1). The alternative strategy of adding Wnts as agonists is not feasible at this time because only palmitoylated forms of Wnts are active and these are extremely difficult and expense to purify. By comparison, many small molecule inhibitors of GSK3β have been described [7] and humanized monoclonal antibodies are available that neutralize the secreted inhibitors, Dkk1, Sfrp1 and Sclerostin.
Wnt Pathway Activation at Fracture Sites
Examination of rodent fracture sites by microarrays and RT-PCR revealed that Wnt pathways contribute to normal fracture healing. Hadjiargyrou and colleagues found that mRNAs for Wnt4, Wnt5a, Fzd2, Lrp5, β-catenin and several other Wnt pathway components were upregulated at the fracture site within 3-5 days of injury, suggesting a role for Wnts in the early stages of healing that involve intramembranous bone formation [8, 9]. These genes were declining towards basal levels, but still elevated, at 14 to 21 days post-fracture, suggesting a role for them during the endochondral ossification stage of fracture healing. Interestingly, transcripts for Lef1 declined during healing [9]. Lef1 inhibits Runx2 dependent activation of some osteoblast genes and might thereby inhibit bone formation by mature osteoblasts [10].
Several other groups also observed increased expression of these and other Wnts and Wnt pathway components at sites of skeletal injury. The Alman laboratory demonstrated that mRNAs for Wnts 4, 5b, 11, and 13, Fzds 1, 2, 4, 5, and 6, and Lrp6 were upregulated within one week of fracture in mice [11]. Wnt5a protein levels were also elevated within the fracture site during the first week of healing, as were protein kinase C levels, suggesting that the “non-canonical” Wnt/calcium pathway was activated. The canonical pathway was also stimulated as β-catenin protein levels rose within four days and remained elevated for two weeks before beginning to decline at three weeks [11]. Importantly, β-catenin protein levels were also elevated in a three-week-old human fracture before returning to basal levels in 12-week-old fractures [11]. The Gerstenfeld/Barnes/Einhorn group reported that mRNAs for Wnts 4, 5a, 5b, 10b and 11, Lrp5, and Lrp6 were upregulated within five days after fracture in a rodent model and remained above baseline levels throughout the three-week analysis [12]. Expression profiles for eleven other Wnts were not changed. This group also found that β-catenin was localized in the nuclei of osteoblastic cells lining trabeculi along the periosteal layer near the fracture site, in maturing chondrocytes of the fracture callus and in cortical bone osteocytes at five days postfracture [12]. Finally, the Helms laboratory found that mRNAs for Wnts 2b, 3a, 5a, 5b, 7a, 11, Fzd4, and Dkk2 were elevated at or adjacent to transcortical defect injuries in murine tibias [13]. Together, these studies demonstrate that many Wnts are expressed during bone repair and regeneration. Moreover, the data suggest that multiple Wnt signaling pathways could be activated in several cell types that are involved in repair.
β-Catenin Stabilization and GSK-3β Inhibition Accelerates Fracture Repair
β-catenin stabilization is a crucial event in the “canonical” signaling pathway, which is activated by a subset of Wnts, including Wnts 1, 3a, 8, 10b. Depending on receptor availability, Wnt5a, can also activate canonical Wnt signaling (reviewed in [5]). In the nucleus, β-catenin cooperates with Lef1/Tcf7 transcription factors to regulate gene expression. As described above, β-catenin expression levels are much higher in newly formed fractures calluses as compared to intact bone in both mice and humans [11, 12]. Studies with transgenic mice expressing a Tcf reporter gene further demonstrate that β-catenin and Tcf7 complexes are active at various stages during fracture repair and bone regeneration, particularly in proliferating chondrocytes and osteoblasts, but not in hypertrophic chondrocytes or inflammatory cells (i.e. neutrophils) [11, 13]. mRNAs for several known Wnt/Tcf7 target genes are also increased in fracture repair sites [8, 9, 14].
Genetically altered mice indicate a direct relationship between β-catenin expression levels and bone formation and repair. Depletion of β-catenin in mesenchymal progenitor cells arrests osteoblast differentiation and favors chondrogenesis; whereas in committed osteoblasts, conditional knockout of β-catenin results in osteopenia and expression of a constitutively stabilized form of β-catenin increases bone mass (reviewed in [15]). The Alman laboratory used some of these mouse models to study the role of β-catenin in fracture repair [11]. Conditional knockout of β-catenin in committed osteoblasts led to a significant reduction in calcified callus formation, along with incomplete bone bridging across the fracture gap. Conversely, transgenic expression of stabilized β-catenin in committed osteoblasts enhanced fracture healing. Interestingly, when β-catenin levels were either up- or down-regulated in all cells of the fracture callus by adenoviral-driven expression of Cre recombinase, a similar radiographic phenotype was observed in both situations, that being no bone bridging at the fracture site [11]. These data suggest that β-catenin makes diverse contributions to the complex orchestration of cellular events that mediate fracture repair.
β-catenin's role as a positive regulator of osteoblast activities makes enhancing its activity an enticing strategy to enhance bone regeneration and repair. Currently, pharmacological activation of β-catenin has been achieved indirectly through inhibition of GSK-3β activity. GSK3β is one of the kinases that phosphorylate β-catenin and target it for degradation (Figure 1). Lithium chloride is a well-known GSK-3β inhibitor and has been used clinically in the treatment of bipolar disorders for over 50 years [16]. Some epidemiological studies indicate that long-term lithium users might have a reduced relative fracture risk [17, 18]. In mouse models of low bone density, inhibition of GSK-3β by lithium chloride or another small molecule inhibitor, LY603281-8, increased trabecular bone mass [19-21]. In elegant experiments, the Alman laboratory tested the ability of lithium chloride to accelerate fracture repair [11]. Oral administration of lithium chloride increased healing and bone volume within fracture calluses in mice when administered four days after the fracture occurred. Notably, mice treated systemically with lithium chloride two weeks prior to fracture showed a significant decrease in callus mineralization, even though β-catenin expression levels were elevated. Mesenchymal precursor cells that failed to differentiate into the osteoblast lineage occupied the fracture sites in these mice. It remains to be determined if materials commonly used to deliver BMP-2 in various orthopedic procedures could also be employed to direct GSK-3β inhibitors to a fracture.
Extracellular Wnt Pathway Inhibitors Delay Fracture Healing
Natural extracellular inhibitors of Wnt signaling are additional targets for new biologic agents that accelerate fracture healing. Known secreted factors block Wnt signaling via two mechanisms (Figure 1). Dkk1 and Sclerostin bind Lrp5/6 and antagonize the receptor complex. In contrast, Sfrps bind the ligand and prevent it from properly associating with membrane-bound receptors. Recent evidence from preclinical models suggest that these factors have a negative impact on fracture repair but are involved in completing the healing process.
Dkk1 Prevents Bone Repair
Dkk1 represses Wnt signaling by recruiting Lrp5/6 into a ternary complex with Kremen1/2 (Figure 1) and thereby preventing Lrp5/6 from associating with Wnts. Dkk1-deficient mice have severe developmental defects and die during embryogenesis [22]; however, Dkk1-haploinsufficient or hypomorphic animals have increased bone mass [23, 24]. Conversely, mice overexpressing Dkk1 in osteoblasts have fewer bone forming cells and are osteopenic [25]. DKK-1 serum levels are elevated in some rheumatoid arthritis patients and individuals with osteolytic tumors [26, 27].
Two studies recently tested the effects of Dkk1 on fracture repair and bone regeneration in skeletally mature (i.e. ∼12 week-old) male mice [11, 13]. Both groups used adenoviruses to deliver Dkk1 to the injury site. The Alman laboratory found that administration of Dkk1 directly to the fracture site reduced β-catenin levels in calluses and prevented chondrogenesis during the first week after fracture [11]. Molecular analyses showed that master genes of chondrogenesis and osteogenesis, Sox9 and Runx2, respectively, were not expressed at the fracture site. The fractures failed to heal and the injury site contained undifferentiated mesenchymal-like tissue. In an independent study, the Helms group injected Dkk1-expressing adenoviruses via the tail vein into transgenic mice expressing a Wnt/β-catenin/Tcf reporter (i.e. TOP-gal mice) immediately before creating a 1 mm-wide transcortical tibial defect [13]. Dkk1 prevented activation of the Wnt/β-catenin/Tcf reporter during the first week of healing and reduced bone regeneration by 84%. In a second set of experiments, this group injected Dkk1-expressing adenoviruses into the tibial musculature two days before generating a skeletal defect in the same area. Once again, Dkk1 blocked Wnt/β-catenin/Tcf reporter activity and bone regeneration [13]. Dkk1 did not prevent angiogenesis or induce cell death at the repair site, but it suppressed the expression of osteogenic genes, including Runx2. Together, these studies demonstrated that Dkk1 prevents the early stages of bone repair by blocking Wnt/β-catenin/Tcf signaling and the differentiation of mesenchymal progenitor cells into chondrocyte or osteoblasts. A third group showed that Dkk1 mRNA levels increase during the second week of fracture healing, suggesting that Dkk1 might be involved in slowing the anabolic response at later stages of repair [12].
On the basis of the above data, inhibiting Dkk1 might be beneficial for bone repair. Indeed, Dkk1-neutralizing monoclonal antibodies stimulated bone formation in two mouse models of myeloma-induced osteolytic disease [28, 29]. Moreover, Diarra and colleagues showed that Dkk1 antibodies not only prevented bone destruction caused by inflammation-induced rheumatoid arthritis, but induced osteophyte formation [26]. The effects of Dkk1 neutralization on fracture repair have not been directly tested; however, Kim and colleagues [13] found that the initial stages of bone regeneration were impaired in transgenic mice overexpressing Lrp5 proteins that contain a point mutation (G171V) which prevents Dkk1 binding and causes high bone mass [30, 31]. This delay coincided with the extensive proliferation of osteoblasts in adjacent uninjured bone, perhaps at the expense of osteoblast migration to and/or differentiation in the fracture site. Interestingly, the initial problems with defect healing were followed by robust repair [13], suggesting that Dkk1 inhibition might be a promising strategy to repair non-union fractures or to accelerate healing. Because Dkk1 is expressed in many tissues, local delivery would produce the fewest concerns. However, the propensity of Dkk1 antibodies to induce osteophytes must be given consideration when designing treatment regimens.
Sfrp1, a Wnt Antagonist, Contributes to Bone Regeneration
Secreted frizzled-related proteins (Sfrp) are decoy receptors and antagonists of Wnts [32] (Figure 1). In a mouse model of inflammation-induced periodontal bone loss/disease, Sfrp1 expression directly correlated with the amount of osteoblast apoptosis, which was partially blocked following treatment with Sfrp1 antibodies [33]. Sfrp1-deficient mice display less osteoblast and osteocyte apoptosis and have increased trabecular bone mass as compared to wildtype animals [34]. Gaur et al found that fracture healing was accelerated and enhanced in these Sfrp1-deficient animals, leading to mechanically stronger bones [35]. Despite the ubiquitous nature of Sfrp1 expression, genomic knockout of Sfrp1 does not result in a multiple tissue phenotype [34]. To the best of our knowledge, Sfrp1-directed therapies have not yet been tested in fracture repair models. However, a specific small molecule inhibitor of Sfrp1 is capable of stimulating ex-vivo bone formation [36] and a commercially available Sfrp1 antibody blocked inflammation-induced periodontal bone loss [33].
Intermittent PTH Stimulates Bone Formation, Regeneration and Repair in Part by Inducing Canonical Wnt Signaling
PTH is a systemic hormone, originally classified as a bone resorptive agent, because of its ability to increase serum calcium levels and stimulate osteoclast activity. Anabolic actions of PTH were first described in rodents in the late 1980's following intermittent, as opposed to continuous, treatment [37]. PTH (amino acids 1-34, Forteo™, Teraparatide) is currently the only clinically approved anabolic therapy used to treat osteoporosis in the United States. New data indicate that PTH and its receptor PTH1R exert their bone anabolic effects in conjunction with several components of the canonical Wnt-signaling pathway. PTH decreased GSK-3β activity in Saos2 cells and reduced Sclerostin (an Lrp5/6 inhibitor) expression in overectomized rats [38, 39]. The anabolic effects of PTH on bone likely result from direct interactions of PTH and osteoblasts. Mice expressing a constitutively active form of PTH receptor (PTH1R) in osteocytes have increased bone mass and reduced Sclerostin levels [40]. PTH signaling in osteoblasts appears to be dependent upon direct interactions involving PTH, PTH1R and Lrp6 [41]. Interestingly, PTH stabilization of cytoplasmic β-catenin required the activation of protein kinase A and appeared to be Wnt-independent [41]. However, in vivo data indirectly support the crucial role that Lrp6, but not Lrp5, plays in mediating PTH dependent increases in bone mass [42].
Several studies showed that PTH treatment increases callus volume, stiffness, and callus bone mineral content in rats that were fractured or subjected to distraction osteogenesis, a technique used to treat non-union fractures in humans[43-45]. The mechanism(s) of this repair process are not understood very well; however, they include activation of Wnt signaling. Kakar and colleagues found that PTH enhanced the expression of several Wnts (Wnts 4, 5a, 5b and 10b), Lrp5 and Lrp6 in the fracture callus [12]. PTH also increased nuclear β-catenin levels in mature chondrocytes and osteoblasts lining the trabecular surfaces. During the second and third weeks of healing, PTH elevated the levels of Dkk1 and Sclerostin transcripts. These data indicate that PTH-mediated bone formation is at least partially Wnt-dependent.
Conclusions
The ability of bone to regenerate and repair itself is a fact that most individuals learn and appreciate at an early age. However, in a small percentage of severe and disabling fractures, repair never occurs and non-union is the result. Signaling cascades that are crucial for skeletal development are reactivated during normal repair. The recent advances in our understanding of the crucial roles that Wnt/β-catenin signaling plays in the development and maturation of osteoblast lineage cells has generated new opportunities to treat non-union fractures and perhaps to accelerate repair. Lithium chloride, which increases β-catenin levels, is clinically approved for mood disorders and might be useful for fracture healing because it improves bone regeneration and repair in rodent models. Preclinical models also support suppression of other Wnt pathway inhibitors (e.g. Dkk1 and Sfrp1) as strategies to improve bone regeneration and fracture repair.
A potential limitation of these strategies is that β-catenin, Dkk1 and Sfpr1 are widely expressed. Thus, agents that inhibit these molecules might be associated with several risks, including tumorigenesis, osteophytes, osteoarthritis, and hypercalcemia (reviewed in [15]]. Sclerostin is a more ideal target for treating fractures because its expression is apparently limited to bone [46]. Sclerostin is the product of the SOST gene, which is genetically inactivated in Sclerosteosis and Van Buchem's disease [47, 48]. Sclerostin repression by genetic knockout and neutralizing antibodies increases systemic bone mass in ovarectimized mice [49, 50], but so far these reagents have not been tested in fracture repair experiments. The available evidence indicates that osteoblasts are the crucial targets of canonical Wnts in the fracture site, but whether Wnt/β-catenin and Lrp5/6 agonists can promote angiogenesis, mesenchymal cell recruitment and chondrogenesis needed to repair clinically severe fractures remains to be determined.
Acknowledgments
Grants AR50074 and AR48147 from the NIAMS/NIH made this publication possible.
References
- 1.Dimitriou R, Tsiridis E, Giannoudis PV. Current concepts of molecular aspects of bone healing. Injury. 2005;36:1392–1404. doi: 10.1016/j.injury.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Tzioupis C, Giannoudis PV. Prevalence of long-bone non-unions. Injury. 2007;38 2:S3–9. doi: 10.1016/s0020-1383(07)80003-9. [DOI] [PubMed] [Google Scholar]
- 3.Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39. doi: 10.1016/j.gene.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 4.Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- 5.van Amerongen R, Mikels A, Nusse R. Alternative wnt signaling is initiated by distinct receptors. Sci Signal. 2008;1:re9. doi: 10.1126/scisignal.135re9. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, He X. Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol. 2008;20:119–125. doi: 10.1016/j.ceb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- 8.Hadjiargyrou M, Lombardo F, Zhao S, et al. Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J Biol Chem. 2002;277:30177–30182. doi: 10.1074/jbc.M203171200. [DOI] [PubMed] [Google Scholar]
- 9.Zhong N, Gersch RP, Hadjiargyrou M. Wnt signaling activation during bone regeneration and the role of Dishevelled in chondrocyte proliferation and differentiation. Bone. 2006;39:5–16. doi: 10.1016/j.bone.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Kahler RA, Westendorf JJ. Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. J Biol Chem. 2003;278:11937–11944. doi: 10.1074/jbc.M211443200. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Whetstone HC, Lin AC, et al. Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 2007;4:e249. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This manuscript demonstrates that Dkk1 inhibits fracture repair while lithium chloride, a GSK3 inhibitor and β-catenin stabilizer, accelerates fracture healing if administered after fracture, but not before injury.
- 12.Kakar S, Einhorn TA, Vora S, et al. Enhanced chondrogenesis and Wnt signaling in PTH-treated fractures. J Bone Miner Res. 2007;22:1903–1912. doi: 10.1359/jbmr.070724. [DOI] [PubMed] [Google Scholar]; • This group found that PTH induced the expression of several Wnt pathway components in osteoblasts at a fracture site.
- 13.Kim JB, Leucht P, Lam K, et al. Bone regeneration is regulated by wnt signaling. J Bone Miner Res. 2007;22:1913–1923. doi: 10.1359/jbmr.070802. [DOI] [PubMed] [Google Scholar]; • This study shows that Dkk1 inhibits the repair of a single trans-cortical defect by preventing osteogenic responses.
- 14.French DM, Kaul RJ, D'Souza AL, et al. WISP-1 is an osteoblastic regulator expressed during skeletal development and fracture repair. Am J Pathol. 2004;165:855–867. doi: 10.1016/S0002-9440(10)63348-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoeppner L, Secreto F, Westendorf J. Wnt signaling as a therapeutic target for bone diseases. Expert Opinion on Therapeutic Targets. 2009 doi: 10.1517/14728220902841961. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livingstone C, Rampes H. Lithium: a review of its metabolic adverse effects. J Psychopharmacol. 2006;20:347–355. doi: 10.1177/0269881105057515. [DOI] [PubMed] [Google Scholar]
- 17.Vestergaard P, Rejnmark L, Mosekilde L. Reduced relative risk of fractures among users of lithium. Calcif Tissue Int. 2005;77:1–8. doi: 10.1007/s00223-004-0258-y. [DOI] [PubMed] [Google Scholar]
- 18.Wilting I, de Vries F, Thio BM, et al. Lithium use and the risk of fractures. Bone. 2007;40:1252–1258. doi: 10.1016/j.bone.2006.12.055. [DOI] [PubMed] [Google Scholar]
- 19.Clement-Lacroix P, Ai M, Morvan F, et al. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A. 2005;102:17406–17411. doi: 10.1073/pnas.0505259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulkarni NH, Onyia JE, Zeng Q, et al. Orally bioavailable GSK-3alpha/beta dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo. J Bone Miner Res. 2006;21:910–920. doi: 10.1359/jbmr.060316. [DOI] [PubMed] [Google Scholar]
- 21.Kulkarni NH, Wei T, Kumar A, et al. Changes in osteoblast, chondrocyte, and adipocyte lineages mediate the bone anabolic actions of PTH and small molecule GSK-3 inhibitor. J Cell Biochem. 2007;102:1504–1518. doi: 10.1002/jcb.21374. [DOI] [PubMed] [Google Scholar]
- 22.Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, et al. Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell. 2001;1:423–434. doi: 10.1016/s1534-5807(01)00041-7. [DOI] [PubMed] [Google Scholar]
- 23.Morvan F, Boulukos K, Clement-Lacroix P, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–945. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 24.MacDonald BT, Joiner DM, Oyserman SM, et al. Bone mass is inversely proportional to Dkk1 levels in mice. Bone. 2007;41:331–339. doi: 10.1016/j.bone.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Sarosi I, Cattley RC, et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–766. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 26.Diarra D, Stolina M, Polzer K, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156–163. doi: 10.1038/nm1538. [DOI] [PubMed] [Google Scholar]; • The report demonstrates that a Dkk1-neutralizing antibody increases bone formation in a mouse model of rheumatoid arthritis.
- 27.Tian E, Zhan F, Walker R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 28.Yaccoby S, Ling W, Zhan F, et al. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109:2106–2111. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heath DJ, Chantry AD, Buckle CH, et al. Inhibiting dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J Bone Miner Res. 2008 doi: 10.1359/jbmr.081104. [DOI] [PubMed] [Google Scholar]
- 30.Ai M, Holmen SL, Van Hul W, et al. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol. 2005;25:4946–4955. doi: 10.1128/MCB.25.12.4946-4955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babij P, Zhao W, Small C, et al. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18:960–974. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 32.Jones SE, Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;24:811–820. doi: 10.1002/bies.10136. [DOI] [PubMed] [Google Scholar]
- 33.Li CH, Amar S. Inhibition of SFRP1 reduces severity of periodontitis. J Dent Res. 2007;86:873–877. doi: 10.1177/154405910708600913. [DOI] [PubMed] [Google Scholar]
- 34.Bodine PV, Zhao W, Kharode YP, et al. The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol. 2004;18:1222–1237. doi: 10.1210/me.2003-0498. [DOI] [PubMed] [Google Scholar]
- 35.Gaur T, Wixted J, Hussain S, et al. Secreted frizzled related protein 1 is a target to improve fracture healing. Presented at the Orthopedic Research Society Annual Meeting; Las Vegas, Nevada. February 21-25, 2009; [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study finds that fracture healing is accelerated in Sfrp1-deficient mice.
- 36.Moore WJ, Kern JC, Bhat R, et al. Modulation of Wnt signaling through inhibition of secreted frizzled-related protein I (sFRP-1) with N-substituted piperidinyl diphenylsulfonyl sulfonamides. J Med Chem. 2009;52:105–116. doi: 10.1021/jm801144h. [DOI] [PubMed] [Google Scholar]
- 37.Hock JM, Gera I, Fonseca J, Raisz LG. Human parathyroid hormone-(1-34) increases bone mass in ovariectomized and orchidectomized rats. Endocrinology. 1988;122:2899–2904. doi: 10.1210/endo-122-6-2899. [DOI] [PubMed] [Google Scholar]
- 38.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–158. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki A, Ozono K, Kubota T, et al. PTH/cAMP/PKA signaling facilitates canonical Wnt signaling via inactivation of glycogen synthase kinase-3beta in osteoblastic Saos-2 cells. J Cell Biochem. 2008;104:304–317. doi: 10.1002/jcb.21626. [DOI] [PubMed] [Google Scholar]
- 40.O'Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3:e2942. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wan M, Yang C, Li J, et al. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008;22:2968–2979. doi: 10.1101/gad.1702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawakami K, Robling AG, Ai M, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–23711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 43.Andreassen TT, Willick GE, Morley P, Whitfield JF. Treatment with parathyroid hormone hPTH(1-34), hPTH(1-31), and monocyclic hPTH(1-31) enhances fracture strength and callus amount after withdrawal fracture strength and callus mechanical quality continue to increase. Calcif Tissue Int. 2004;74:351–356. doi: 10.1007/s00223-003-0093-6. [DOI] [PubMed] [Google Scholar]
- 44.Seebach C, Skripitz R, Andreassen TT, Aspenberg P. Intermittent parathyroid hormone (1-34) enhances mechanical strength and density of new bone after distraction osteogenesis in rats. J Orthop Res. 2004;22:472–478. doi: 10.1016/j.orthres.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 45.Komatsu DE, Brune KA, Liu H, et al. Longitudinal In Vivo Analysis of the Region Specific Efficacy of PTH in a Rat Cortical Defect Model. Endocrinology. 2008 doi: 10.1210/en.2008-0814. [DOI] [PubMed] [Google Scholar]
- 46.van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 48.Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2008 doi: 10.1359/jbmr.081206. In press. [DOI] [PubMed] [Google Scholar]; • This is the first published report showing that a Sclerostin-neutralizing antibody enhances bone formation.