

Abstract
Human immunodeficiency virus-associated neurological disease (HAND) still causes significant morbidity, despite success reducing viral loads with combination antiretroviral therapy. The dopamine (DA) system is particularly vulnerable in HAND. We hypothesize that early, “reversible” DAergic synaptic dysfunction occurs long before DAergic neuron loss. As such, aging human immunodeficiency virus (HIV)-infected individuals may be vulnerable to other age-related neurodegenerative diseases like Parkinson's disease (PD), underscoring the need to understand shared molecular targets in HAND and PD. Previously, we reported that the neurotoxic HIV-1 transactivating factor (Tat) acutely disrupts mitochondrial and endoplasmic reticulum calcium homeostasis via ryanodine receptor (RyR) activation. Here, we further report that Tat disrupts DA transporter (DAT) activity and function, resulting in increased plasma membrane (PM) DAT and increased DAT Vmax, without changes in Km or total DAT protein. Tat also increases calpain protease activity at the PM, demonstrated by total internal reflection fluorescence microscopy of a cleavable fluorescent calpain substrate. Tat-increased PM DAT and calpain activity are blocked by the RyR antagonists ryanodine and dantrolene, the calpain inhibitor calpastatin, and by a specific inhibitor of GSK-3β. We conclude that Tat activates RyRs via a calcium- and calpain-mediated mechanism that upregulates DAT trafficking to the PM, and is independent of DAT protein synthesis, reinforcing the feasibility of RyR and GSK-3β inhibition as clinical therapeutic approaches for HAND. Finally, we provide key translational relevance for these findings by highlighting published human data of increased DAT levels in striata of HAND patients and by demonstrating similar findings in Tat-expressing transgenic mice.
Introduction
Human immunodeficiency virus-associated neurological disease (HAND) remains a significant source of morbidity despite efficacious reduction of viral load by combination antiretroviral therapy (cART). Onset of HAND correlates with virus load within the brain and CSF, and with CD4 count. Although cART has reduced the incidence of HAND, it cannot always prevent HAND (Masliah et al., 2000) or reduce HAND severity when administered to cART-naive patients (Chang et al., 2003). With cART-treated human immunodeficiency virus-positive (HIV+) patients living longer, the prevalence of HAND continues to increase, highlighting the importance of understanding HAND pathogenetic mechanisms as they may synergize with, or increase susceptibility to, comorbidly evolving neurological disease in aging HIV+ populations, for instance Parkinson's disease (PD).
The dopaminergic (DAergic) nigrostriatal pathway is notably afflicted in HAND, and 42.8% of HIV patients presenting with parkinsonism show direct effects of HIV-1 on the basal ganglia (Mattos et al., 2002), with the greatest neuronal loss occurring in the DAergic basal ganglia and substantia nigra (SN) (Reyes et al., 1991). The chemical structure of DA renders it easily auto-oxidized into electrophilic quinone-type species, making DAergic neurons particularly susceptible to oxidative stress. This may result in selective vulnerability of DAergic neurons in diseases such as PD and HAND. However, postmortem HIV and HIV encephalitis (HIVE) brains can present with considerable evidence of DA synaptic dysfunction without much evidence of frank DAergic neuron loss (Gelman et al., 2006; Silvers et al., 2006), and HAND symptoms referable to basal ganglia deficits are improved with cART (Hersh et al., 2001). These findings suggest many early DAergic synaptic deficits in HAND are reversible and thus amenable to early therapeutic intervention.
Likewise, HIV secretory neurotoxins decrease synaptic density and increase vacuolation of dendritic spines in affected brain regions of HIV patients (Wiley et al., 1986; Masliah et al., 1997; Everall et al., 1999; Sá et al., 2004), and these synaptic alterations correlate far better with premortem neuropsychological assessments of cognitive function than does frank neuronal cell loss (Everall et al., 1994). Although cART can ameliorate or even reverse the course of HAND in some HIV-infected persons (Gendelman et al., 1998), the severity of neurocognitive impairment at cART initiation is the strongest predictor of persistent neurological deficits despite ongoing cART (Tozzi et al., 2007). Work from our and others' laboratories has shown that the phospholipid mediator platelet-activating factor (PAF) and Tat can induce reversible synaptic dysfunction before cell death (Perry et al., 2005a; Lu et al., 2007; Kim et al., 2008), and elevated levels of PAF may render “normal” physiological synaptic activity excitotoxic (Bellizzi et al., 2005).
Together, these results suggest HAND symptomatology may in part arise from reversible synaptic dysfunctions, which, if left unchecked, ultimately result in neuronal cell loss and permanent deficit (Perry et al., 2005b; Bellizzi et al., 2006). Herein, we investigated whether DAergic synapses might exhibit similar reversible deficits in response to the HIV neurotoxin Tat, which has well established neurotoxic effects on DAergic pathways. We report Tat significantly elevates plasma membrane (PM)-localized DA transporter (DAT) and DAT uptake activity, by a GSK-3β-regulated pathway that modulates ryanodine receptor (RyR)- and calpain-associated control of DAT.
Materials and Methods
Reagents
The recombinant HIV-1 Tat1-72 was generously provided by the laboratory of Dr. Avindra Nath (The Johns Hopkins University, Baltimore, MD) and by Dr. Phil Ray (University of Kentucky, Lexington, KY). In these experiments, we used heat-inactivated aliquots of the above Tat protein as the control for Tat, to control for nonspecific effects (e.g., pyrogenic effects) not related to the functional protein activity of Tat, as previously described (Kim et al., 2008). In addition, no significant differences between heat-inactivated Tat control, and vehicle control, were found in our assays. Therefore, having established this in these and previous studies, in some cases vehicle control alone is displayed, where described.
For these studies, Tat was used at 120 nm (∼1 μg/ml) concentration unless otherwise described, a concentration that closely approximates (or is lower than) doses used in similar studies of DAergic, synaptic, and nervous system function, using equivalently prepared Tat (Orsini et al., 1996; Gurwell et al., 2001; Aksenova et al., 2006; Aprea et al., 2006; Wallace et al., 2006; Zhong et al., 2008; Zhu et al., 2009). We and others have determined that doses on this order of magnitude (50–500 nm) represent the minimum Tat doses required to elicit modest (10–20%) but statistically significant increases in cell death and synapse loss over extended 24 h treatments (Maggirwar et al., 1999; Ramirez et al., 2001; Perry et al., 2005a; Aksenova et al., 2006), while at the same time eliciting acute intracellular metabolic effects without cell death over abbreviated 30 min to 1 h treatments (Perry et al., 2005a; Wallace et al., 2006; this study), thus making 120 nm Tat a suitable dose to model early effects of HAND on dopaminergic neurons.
Indeed, soluble Tat levels in HIV+ patient sera have also been measured up to 40 ng/ml (Westendorp et al., 1995; Xiao et al., 2000), despite that the interactions of Tat with endogenous glycosaminoglycans and heparin sulfates may lower its measurable concentration in vivo (Chang et al., 1997; Xiao et al., 2000). Therefore, Tat concentrations surrounding HIV-infected cells are likely much higher than sera concentrations (Hayashi et al., 2006). Moreover, recombinantly produced Tat—using a commonly used, highly standardized, and reproducible production procedure—is considerably less potent than Tat released from Tat-expressing astrocytic cells (Li et al., 2008), suggesting that comparatively higher doses of this commonly used recombinant Tat protein are required to adequately mimic the effects of Tat constitutively produced in the HIV-infected brain. Additionally, the strong affinity of Tat for other proteins and glass/plastic surfaces, its temperature sensitivity, and its susceptibility to oxidation, make it impossible to determine what proportion of the initial Tat dose actually reaches the experimental specimen, leading to underestimation of Tat functions in vitro (Nath et al., 2000). Finally, in the context of HIV infection, many of the effects of Tat may occur over long-term chronic exposures. Chronic low-dose effects of any reagent in vivo are often appropriately modeled in vitro by proportionately higher doses of that same reagent over more acute time frames. In this context, many studies have found Tat concentrations on the order of magnitude used herein, to be appropriate doses by which to model, translate, and evaluate in vivo effects of Tat in vitro. For the sum of these reasons, we use 120 nm Tat for these experiments herein. Despite these caveats, in many cases we were also able to achieve similar but sometimes less robust effects with considerably lower Tat doses (∼12 nm/∼100 ng/ml) (data not shown), but elected to use the 120 nm dose throughout for consistency of the response.
PC12-hDAT cells were kindly provided by Dr. Haley Melikian (University of Massachusetts, Worcester, MA), and eGFP-hDAT construct was kindly provided by Dr. Susan Amara (University of Pittsburgh, Pittsburgh, PA). B27 supplement (with and without antioxidants), Neurobasal media, and ASP+ were purchased from Invitrogen. The cell-permeable GSK inhibitor VIII (AR-AO14418) and calpastatin peptide (208902) were purchased from Calbiochem/EMD Biosciences. Antibodies for Western blots were purchased from Santa Cruz Biotechnology and Millipore Bioscience Research Reagents. All other chemicals and reagents were purchased from Sigma-Aldrich, except as noted.
Cell culture
Primary neuronal cell culture.
Cultures of primary rat mesencephalic neurons containing SN and the DA-rich rostral mesencephalon were prepared from embryonic day 12 (E12) to E13 rodent SN and rostral mesencephalon in accordance with a protocol modified from that of Brewer (Brewer et al., 1993; Brewer, 1995) as follows. Briefly, primary neuronal cultures were harvested and prepared from embryonic day 12–13 Sprague Dawley rat pups. The above areas were isolated from a litter of E12–E13 rats, and the meninges and extraneous tissue were removed. Tissue was incubated in 2 ml of Ca2+/Mg2+-free HBSS (HBSS with 10 mm HEPES, pH 7.3) with gentamicin (50 μg/ml) and 0.25% trypsin for 15 min at 37°C. The cells were centrifuged at 1000 rpm for 5 min, washed twice with HBSS (with Ca2+/Mg2+), and then dissociated in Neurobasal media supplemented with glutamate, gentamicin, and B27 supplement plus antioxidants (Invitrogen) by 10 passages through a 0.9 mm bore pipette tip. Dissociated cells were counted using the trypan blue viability assay and were plated on poly-d-lysine-coated cell culture plastic and incubated in a humidified atmosphere of 5% CO2/95% air at 37°C. In addition, cAMP (1 mm) and ascorbic acid (100 μm) were added for the first 3 d of culture to improve yield and survival of DA neurons. Fetal bovine serum (FBS) (irradiated, defined FBS; HyClone/Thermo Fisher Scientific) was added at 2% for the first 3 d also to improve DA neuron survival, and then diluted out thereafter. Cultures were used for experiments at days in vitro 11–14 unless otherwise noted.
PC12 cell culture.
PC12 and PC12-hDAT cells were cultured as previously described (Daniels and Amara, 1999; Loder and Melikian, 2003), in DMEM (high glucose) with 10% fetal bovine serum (HyClone; defined, gamma-irradiated), nonessential amino acids, and penicillin–streptomycin.
DAT activity assays
3H (radioactive) DA uptake assay.
Changes in DA uptake by the DAT were assessed in primary mesencephalic cultures in response to 120 nm Tat treatment, as previously described (Prochiantz et al., 1979; Dal Toso et al., 1988; Engele et al., 1989; Fiszman et al., 1991; Dalia et al., 1993; Bennett et al., 1998; Salum et al., 2008). Briefly, mesencephalic neurons in 12- or 24-well plates were incubated with 120 nm Tat under normal culture conditions for 24 h; for the acute 15 and 30 min treatments, DA uptake assay was performed during Tat administration. Background (i.e., DA uptake that was not specific to DAT) was determined by incubating both control and Tat conditions with and without the specific DAT antagonist 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine hydrochloride (GBR-12909) (1 μm). Wells were then washed two times for 3 min each time with preheated 37°C HBSS with Ca+ and Mg+, containing 100 μm ascorbic acid (to protect against DA toxicity) and 50 μm pargyline (monoamine oxidase inhibitor that prevents endogenous DA catabolism). Washes were removed and replaced with 50 nm tritiated (“hot”) DA (3H-DA) and 450 nm cold DA in the same wash buffer, and incubated at 37°C for 20 min. Our own (see Fig. 2A) and previous (Dal Toso et al., 1988; Engele et al., 1989; Bennett et al., 1998) studies have demonstrated DAT uptake activity in cell culture to be linear over this time period. Reactions were stopped in ice-cold wash buffer. Cells were then lysed on ice for 30 min with 1% SDS in the same ice-cold wash buffer (250 μl/well for 24-well plate, 500 μl/well for 12-well plate), and then triturated to lift cells. Equal amounts of lysate protein were added to 5 ml of aqueous scintillation fluid and amount of 3H-DA uptake was read by scintillation counter. Readout was given in background corrected counts per minute (CCPM). For each condition, nonspecific DA uptake (+GBR condition) was subtracted from total DA uptake (−GBR condition), to yield specific DA uptake, which was then normalized to control, and data were expressed as normalized mean CCPM ± SEM for each condition. Mean CCPM (i.e., DAT-specific DA uptake) was obtained from N = 4 experiments, and significance was determined by Student's t test at p < 0.05.
Figure 2.

Tat increases DAT Vmax but not Km. A, PC12-hDAT cells were pretreated for 30 min with 120 nm Tat or control, followed immediately by measurement of DAT uptake activity. The 120 nm Tat significantly increased DAT-specific uptake versus control, as assessed by the fluorescence ASP+ substrate assay. No significant differences in vehicle control, versus 120 nm heat-inactivated Tat control, have been found for DAT uptake activity or membrane DAT levels (see Figs. 4 and 5 below, and supplemental Fig. S1, available at www.jneurosci.org as supplemental material), demonstrating specificity of the effects of Tat on DAT activity. For each condition (i.e., ±Tat) at each kinetic time point, nonspecific ASP+ uptake in the presence of the DAT-specific antagonist GBR-12909 was subtracted from total uptake (i.e., ±Tat without GBR-12909), to generate DAT-specific uptake displayed. Fitting fourth-degree polynomial curves to the data points illustrated sigmoid-shaped three-phase ASP+ accumulation versus time, with linear DAT substrate uptake in the range of ∼10–40 min (see Results for discussion). B, Nonlinear regression analysis of this DAT-specific ASP+ uptake data indicated that 30 min of 120 nm Tat treatment increased Vmax (*p < 0.016, two-tailed paired t test) but not Km versus control. For each individual experimental run, data over the linear ASP+ uptake range were plotted and modeled by Michaelis–Menten kinetics, fitting curves to the data with the equation Y = (Vmax*X)/(Km + X). Km and Vmax values were obtained from curves fit to each experimental run then averaged, over six treatment runs. These average Km and Vmax values are displayed ± SEM, with curves from these mean values plotted to the above equation. The 120 nm Tat treatment over 30 min changed Vmax but not Km.
Fluorescent (ASP+) DAT activity assay.
For kinetic analysis of Tat-induced changes in DAT uptake activity, a fluorescent assay was adopted in the DAergic PC12 cell line, stably transfected with the human DA transporter (PC12-hDAT) (kind gift from Dr. Haley Melikian) (Loder and Melikian, 2003). In this assay, the fluorescence substrate 4-(4-(dimethylamino)styryl)-N-methylpyridinium iodide (4-Di-1-ASP) (herein referred to as ASP+) (Invitrogen; D288) was used. ASP+ is a fluorescent dye with high affinity for the DA transporter that has been extensively validated for use as a nonradioactive, fluorescent indicator of DA transporter-specific uptake activity (Schwartz et al., 2003; Mason et al., 2005).
This assay was performed essentially as the radioactive assay above, using ASP+ uptake in the presence of ±GBR-12909 to determine DAT-specific uptake activity. Briefly, PC12-hDAT cells were cultured as above, and then treated in replicate (four to eight replicates/condition) with control and Tat (120 nm). After treatment, medium was removed, and ASP+ was added in HBSS buffer including 8 μm trypan blue as masking agent. Mean fluorescent units were read in a fluoromark plate reader (Bio-Rad), and kinetic analysis was performed by nonlinear regression analysis using GraphPad Prism or Synergy Kaleidagraph software for calculation of Km and Vmax values.
Immunoblots
Primary neuronal cultures or PC12-hDAT cells were treated as described in the figure legends, and immunoblots were prepared and performed as described previously (Norman et al., 2007, 2008), with the following exceptions: DAT antibodies were found to be extremely sensitive to the effects of heating the lysates to 100°C, as this appeared to alter the conformation of critical epitopes sufficiently to prevent DAT antibody binding. Therefore, the lysates were not heated to 100°C before loading the gel. Antibodies were used as described in the figure legends. For relative quantification of the signal intensity between bands, Western blots were digitally photographed using a uniform-intensity illuminator and monochrome digital CCD (Interfocus Imaging). Bands were quantified by summing the total value of all pixels within the band area, using ImageJ, and then normalized to control value for group analysis across multiple runs. Two replicates for each condition were run in separate lanes to ensure consistency of results.
Cellular subfractionation for immunoblots
Plasma membrane and cytosolic components were separated from total cellular protein using a PEG (polyethylene glycol) and dextran T-500-based centrifugal gradient kit, according to the manufacturer's directions, resulting in plasma membrane purity >90% (BioVision Membrane Protein Extraction kit).
Total internal reflection fluorescence live imaging
Total internal reflection fluorescence (TIRF) studies used PC12 cells cultured as above on glass-bottom Petri dishes (MatTek), then transfected with eGFP-hDAT DNA using the Lipofectamine 2000 (Invitrogen) protocol and returned to the incubator for 2 d to maximize expression. Cells were removed from the incubator and placed in Hibernate (BrainBits) media (intended for pH equilibration in ambient CO2 levels) with or without treatment for the imaging procedure. Under these conditions, cells are stable at ambient air CO2 concentrations for several hours. On treatment with control or Tat, cells were immediately taken to the microscope, a random field of eGFP-hDAT-transfected PC12 cells was obtained, and images under TIRF microscopy (TIRFM) were taken at a rate of one image (500 ms exposure) per 30 s for 30 min using a QImaging Retiga Exi cooled CCD camera and MetaMorph (Molecular Devices/MDS Analytical Technologies) software for automated image acquisition. Hardware included 488 laser excitation with Melles Griot tunable argon laser, Olympus TIRF illuminator on an Olympus IX70 microscope with Olympus 60×, 1.45 numerical aperture TIRF objective, Chroma z488/10× clean-up filter and dichroic mirror, with laser illumination custom-synchronized with camera exposures via the I/O port of the camera. Using MetaMorph software, fluorescence intensities were analyzed by placing 100 pixel square regions of interest (ROIs) in random locations throughout each of the transfected cells under study, and averaging the mean signal over all ROIs for each image series, normalized to the starting fluorescence intensity value for each image series.
TIRF imaging of membrane calpain activity was performed in the same fashion, except that cells were preloaded for 30 min at room temperature with the cell-permeable fluorescent calpain substrate bis-(CBZ-l-alanyl-l-alanine amide) rhodamine 110 (BCAA-R110) (10 μm) (AnaSpec) in Ca2+-free Hibernate buffer containing 1 μm calpastatin (loading buffer was Ca2+-free plus calpastatin to prevent premature cleavage of the BCAA-R110 probe before experimental treatment). After BCAA-R110 loading, dye was replaced with Hibernate (with calcium) plus experimental treatment, and TIRF imaged as above.
Data and statistical analysis
The raw data were analyzed from three to five independent experiments and expressed as the mean ± SEM. Where applicable, the percentage of control and percentage of control SEM for each treatment condition were calculated by dividing the raw means and raw SEMs by the control condition raw mean. Student's t test using a two-tailed distribution and unequal variance was used to compare data. A probability of p < 0.05 was considered statistically significant. Replicates for each condition were run in separate lanes to ensure consistency of results.
In vivo studies
Tat-transgenic mice were kindly provided by Dr. Johnny He (Indiana University School of Medicine, Indianapolis, IN) (Kim et al., 2003). Six-week-old Tat-transgenic mice with Tat under the control of a doxycycline-inducible glial fibrillary acidic protein (GFAP) promoter, or wild-type (WT) control mice, were treated intraperitoneally with 80 mg/kg doxycycline in saline, once daily for 7 d. Tat-Tg and WT control animals were then killed under anesthesia in accordance with Institutional Animal Care and Use Committee guidelines, followed by cardiac perfusion with PBS, and then 4% paraformaldehyde (PFA) chased with 0.05 m sodium phosphate; brains were removed and postfixed in 4% PFA overnight, followed by storage at 4°C in PBS. Forty-micrometer coronal sections from striatum were cut on a vibratome, followed by free-floating immunocytochemistry (ICC) procedure with anti-DAT (1:1000; Millipore Bioscience Research Reagents; MAB369) and anti-synaptophysin (1:250; Millipore Bioscience Research Reagents; MAB5258) antibodies. After ICC, sections were mounted, and analyzed for DAT and synaptophysin expression levels in each region by computerized image capture and analysis. A total of ≥50 × 40 μm coronal striatal sections were analyzed for each antibody, over multiple runs (two brains/group). For each section, background was normalized, ROIs were manually drawn around striatum, and mean fluorescent intensity for DAT and synaptophysin labeling was calculated for each ROI. To normalize DAT content to total presynaptic volume for each slice, intensity data were further expressed as mean DAT intensity/mean synaptophysin intensity for each ROI, and these values were then averaged over all sections ± SEM. MetaMorph software was used for image acquisition and analysis.
Results
Tat increases DAT-specific DA uptake in mesencephalic cultures
Tat is excitatory and excitotoxic (Cheng et al., 1998; Haughey et al., 2001; Perry et al., 2005a), and toxic to DAergic populations and imparts changes in DAergic function (Bansal et al., 2000; Aksenov et al., 2001, 2008; Aksenova et al., 2006; Wallace et al., 2006; Silvers et al., 2007). Therefore, we hypothesized that Tat might disrupt DAT activity, leading to permanent neuronal loss and neurological deficit, consequent to unsustainable metabolic demands and/or enhanced auto-oxidative DA toxicity presynaptically, or overstimulation/understimulation of postsynaptic striatal connections. We found that treatment of mesencephalic neurons with 120 nm Tat robustly increased DAT-specific DA uptake versus control (Fig. 1A). This effect was greatest acutely, as DA uptake was markedly elevated fourfold to fivefold after 30 min Tat treatment (p < 0.02). After 24 h Tat treatment, this effect had declined to a more modest twofold increase in DA uptake (p < 0.04). These results suggest that the actions of Tat on DAT activity in vitro begin acutely, persist for at least 24 h during prolonged Tat exposure, and may be in part responsible to the effects of Tat on DAergic systems in the context of HAND.
Figure 1.
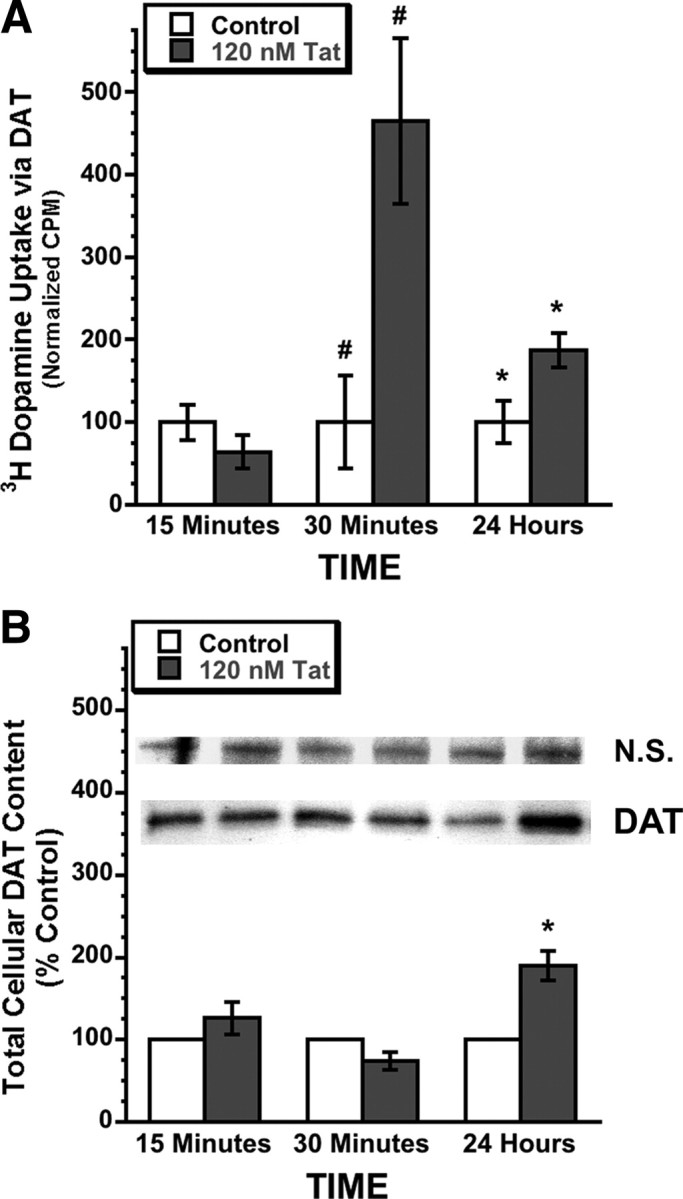
Tat acutely increases DAT-specific DA uptake without equivalent total DAT protein increase in rodent mesencephalic neurons. A, Treatment of rat mesencephalic neurons with 120 nm Tat for 15 min, 30 min, or 24 h, caused significant increases in DAT specific DA uptake versus vehicle control at 30 min (#p < 0.02) and 24 h (*p < 0.04) (two-tailed unpaired t test). No significant differences in vehicle control, versus 120 nm heat-inactivated Tat control, have been found for DAT uptake activity or membrane DAT levels (see Figs. 4 and 5 below, and supplemental Fig. S1, available at www.jneurosci.org as supplemental material), demonstrating specificity of the effects of Tat on DAT activity. For each condition (i.e., ±Tat), non-DAT-specific DA uptake in the presence of the DAT-specific antagonist GBR-12909 was subtracted from total uptake (i.e., ±Tat without GBR-12909) to generate DAT-specific uptake displayed. To facilitate comparisons across groups and to subsequent immunoblot data (B), the mean 3H-DA uptake count per minute (CPM) ± SEM values of each time-matched control group were normalized to “100.” N = 4. B, Despite a large increase in DA uptake at 30 min of 120 nm Tat, total DAT protein levels did not change at this time point. By 24 h 120 nm Tat treatment, the increase in total DAT protein (190 ± 18%; *p < 0.04 vs time-matched control, two-tailed paired t test) was similar to the increase in DA uptake at this same time point (187 ± 21 normalized CPM; *p < 0.04 vs time-matched control, two-tailed unpaired t test) (A, 24 h Tat treatment). Equal total protein amounts were loaded (Bradford assay), and a nonspecific band was used as loading control (Ditzel et al., 2003; Lin et al., 2007). After background correction, integrated band densities were averaged across several blots and expressed in graphical form ± SEM. Images represent sample bands for the bar immediately below each band image. N = 3.
Acute Tat treatment does not change DAT protein levels in mesencephalic cultures
Increased DA uptake via DAT could be accomplished in several ways, including increased production of DAT protein (i.e., leading to increased membrane DAT levels), increased rate of DAT substrate turnover across the membrane (i.e., individual DAT molecules are “running” faster), or changes in DAT trafficking to/from the cell membrane without changes in total intracellular DAT levels. To address these possibilities, we first measured total cellular DAT protein in primary mesencephalic cultures at time points matching the DA uptake studies in Figure 1A. We found no increase in DAT protein at 30 min Tat treatment (Fig. 1B), when DA uptake was most elevated (Fig. 1A), suggesting that the fourfold to fivefold increase in DA uptake 30 min after Tat addition was not attributable to increased translational expression of DAT. Therefore, we speculated that the increased DA uptake at this acute time point resulted from changes in DAT membrane trafficking independent of DAT protein levels. In contrast, after 24 h Tat treatment, total DAT protein levels were increased approximately twofold (p < 0.04) (Fig. 1B), which closely approximated the twofold increase in DAT uptake at this same time point (Fig. 1A), suggesting that, with 24 h continuous exposure to 120 nm Tat, increased DAT protein expression might be at least in part responsible for the increased DA uptake observed at this longer time point. In other words, increased protein translation may eventually “catch up” to acute synaptic DAT demands that, in the short term, are satisfied exclusively by altered DAT kinetics (e.g., changes in DAT trafficking and/or substrate turnover), rather than by a generalized increase in DAT protein expression.
Because both DA uptake and total DAT protein were elevated at 24 h 120 nm Tat treatment (albeit a smaller increase in DA uptake, compared with 30 min), to avoid the potentially confounding variable of changes in DAT protein translation as they affect intracellular DAT pools, and because we wanted to investigate changes in DAT trafficking/kinetics commensurate with the timescale of synaptic transmission and adaptability before changes in protein expression, the remainder of this report focuses on this acute 30 min time point. We hypothesized that changes in DAT membrane trafficking, rather than increased DAT expression, were likely to satisfy these elevated near-term demands for DAT at the synapse.
Acute Tat treatment increases DAT Vmax but not Km
To test this hypothesis further, next we investigated whether Tat was altering kinetics of DA uptake. For these and studies that follow, we used PC12 cells transiently or stably transfected with the human DA transporter (PC12-hDAT; kind gift from Dr. Haley Melikian), a dopaminergic model system that provides ease of genetic manipulation, reproducibly high yields of hDAT+ cells, and that has been well characterized for investigations of DAT membrane trafficking (Melikian and Buckley, 1999; Loder and Melikian, 2003). Moreover, this model system obviates technical limitations associated with obtaining yields of primary rodent DAergic neurons sufficient for kinetic analyses. To investigate kinetics of DA uptake in this model system, we used a well characterized nonradioactive DAT uptake activity assay. This assay relies on uptake of the fluorescent probe 4-Di-1-ASP (Invitrogen; D-288) (ASP+), which has a high affinity for DAT (Schwartz et al., 2003; Mason et al., 2005). In our own experience, and in the experience of laboratories that have developed this as a nonisotopic alternative to measuring DA uptake (Schwartz et al., 2003; Mason et al., 2005), this assay faithfully replicates kinetics of DAT-mediated uptake, and a slightly modified version of this assay has since been made commercially available in “kit” form (part numbers R8173, R8174; MDS Analytical Technologies). In our experiments, nonspecific uptake was determined in the presence of the specific DAT antagonist GBR-12909, and this value was subtracted from uptake in the minus GBR-12909 condition, to yield DAT specific uptake. As expected, GBR-12909 exhibited dose-dependent effects against ASP+ uptake in this model system, and untransfected (hDAT-negative) PC12 cells showed significantly reduced ASP+ uptake versus their PC12-hDAT counterparts (data not shown).
Because we were particularly interested in investigating changes in DAT activity over acute time periods, before significant changes in protein translation occur (Fig. 1B), we pretreated PC12-hDAT cells for 30 min at 37°C with either control or 120 nm Tat, followed immediately by the ASP+ assay (see Materials and Methods). From these studies, we found that 30 min of 120 nm Tat treatment significantly increased DAT uptake of ASP+, when compared with control. Figure 2A illustrates this effect, during a ∼50 min uptake experiment at a single ASP+ concentration (12 μm). DAT uptake activity is best modeled as analogous to enzyme activity (Schenk et al., 2005). Our data validated this assumption, exhibiting sigmoid shaped three-phase ASP+ accumulation versus time, best fit by fourth-degree polynomial curves demonstrating linear DAT substrate uptake in the range of ∼10–40 min (Fig. 2A). In contrast to more reductionist model systems (e.g., synaptosomes), which may have more rapidly saturable DA uptake because of higher frequencies of DAT/substrate interactions, linear dopamine uptake over this time range has been commonly described in DAergic whole-cell systems (Dal Toso et al., 1988; Engele et al., 1989; Bennett et al., 1998). Additional kinetic analysis of this effect showed that Tat increased the maximal velocity (Vmax) of DAT ASP+ uptake versus control (5.7 vs 4.3 RFU/s respectively; p < 0.016, two-tailed paired t test), without changing the Michaelis constant (Km, or the substrate concentration that achieves half-maximal rate) (Fig. 2B).
Acute Tat treatment increases plasma membrane DAT but not total DAT levels
Given that DAT uptake activity is tightly regulated by DAT trafficking between intracellular pools and the plasma membrane (Melikian and Buckley, 1999; Buckley et al., 2000; Loder and Melikian, 2003; Melikian, 2004), we reasoned that the most likely explanation for increased Vmax without any change in Km was attributable to increased concentration of DAT at the plasma membrane, which would in turn increase maximal velocity of DAT-mediated uptake. To test this hypothesis, we again pretreated PC12-hDAT cells for 30 min at 37°C with either vehicle control or 120 nm Tat, and then performed fractionated Western blot analysis for DAT in total protein, cytoplasmic, and plasma membrane cellular subfractions. This analysis showed that, after 30 min Tat treatment, there was a significant increase in plasma membrane DAT (∼177% vs control; p < 0.05, two-tailed paired t test), as well as a smaller but also significant decrease in cytoplasmic DAT (∼86% vs control; p < 0.02), findings consistent with a redistribution of DAT to the PM. Total protein, however, was not changed (Fig. 3A). These effects were specific to functional Tat activity, as 120 nm heat-inactivated Tat, in contrast, did not demonstrate any ability to alter plasma membrane or cytoplasmic DAT levels when compared with vehicle control (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Figure 3.

Tat increases membrane DAT and decreases cytoplasmic DAT, without changing total DAT. A, The 120 nm Tat treatment of PC12-hDAT cells for 30 min significantly increased PM DAT (177%; *p < 0.05 vs control, two-tailed paired t test), but not total DAT protein, as assessed by Western blot. Cytoplasmic (Cyto) DAT is also slightly but significantly down (86%; *p < 0.02 vs control, two-tailed paired t test), as would be expected with increased trafficking of DAT to the membrane. After background correction, integrated band densities were averaged across several blots and expressed in graphical form ± SEM. Images represent sample bands for the bar immediately below each band image. N = 4. B, PC12-hDAT cells were treated as in A (120 nm Tat for 30 min), immunoprobed with DAT antibody (Alexa Fluor 488 secondary antibody), mounted under water, and then subject to TIRF microscopy to isolate membrane DAT. Qualitative visual representations of anti-DAT staining in control versus Tat-treated PC12-hDATcells are shown above; quantitative real-time TIRFM of membrane DAT in PC12 cells is shown in Figure 4 below and in supplemental Movies 1 and 2 (available at www.jneurosci.org as supplemental material).
Several additional experiments provided additional visual confirmation that membrane DAT was increased in Tat-treated cultures versus controls. First, we performed the same treatments in the PC12-hDAT cells, and then immediately after treatment, fixed the cultures with 4% paraformaldehyde and performed immunocytochemistry for hDAT with Alexa Fluor 488 as the secondary label (the exogenous human DAT expressed in these cells is DAT only, with no fluorescent fusion reporter protein, thus necessitating immunocytochemical detection), and viewed them under TIRFM. By reflecting a 488 nm laser-based excitation source at incident angle against the underside of the glass coverslip on which the cells are cultured, TIRFM uses the resulting evanescent wave to effectively optically isolate fluorescent events within ∼100 nm (Z-direction) of the basilar plasma membrane, thus allowing us to visualize only those DAT that are at or near the cytoplasmic border of the plasma membrane. Congruent with the immunoblot results in Figure 3A, membrane expression of DAT was significantly increased in the Tat-treated PC12-hDAT cultures, as evidenced with TIRFM visualization (Fig. 3B).
Since our stably transfected hDAT-PC12 cells did not contain a fluorescent reporter protein, to observe these changes live, we transiently transfected the dopamine-producing PC12 cell line with plasmid DNA encoding an enhanced green fluorescent protein-human DAT (eGFP-hDAT) fusion protein (kind gift from Dr. Susan Amara) and observed membrane activity of the eGFP-hDAT signal in real-time using TIRFM. In this model system, in response to 30 min treatment with 120 nm Tat, we also observed DAT levels increasing at the membrane in real time [Fig. 4, compare Tat treatment (red trace) with heat-inactivated Tat control (black trace)]. These graphical results are further highlighted by representative TIRFM image series of these eGFP-hDAT transfected PC12 cells, showing a rise in membrane eGFP-hDAT signal during 30 min treatment with 120 nm Tat (supplemental Movie 1, available at www.jneurosci.org as supplemental material) versus a slight decline in membrane eGFP-hDAT in 30 min control treated cells (supplemental Movie 2, available at www.jneurosci.org as supplemental material), as summarized in Figure 4. Images were captured at 2 frames/min for 30 min (∼61 frames total), and then played back at ∼7 fps. A frame-by-frame graph is overlaid on each movie, representing the normalized averaged eGFP-hDAT intensity value over the cell area for each frame.
Figure 4.
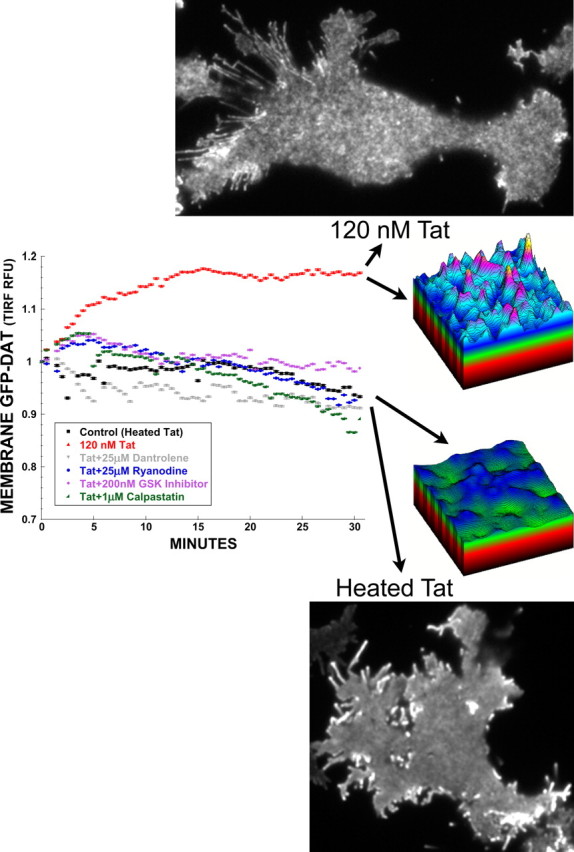
Tat activates RyR, calpain, and GSK-3β to increase membrane DAT. PC12 cells transfected with eGFP-hDAT, and then acutely treated with 120 nm Tat over 30 min, showed increased membrane DAT signal under TIRF microscopy. This increased translocation of DAT to the plasma membrane, as evidenced by increased average membrane GFP-DAT signal (graph, compare black 120 nm heat-inactivated Tat control vs red 120 nm Tat), and by enhanced punctate topography reflecting elevated surface DAT (compare Tat vs heated Tat control images, and topographic representations), could be ameliorated by the RyR antagonists dantrolene and ryanodine (25 μm), the GSK-3β inhibitor N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea (200 nm), and the endogenous calpain inhibitor calpastatin (1 μm) (27 aa cell-permeable peptide from calpastatin exon 1B), thus implicating ryanodine receptors, GSK-3β activation, and calpain protease activity, respectively, in the increased membrane DAT response. Three-dimensional topographic representations are taken from a representative ROI of the images shown. Graphical traces were obtained by averaging membrane eGFP-hDAT intensity under TIRFM, from ≥30 cells collected over multiple 30 min experimental treatment runs, and then normalized to 1 ± SEM. Error bars are present but small because relative fluorescent intensities were measured on a pixel-by-pixel basis over the cell areas.
Together, these results support a mechanism whereby Tat acutely increases DAT expression at the plasma membrane by affecting trafficking mechanisms rather than increasing intracellular DAT protein levels.
DAT molecules are impaired despite increased plasma membrane levels of DAT and increased Vmax
DAT activity conforms to Michaelis–Menten kinetics and can be modeled as follows (Schenk et al., 2005):
 |
where DA represents dopamine; T, transporter; DA-T, DA bound to T; κ1, binding constant; κ2, dissociation constant; and κ3, translocation constant. Fitting our data using nonlinear regression analysis of specific DAT uptake activity in PC12-hDAT cells, we found Tat treatment increased Vmax by a factor of ∼1.33 (5.693 Tat vs 4.266 control). However, DAT concentration at the membrane in these same cells increased by a factor of 1.77 in the Tat-treated condition (Fig. 3A). Since Vmax = κ3[DAT] [i.e., the product of the translocation constant (i.e., how quickly DAT transports each DA molecule across the membrane) and the concentration of DAT at the membrane], by substituting our relative values into this equation (units are omitted for simplicity) as follows:
 |
Thus, we can infer that in the Tat-treated cultures, although Vmax was increased, the translocation constant κ3 instead declined by ∼25%. In other words, Tat slows down the rate at which DAT transports each molecule of substrate across the membrane, but Vmax is slightly increased overall because of (a compensatory?) increase in DAT concentration at the membrane. This observation may help explain differences between our observed Tat effects on DAT and those reported previously (Wallace et al., 2006; Zhu et al., 2009) (see Discussion).
Tat-induced rise in plasma membrane DAT is prevented by ryanodine receptor antagonism, and calpain or GSK-3β inhibition
Previously, we showed that ryanodine receptors mediate several intracellular effects of Tat by regulating calcium release from mitochondria and endoplasmic reticulum (Norman et al., 2007, 2008). Calpains, in turn, are ubiquitous Ca2+-activated proteases involved in numerous integral cell functions including proteolytic regulation of cytoskeletal dynamics, and which are also increasingly being implicated as key mediators of neurodegenerative disease (for review, see Liu et al., 2008). GSK-3β mediates the neurotoxic effects of Tat (Maggirwar et al., 1999; Sui et al., 2006a) and platelet-activating factor (Tong et al., 2001) in models of HAND, and furthermore, we have shown that GSK-3β is recruited to membrane lipid rafts, and GSK-3β inhibition blocks specific phosphorylation of raft-associated Tau (Sui et al., 2006b). Membrane DAT trafficking may also be regulated at lipid rafts (Adkins et al., 2007). Finally, calpain cleaves GSK-3β to augment its kinase activity (Goñi-Oliver et al., 2007). Therefore, we wanted to determine whether inhibition of (1) ryanodine receptor activity with the RyR antagonists ryanodine or dantrolene, or (2) calpain activity by a cell-permeable calpastatin peptide (item 208902; Calbiochem/EMD Biosciences), or (3) GSK-3β kinase activity with the specific GSK-3β inhibitor N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea (GSK-3β inhibitor VIII; Calbiochem/EMD Biosciences), prevented the Tat-induced rise in plasma membrane DAT levels.
Again, tracking the eGFP-hDAT signal at the plasma membrane with real-time TIRFM, we found that the increased membrane DAT levels induced by 30 min treatment with 120 nm Tat could be prevented by coapplication of (1) ryanodine or dantrolene (25 μm); (2) a 27 aa cell-permeable peptide from exon 1B of the endogenous calpain inhibitor calpastatin (1 μm) (Maki et al., 1989; Eto et al., 1995) (Calbiochem/EMD; 208902); or (3) the GSK-3β inhibitor N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea (200 nm) (Calbiochem/EMD; GSK-3β inhibitor VIII) (Fig. 4). These results indicate that Tat-mediated activation of RyRs, with accompanying changes in calpain and GSK-3β activity, alter DAT trafficking kinetics at the plasma membrane.
Tat activates RyRs to increase calpain protease activity at the plasma membrane
Having shown RyR receptor antagonism, and calpain inhibition, prevent Tat-induced increases in membrane DAT (Fig. 4), we hypothesized a mechanism whereby Tat activates RyRs to increase intracellular calcium (Norman et al., 2007, 2008), thus activating calpain proteases. We additionally hypothesized that GSK-3β activity is linked to calpain signaling to further regulate membrane DAT trafficking. Therefore, using the cell-permeable fluorescent calpain substrate BCAA-R110 (AnaSpec), which has been validated as an indicator of calpain protease activity in live cells (Gitler and Spira, 1998), we measured calpain activity at the plasma membrane, under the same treatment conditions as in Figure 4 above. This substrate is nonfluorescent until cleaved by proteases at the amide bond, thus releasing the rhodamine 110 fluorophore, rendering it available for excitation. Imaging this dye by TIRFM, under the same treatment conditions as in Figure 4, we show that 30 min treatment of PC12 cells with 120 nm Tat increases calpain protease activity at the plasma membrane versus control cells (Fig. 5). Furthermore, just as 25 μm dantrolene, 25 μm ryanodine, 1 μm cell-permeable calpastatin peptide, and 200 nm N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea restore plasma membrane DAT to control levels (Fig. 4), these treatments also restore membrane calpain activity to control levels (Fig. 5). Together, these results suggest membrane-localized control of DAT trafficking by RyR- and GSK-3β-mediated activation of calpain proteases located at or near the plasma membrane.
Figure 5.

Tat increases calpain protease activity at the plasma membrane via RyR and GSK-3β pathways. Tat also increased membrane-proximal calpain protease activity in PC12 cells, as shown by TIRFM imaging of the cell-permeable fluorescent calpain substrate BCAA-R110. This substrate is nonfluorescent until cleaved by proteases at the amide bond, thus releasing the rhodamine 110 fluorophore and rendering it available for excitation. Imaging this dye by TIRFM, under the same treatment conditions as in Figure 4, we show that 30 min treatment of PC12 cells again with 120 nm Tat increases calpain protease activity at the plasma membrane versus control cells treated with 120 nm heat-inactivated Tat. Just as 25 μm dantrolene, 25 μm ryanodine, 1 μm calpastatin peptide, and 200 nm N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea prevent 120 nm Tat-induced increases in plasma membrane DAT (Fig. 4), these treatments also prevent Tat-induced increases in membrane calpain activity (Fig. 5). Together, these results suggest membrane-localized control of DAT trafficking by RyR- and GSK-3β-mediated activation of membrane-proximal calpain proteases. After 30 min treatment with 120 nm Tat, mean fluorescent BCAA-R110 signal intensity at the membrane was quantified from the cell membrane surface area of ≥30 cells collated over multiple experimental treatment runs and imaged under TIRFM, and then averaged and expressed as raw intensity (arbitrary fluorescent units) ± SEM.
DAT is elevated at striatal synapses in HAND patients, and in Tat-expressing transgenic mice
Above, we presented data on Tat-induced changes in DAT trafficking and function during acute time frames in situ, because we wanted to investigate changes in DAT kinetics commensurate with the timescale of synaptic transmission and adaptability before changes in protein expression, to better understand mechanisms for DAergic deficits before frank loss of synapses or neurons. However, it is important to also correlate these findings with in vivo evidence of DAergic dysfunction in the context of HAND.
Our in situ studies suggest that Tat increases membrane DAT levels and functional uptake via alterations in DAT trafficking behavior, without increased production of DAT. Perhaps the best translational in vivo correlate for these findings comes from observations made in postmortem brain tissue from HAND patients. In these studies, Gelman et al. (2006) found significantly increased DAT expression in the striatum in postmortem brain tissue with neuropathological confirmation of HIVE [Gelman et al. (2006), their Figs. 1, 3], which the authors concluded did not result from generalized increases in synaptic protein expression or synaptic density. Other studies found no significant change (with a slight downward trend) in DAT levels in substantia nigra of HAND patients versus controls [Silvers et al. (2006), their Fig. 2]. Together, these studies suggest that, in HAND, increased striatal DAT expression may result from increased translocation of DAT from mesencephalic neuronal cell bodies, to distal striatal presynaptic sites, without significant changes in total DAT protein expression—thus representing a potential clinically relevant in vivo correlate for our in situ findings of increased membrane DAT expression, caused by altered DAT trafficking rather than increased protein expression.
To further support these clinical findings, and our own in situ data, we next examined synaptic DAT levels in the striatum of Tat-expressing transgenic mice. These mice express a doxycycline-inducible Tat gene driven by the GFAP promoter, for regulatable Tat expression by astrocytes (Kim et al., 2003). Tat is released from HIV-infected microglia, macrophages, and astrocytes in the CNS, and release of soluble neurotoxins from activated immune cells likely contributes significantly to the neuropathology of HAND (Chang et al., 1997; Nath and Geiger, 1998; Nath, 2002; Rumbaugh and Nath, 2006). These doxycycline-induced Tat-transgenic mice, in turn, develop behavioral changes and neurological abnormalities including tremor, ataxia, and cognitive and motor deficits, and exhibit several neuropathologies including astrocytosis, increased infiltration of activated monocytes, neuronal apoptosis, and synaptic damage (Kim et al., 2003)—all of which closely mirror HAND neuropathological features. Thus, these mice model at least some of the features of HAND, and we now further extend the translational relevance of this model system, with novel studies of the effects of Tat on DAergic synapses in these mice.
In these studies, we found significantly higher DAT levels in the striatum of doxycycline-induced Tat-expressing mice (∼35% increase relative to control), versus their non-Tat-expressing control counterparts (Fig. 6). This occurred despite a moderate decline in total presynaptic density in the Tat Tg mice (∼87% vs WT control; p < 0.0004), as assessed by synaptophysin staining (a reliable method for quantifying total synaptic density) (Masliah et al., 1989; Calhoun et al., 1996). Together these observations suggest that synaptic DAT levels are significantly elevated per synapse in these Tat Tg mice (Fig. 6), findings that are correlative with the findings in postmortem HAND striatum as discussed above (Gelman et al., 2006), and that we posit reflect increased anterograde DAT trafficking to striatal synapses in vivo independent of gross changes in DAT protein synthesis. Future real-time in vivo and ex vivo studies will seek to further confirm this hypothesis.
Figure 6.

Striatal DAT expression is increased in Tat transgenic mice. Tat-expressing transgenic mice exhibited significantly increased striatal DAT content versus WT control mice. To normalize DAT content to synaptic volume, relative DAT intensities were normalized to synaptophysin for each ROI, and presented ± SEM as described in Materials and Methods (*p < 0.0014 Tat Tg vs WT, two-tailed paired t test).
In total, these observations that Tat increases synaptic DAT in vitro and in vivo, with upregulation of high-affinity transporter capacity independent of nerve terminal density, suggest that exposure of DAergic nerve terminals to Tat may alter DAergic tone by (1) increasing presynaptic DA turnover, (2) creating a postsynaptic undersupply of DA, and/or (3) increasing oxidative stress in the nerve terminal.
Discussion
cART controls active viral replication, but once proviral DNA forms, cART does not prevent production of early viral proteins Tat, Nef, and Rev in infected cells (Rumbaugh et al., 2008; Li et al., 2009). These proteins may be excreted or, like Tat, actively secreted from infected cells, including glia (Chang et al., 1997; Nath, 2002). Indeed, soluble Tat levels in HIV+ patient CSF and sera have been measured up to 40 ng/ml (Westendorp et al., 1995; Xiao et al., 2000). Tat is also detectable in AIDS brains with progressive multifocal leukoencephalopathy, including in uninfected cells, suggesting that Tat can propagate effects on uninfected “bystander” brain cells (Del Valle et al., 2000), which may initiate the concatenative effects of Tat on glial cell activation and neurotoxicity (Rumbaugh and Nath, 2006). Moreover, a recent study showed that up to 19% of astrocytes in HAND patient brains are HIV-infected, with frequency of astrocyte infection correlating with neuropathological severity and proximity to perivascular macrophages (Churchill et al., 2009), suggesting that the number of HIV+ cells in the brain is far more than originally anticipated, with direct consequences for HAND progression and severity. Together, these factors likely contribute to the ongoing ability of viral proteins to cause HAND despite cART, and underpin our rationale for investigating the synapse-modifying abilities of Tat in the brain.
We demonstrate here that Tat alters DAergic function by previously unreported mechanisms. In rodent mesencephalic neurons and hDAT-transfected PC12 cells treated with 120 nm Tat, DAT-specific DA uptake was significantly elevated within 30 min, with increased membrane DAT levels caused by redistribution of intracellular DAT to the plasma membrane, and consequent increases in Vmax (Figs. 1–4). However, there was no increase in DAT protein synthesis until 24 h Tat exposure (Fig. 1). Likewise, DAT was elevated at striatal synaptic terminals of doxycycline-induced Tat Tg mice versus controls (Fig. 6), which correlates with similar findings in postmortem tissue from HAND patients (Gelman et al., 2006).
Providing key translational congruence with our results, Gelman et al. (2006) found significantly increased striatal DAT expression (i.e., on the presynaptic afferent DA inputs from the SN), in postmortem brain tissue with neuropathological confirmation of HIVE. TH (tyrosine hydroxylase) expression, in contrast, was decreased, suggesting downregulated DA production compensatory to increased DA uptake. Others found no differences in SN (i.e., cell body) DAT in HAND versus controls (Silvers et al., 2006). Another study found decreased DA content, but stable levels of the DA metabolite homovanillic acid (HVA), in postmortem HAND brains versus uninfected controls (Kumar et al., 2009). Stable HVA despite reduced DA may reflect the balance of downregulated DA production, compensatory to increased DA turnover, which leaves net DA breakdown (and thus HVA) unchanged. We hypothesize these postmortem findings, congruent with our findings herein, suggest total cellular DAT content is not greatly changed in the DAergic SN neurons of HAND versus control (Silvers et al., 2006). We further speculate that synaptic DAT (i.e., translocation of DAT from the SN cell body to the striatal synaptic membrane) is increased in HAND. Indeed, much of total cellular DAT is normally located intracellularly, with regulation of functional DAT uptake activity accomplished mostly by altering DAT membrane trafficking/recycling, rather than gross changes in protein expression (Figs. 2–4) (Melikian and Buckley, 1999). Although no model system by itself can completely capture the continuum of neurodegenerative disease or DAergic dysfunction encompassed by HAND, our in situ and in vivo data herein mesh well with existing postmortem data.
The sum of these data, together with previous work demonstrating that the HIV mediators Tat and PAF increase neuronal metabolic and synaptic activity (Haughey et al., 2001; Bellizzi et al., 2005; Perry et al., 2005a; Lu et al., 2007), suggests DAergic tone may be increased in HAND. Bolstering this concept, PAF also increases membrane DAT by mechanisms similar to those shown here for Tat (our unpublished data). We further posit that increased membrane DAT levels together with a slower DA translocation constant (κ3) (see Results) may model presynaptic nerve terminals under bioenergetic stress, further evidenced by other Tat-induced impairments of synapses, mitochondria, and endoplasmic reticulum (ER) (Bellizzi et al., 2005; Perry et al., 2005a; Norman et al., 2007, 2008). Likewise, others found that 48 h of 50 nm Tat1-72 reduced [3H]WIN 35428 [methyl(1R,2S,3S,5S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate] ligand binding to DAT in rat midbrain neurons, yet without changing total DAT protein despite significant cell death (Aksenova et al., 2006). These findings remain consistent with a mechanism whereby Tat interferes with DAT function (as evidenced by impaired ligand binding), but relative DAT protein levels (per cell) are stable or even increased despite synapse loss or neuronal death. Also similar to our results, 30 min treatment of rat striatal synaptosomes with 60 nm or 1 μm Tat did not change Km or total synaptosomal DAT levels (Wallace et al., 2006; Zhu et al., 2009). Yet contrasting with our results, Tat decreased DAT Vmax in this synaptosome model. However, synaptosomes contain only ∼7% of the total DAT content of DA neurons (Johnson et al., 2005) and lack the more distal (extrasynaptic) molecular machinery that also influences DAT membrane trafficking. Therefore, the DAT translocation constant (κ3) impacts Vmax more heavily in synaptosomes than in whole cells, which may explain how Tat lowers Vmax in synaptosomes (Wallace et al., 2006; Zhu et al., 2009) but increases Vmax in intact cells (Fig. 2). Indeed, increased membrane DAT may be a compensatory response to decreased transporting efficiency of individual DATs.
Increased synaptic DAT and DA turnover/reuptake (Gelman et al., 2006; this study) seems paradoxical given that parkinsonian-like symptoms in HAND (for review, see Berger and Arendt, 2000; Koutsilieri et al., 2002; Tse et al., 2004), which can manifest despite cART (Mirsattari et al., 1998; Shimohata et al., 2006; Tisch and Brew, 2009), have previously been thought to result from decreased DAergic tone (i.e., loss of DAT-containing DAergic synapses). However, chronically increased DAT motility and DA uptake or DA turnover, as our data suggest, may progressively damage DAergic nerve terminals because of unsustainable metabolic demands and enhanced auto-oxidative DA toxicity from excessive intracellular DA accumulation. This in turn may ultimately lead to degeneration of DAergic synapses and/or cell bodies (Reyes et al., 1991; Everall et al., 1995; Gelbard et al., 1995; Itoh et al., 2000). However, more recent analyses of postmortem HIV+ brains (cART status was not reported) found significant alterations in DAergic markers without evidence of synapse or cell body loss (Gelman et al., 2006; Silvers et al., 2006). Functional alterations at DAergic synapses, for example increased DA reuptake, might still cause parkinsonian symptoms by depriving postsynaptic striatal connections of excitatory stimuli. Additional investigations are required to clarify the mechanisms by which DAergic symptoms develop in HAND, particularly in the current cART era, in which DAergic neuronal and synaptic dropout may be less common.
Together, our current and previous (Norman et al., 2007, 2008) data suggest Tat activates RyRs to elevate intracellular calcium, thus activating calpain proteases, which in turn regulate membrane DAT levels (see data herein) (Fig. 7, diagram). Calpain proteases are known to mediate postsynaptic physiology (Liu et al., 2008), but this is the first report implicating membrane-localized calpain protease activity in regulating membrane DAT (Figs. 4, 5). [Both the fluorescent probe for calpain activity and the calpain-blocking calpastatin peptide are cell permeable (Eto et al., 1995; Gitler and Spira, 1998) (results herein), thus allowing us to assess intracellular calpain activity.] Details of this mechanism require additional investigation, but calpains critically regulate both cytoskeletal elements and α-synuclein (Dufty et al., 2007), either of which can regulate trafficking of DAT (Lee et al., 2001; Wersinger and Sidhu, 2003) or other transporters (Jeannotte and Sidhu, 2008).
Figure 7.

Diagram representation of mechanisms leading to Tat-increased membrane DAT. Shown are demonstrated and hypothesized mechanisms leading to increased membrane DAT in our model systems. The red arrows and/or text indicate mechanisms and pathways we have demonstrated herein and/or previously (Norman et al., 2007, 2008). The black arrows and/or text are known relationships or pathways from previous work or the literature. The hashed lines represent pathways or mechanisms hypothesized based on our results herein and presently under investigation. GSK-3β may also act upstream of (rather than in parallel with) calpain activation and/or upstream of UPR activation (data not shown). “Fod” represents Fodrin, an actin-to-membrane-anchoring calpain substrate, that we speculate may be candidate intermediary for calpain-regulated control of membrane DAT levels. The “arrowed” electron microscopy images show RyR labeling on neuronal ER and mitochondria, as previously demonstrated (Norman et al., 2007, 2008). See Discussion for additional details.
GSK-3β inhibition prevented Tat-induced increases in membrane DAT and membrane calpain activity, highlighting mechanisms by which GSK-3β inhibitors may confer neuroprotective benefits for DAergic symptoms in HAND (Dewhurst et al., 2007; Ances et al., 2008). This observed blockade effect was unlikely to be attributable to nonspecific effects of AR-A014418 on calpain cdk2/cdk5 pathways, as the IC50 of AR-A014418 on these pathways is >100 μm (Bhat et al., 2003), whereas our studies used 200 nm. AR-A014418 also prevents DAergic cell death and loss of striatal DA in an in vivo rodent model of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) toxicity (Wang et al., 2007), further emphasizing potential overlaps in DAergic dysfunctions characterizing HAND and PD. How GSK-3β interacts with calpain signaling to achieve these effects remains unclear; however, GSK-3β has been implicated as an upstream trigger of ER and unfolded protein response (UPR) stress conditions (Huang et al., 2009), which may in turn activate calpain proteases. GSK-3β and calpain also share many ubiquitous substrates, including tau and β-catenin (Liu et al., 2008; Vosler et al., 2008). We hypothesize that, when GSK-3β activity is inhibited, this increases substrate availability to calpain, thus competitively diminishing the effects of calpain on DAT kinetic parameters (Fig. 7). Future studies will seek to further clarify these mechanisms by which RyR, calpain, and GSK-3β interact with DAT to promote synaptic dysfunction in HAND, potentially yielding additional therapeutic options for treatment of this and related neurological diseases.
Footnotes
This work was supported by National Institutes of Health Grants R21MH084718 and R21DA030256 (S.W.P.), R01NS054578 and R01NS066801 (S.B.M.), and R01MH056838 and PO1MH64570 (H.A.G.).
H.A.G. and S.B.M. hold a patent filing related to treatment of HAND using glycogen synthase kinase inhibitors.
References
- Adkins EM, Samuvel DJ, Fog JU, Eriksen J, Jayanthi LD, Vaegter CB, Ramamoorthy S, Gether U. Membrane mobility and microdomain association of the dopamine transporter studied with fluorescence correlation spectroscopy and fluorescence recovery after photobleaching. Biochemistry. 2007;46:10484–10497. doi: 10.1021/bi700429z. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Hasselrot U, Bansal AK, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM. Oxidative damage induced by the injection of HIV-1 Tat protein in the rat striatum. Neurosci Lett. 2001;305:5–8. doi: 10.1016/s0304-3940(01)01786-4. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Silvers JM, Mactutus CF, Booze RM. Different effects of selective dopamine uptake inhibitors, GBR 12909 and WIN 35428, on HIV-1 Tat toxicity in rat fetal midbrain neurons. Neurotoxicology. 2008;29:971–977. doi: 10.1016/j.neuro.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenova MV, Silvers JM, Aksenov MY, Nath A, Ray PD, Mactutus CF, Booze RM. HIV-1 Tat neurotoxicity in primary cultures of rat midbrain fetal neurons: changes in dopamine transporter binding and immunoreactivity. Neurosci Lett. 2006;395:235–239. doi: 10.1016/j.neulet.2005.10.095. [DOI] [PubMed] [Google Scholar]
- Ances BM, Letendre SL, Alexander T, Ellis RJ. Role of psychiatric medications as adjunct therapy in the treatment of HIV associated neurocognitive disorders. Int Rev Psychiatry. 2008;20:89–93. doi: 10.1080/09540260701877670. [DOI] [PubMed] [Google Scholar]
- Aprea S, Del Valle L, Mameli G, Sawaya BE, Khalili K, Peruzzi F. Tubulin-mediated binding of human immunodeficiency virus-1 Tat to the cytoskeleton causes proteasomal-dependent degradation of microtubule-associated protein 2 and neuronal damage. J Neurosci. 2006;26:4054–4062. doi: 10.1523/JNEUROSCI.0603-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000;879:42–49. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- Bellizzi MJ, Lu SM, Masliah E, Gelbard HA. Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J Clin Invest. 2005;115:3185–3192. doi: 10.1172/JCI25444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi MJ, Lu SM, Gelbard HA. Protecting the synapse: evidence for a rational strategy to treat HIV-1 associated neurologic disease. J Neuroimmune Pharmacol. 2006;1:20–31. doi: 10.1007/s11481-005-9006-y. [DOI] [PubMed] [Google Scholar]
- Bennett BA, Hollingsworth CK, Martin RS, Childers SR, Ehrenkaufer RE, Porrino LJ, Davies HM. Prolonged dopamine and serotonin transporter inhibition after exposure to tropanes. Neuropharmacology. 1998;37:123–130. doi: 10.1016/s0028-3908(97)00194-9. [DOI] [PubMed] [Google Scholar]
- Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol. 2000;14:214–221. doi: 10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, Nilsson Y, Radesäter AC, Jerning E, Markgren PO, Borgegård T, Nylöf M, Giménez-Cassina A, Hernández F, Lucas JJ, Díaz-Nido J, Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Buckley KM, Melikian HE, Provoda CJ, Waring MT. Regulation of neuronal function by protein trafficking: a role for the endosomal pathway. J Physiol. 2000;525:11–19. doi: 10.1111/j.1469-7793.2000.t01-2-00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR. Comparative evaluation of synaptophysin-based methods for quantification of synapses. J Neurocytol. 1996;25:821–828. doi: 10.1007/BF02284844. [DOI] [PubMed] [Google Scholar]
- Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11:1421–1431. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, Witt MD, Ames N, Walot I, Jovicich J, DeSilva M, Trivedi N, Speck O, Miller EN. Persistent brain abnormalities in antiretroviral-naive HIV patients 3 months after HAART. Antivir Ther. 2003;8:17–26. [PubMed] [Google Scholar]
- Cheng J, Nath A, Knudsen B, Hochman S, Geiger JD, Ma M, Magnuson DS. Neuronal excitatory properties of human immunodeficiency virus type 1 Tat protein. Neuroscience. 1998;82:97–106. doi: 10.1016/s0306-4522(97)00174-7. [DOI] [PubMed] [Google Scholar]
- Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, Gorry PR. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66:253–258. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- Dalia A, Neff NH, Hadjiconstantinou M. GM1 ganglioside improves dopaminergic markers of rat mesencephalic cultures treated with MPP+ J Neurosci. 1993;13:3104–3111. doi: 10.1523/JNEUROSCI.13-07-03104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Toso R, Giorgi O, Soranzo C, Kirschner G, Ferrari G, Favaron M, Benvegnù D, Presti D, Vicini S, Toffano G. Development and survival of neurons in dissociated fetal mesencephalic serum-free cell cultures: I. Effects of cell density and of an adult mammalian striatal-derived neuronotrophic factor (SDNF) J Neurosci. 1988;8:733–745. doi: 10.1523/JNEUROSCI.08-03-00733.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GM, Amara SG. Regulated trafficking of the human dopamine transporter. Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. J Biol Chem. 1999;274:35794–35801. doi: 10.1074/jbc.274.50.35794. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:221–228. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2:93–96. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, Deas E, Meier P. Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat Cell Biol. 2003;5:467–473. doi: 10.1038/ncb984. [DOI] [PubMed] [Google Scholar]
- Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engele J, Pilgrim C, Reisert I. Sexual differentiation of mesencephalic neurons in vitro: effects of sex and gonadal hormones. Int J Dev Neurosci. 1989;7:603–611. doi: 10.1016/0736-5748(89)90019-1. [DOI] [PubMed] [Google Scholar]
- Eto A, Akita Y, Saido TC, Suzuki K, Kawashima S. The role of the calpain-calpastatin system in thyrotropin-releasing hormone-induced selective down-regulation of a protein kinase C isozyme, nPKC epsilon, in rat pituitary GH4C1 cells. J Biol Chem. 1995;270:25115–25120. doi: 10.1074/jbc.270.42.25115. [DOI] [PubMed] [Google Scholar]
- Everall I, Barnes H, Spargo E, Lantos P. Assessment of neuronal density in the putamen in human immunodeficiency virus (HIV) infection. Application of stereology and spatial analysis of quadrats. J Neurovirol. 1995;1:126–129. doi: 10.3109/13550289509111017. [DOI] [PubMed] [Google Scholar]
- Everall IP, Glass JD, McArthur J, Spargo E, Lantos P. Neuronal density in the superior frontal and temporal gyri does not correlate with the degree of human immunodeficiency virus-associated dementia. Acta Neuropathol. 1994;88:538–544. doi: 10.1007/BF00296490. [DOI] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszman ML, Zuddas A, Masana MI, Barker JL, di Porzio U. Dopamine synthesis precedes dopamine uptake in embryonic rat mesencephalic neurons. J Neurochem. 1991;56:392–399. doi: 10.1111/j.1471-4159.1991.tb08164.x. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, James HJ, Sharer LR, Perry SW, Saito Y, Kazee AM, Blumberg BM, Epstein LG. Apoptotic neurons in brains from paediatric patients with HIV-1 encephalitis and progressive encephalopathy. Neuropathol Appl Neurobiol. 1995;21:208–217. doi: 10.1111/j.1365-2990.1995.tb01052.x. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Spencer JA, Holzer CE, 3rd, Soukup VM. Abnormal striatal dopaminergic synapses in National NeuroAIDS Tissue Consortium subjects with HIV encephalitis. J Neuroimmune Pharmacol. 2006;1:410–420. doi: 10.1007/s11481-006-9030-6. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Zheng J, Coulter CL, Ghorpade A, Che M, Thylin M, Rubocki R, Persidsky Y, Hahn F, Reinhard J, Jr, Swindells S. Suppression of inflammatory neurotoxins by highly active antiretroviral therapy in human immunodeficiency virus-associated dementia. J Infect Dis. 1998;178:1000–1007. doi: 10.1086/515693. [DOI] [PubMed] [Google Scholar]
- Gitler D, Spira ME. Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron. 1998;20:1123–1135. doi: 10.1016/s0896-6273(00)80494-8. [DOI] [PubMed] [Google Scholar]
- Goñi-Oliver P, Lucas JJ, Avila J, Hernández F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–563. doi: 10.1016/s0306-4522(00)00461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab. 2006;26:1052–1065. doi: 10.1038/sj.jcbfm.9600254. [DOI] [PubMed] [Google Scholar]
- Hersh BP, Rajendran PR, Battinelli D. Parkinsonism as the presenting manifestation of HIV infection: improvement on HAART. Neurology. 2001;56:278–279. doi: 10.1212/wnl.56.2.278. [DOI] [PubMed] [Google Scholar]
- Huang WC, Lin YS, Chen CL, Wang CY, Chiu WH, Lin CF. Glycogen synthase kinase-3beta mediates endoplasmic reticulum stress-induced lysosomal apoptosis in leukemia. J Pharmacol Exp Ther. 2009;329:524–531. doi: 10.1124/jpet.108.148122. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mehraein P, Weis S. Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathol. 2000;99:376–384. doi: 10.1007/s004010051139. [DOI] [PubMed] [Google Scholar]
- Jeannotte AM, Sidhu A. Regulated interactions of the norepineprhine transporter by the actin and microtubule cytoskeletons. J Neurochem. 2008;105:1668–1682. doi: 10.1111/j.1471-4159.2008.05258.x. [DOI] [PubMed] [Google Scholar]
- Johnson LA, Furman CA, Zhang M, Guptaroy B, Gnegy ME. Rapid delivery of the dopamine transporter to the plasmalemmal membrane on amphetamine stimulation. Neuropharmacology. 2005;49:750–758. doi: 10.1016/j.neuropharm.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsilieri E, Sopper S, Scheller C, ter Meulen V, Riederer P. Parkinsonism in HIV dementia. J Neural Transm. 2002;109:767–775. doi: 10.1007/s007020200063. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Fernandez JB, Singer EJ, Commins D, Waldrop-Valverde D, Ownby RL, Kumar M. Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol. 2009;15:257–274. doi: 10.1080/13550280902973952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, Shankar SK, Mahadevan A, Satishchandra P, Nath A. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. 2008;28:12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotox Res. 2009;16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal. 2008;1:re1. doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- Loder MK, Melikian HE. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J Biol Chem. 2003;278:22168–22174. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SM, Tong N, Gelbard HA. The phospholipid mediator platelet-activating factor mediates striatal synaptic facilitation. J Neuroimmune Pharmacol. 2007;2:194–201. doi: 10.1007/s11481-007-9064-4. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- Maki M, Bagci H, Hamaguchi K, Ueda M, Murachi T, Hatanaka M. Inhibition of calpain by a synthetic oligopeptide corresponding to an exon of the human calpastatin gene. J Biol Chem. 1989;264:18866–18869. [PubMed] [Google Scholar]
- Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett. 1989;103:234–239. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Masliah E, DeTeresa RM, Mallory ME, Hansen LA. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS. 2000;14:69–74. doi: 10.1097/00002030-200001070-00008. [DOI] [PubMed] [Google Scholar]
- Mason JN, Farmer H, Tomlinson ID, Schwartz JW, Savchenko V, DeFelice LJ, Rosenthal SJ, Blakely RD. Novel fluorescence-based approaches for the study of biogenic amine transporter localization, activity, and regulation. J Neurosci Methods. 2005;143:3–25. doi: 10.1016/j.jneumeth.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Mattos JP, Rosso AL, Correa RB, Novis SA. Movement disorders in 28 HIV-infected patients. Arq Neuropsiquiatr. 2002;60:525–530. [PubMed] [Google Scholar]
- Melikian HE. Neurotransmitter transporter trafficking: endocytosis, recycling, and regulation. Pharmacol Ther. 2004;104:17–27. doi: 10.1016/j.pharmthera.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsattari SM, Power C, Nath A. Parkinsonism with HIV infection. Mov Disord. 1998;13:684–689. doi: 10.1002/mds.870130413. [DOI] [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186(Suppl 2):S193–S198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Nath A, Geiger J. Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog Neurobiol. 1998;54:19–33. doi: 10.1016/s0301-0082(97)00053-1. [DOI] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. [PubMed] [Google Scholar]
- Norman JP, Perry SW, Kasischke KA, Volsky DJ, Gelbard HA. HIV-1 trans activator of transcription protein elicits mitochondrial hyperpolarization and respiratory deficit, with dysregulation of complex IV and nicotinamide adenine dinucleotide homeostasis in cortical neurons. J Immunol. 2007;178:869–876. doi: 10.4049/jimmunol.178.2.869. [DOI] [PubMed] [Google Scholar]
- Norman JP, Perry SW, Reynolds HM, Kiebala M, De Mesy Bentley KL, Trejo M, Volsky DJ, Maggirwar SB, Dewhurst S, Masliah E, Gelbard HA. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS One. 2008;3:e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini MJ, Debouck CM, Webb CL, Lysko PG. Extracellular human immunodeficiency virus type 1 Tat protein promotes aggregation and adhesion of cerebellar neurons. J Neurosci. 1996;16:2546–2552. doi: 10.1523/JNEUROSCI.16-08-02546.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA. HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol. 2005a;174:4333–4344. doi: 10.4049/jimmunol.174.7.4333. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Gelbard HA. Adjunctive therapies for HIV-1 associated neurologic disease. Neurotox Res. 2005b;8:161–166. doi: 10.1007/BF03033827. [DOI] [PubMed] [Google Scholar]
- Prochiantz A, di Porzio U, Kato A, Berger B, Glowinski J. In vitro maturation of mesencephalic dopaminergic neurons from mouse embryos is enhanced in presence of their striatal target cells. Proc Natl Acad Sci U S A. 1979;76:5387–5391. doi: 10.1073/pnas.76.10.5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB. Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem. 2001;78:874–889. doi: 10.1046/j.1471-4159.2001.00467.x. [DOI] [PubMed] [Google Scholar]
- Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R. Nigral degeneration in acquired immune deficiency syndrome (AIDS) Acta Neuropathol. 1991;82:39–44. doi: 10.1007/BF00310921. [DOI] [PubMed] [Google Scholar]
- Rumbaugh JA, Nath A. Developments in HIV neuropathogenesis. Curr Pharm Des. 2006;12:1023–1044. doi: 10.2174/138161206776055877. [DOI] [PubMed] [Google Scholar]
- Rumbaugh JA, Steiner J, Sacktor N, Nath A. Developing neuroprotective strategies for treatment of HIV-associated neurocognitive dysfunction. Futur HIV Ther. 2008;2:271–280. doi: 10.2217/17469600.2.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sá MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM. Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol. 2004;107:97–110. doi: 10.1007/s00401-003-0781-3. [DOI] [PubMed] [Google Scholar]
- Salum C, Raisman-Vozari R, Michel PP, Gomes MZ, Mitkovski M, Ferrario JE, Ginestet L, Del Bel EA. Modulation of dopamine uptake by nitric oxide in cultured mesencephalic neurons. Brain Res. 2008;1198:27–33. doi: 10.1016/j.brainres.2007.12.054. [DOI] [PubMed] [Google Scholar]
- Schenk JO, Wright C, Bjorklund N. Unraveling neuronal dopamine transporter mechanisms with rotating disk electrode voltammetry. J Neurosci Methods. 2005;143:41–47. doi: 10.1016/j.jneumeth.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Schwartz JW, Blakely RD, DeFelice LJ. Binding and transport in norepinephrine transporters. Real-time, spatially resolved analysis in single cells using a fluorescent substrate. J Biol Chem. 2003;278:9768–9777. doi: 10.1074/jbc.M209824200. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Takadou Y, Terajima K, Tsukada H, Gejo F, Tanaka K, Nishizawa M. A patient with HIV encephalopathy presenting with parkinsonism during HAART therapy (in Japanese) No To Shinkei. 2006;58:495–499. [PubMed] [Google Scholar]
- Silvers JM, Aksenov MY, Aksenova MV, Beckley J, Olton P, Mactutus CF, Booze RM. Dopaminergic marker proteins in the substantia nigra of human immunodeficiency virus type 1-infected brains. J Neurovirol. 2006;12:140–145. doi: 10.1080/13550280600724319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvers JM, Aksenova MV, Aksenov MY, Mactutus CF, Booze RM. Neurotoxicity of HIV-1 Tat protein: involvement of D1 dopamine receptor. Neurotoxicology. 2007;28:1184–1190. doi: 10.1016/j.neuro.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Sniderhan LF, Fan S, Kazmierczak K, Reisinger E, Kovács AD, Potash MJ, Dewhurst S, Gelbard HA, Maggirwar SB. Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3beta to antagonize nuclear factor-kappaB survival pathway in neurons. Eur J Neurosci. 2006a;23:2623–2634. doi: 10.1111/j.1460-9568.2006.04813.x. [DOI] [PubMed] [Google Scholar]
- Sui Z, Kovács AD, Maggirwar SB. Recruitment of active glycogen synthase kinase-3 into neuronal lipid rafts. Biochem Biophys Res Commun. 2006b;345:1643–1648. doi: 10.1016/j.bbrc.2006.05.087. [DOI] [PubMed] [Google Scholar]
- Tisch S, Brew B. Parkinsonism in HIV-infected patients on highly active antiretroviral therapy. Neurology. 2009;73:401–403. doi: 10.1212/WNL.0b013e3181b04b0d. [DOI] [PubMed] [Google Scholar]
- Tong N, Sanchez JF, Maggirwar SB, Ramirez SH, Guo H, Dewhurst S, Gelbard HA. Activation of glycogen synthase kinase 3 beta (GSK-3beta) by platelet activating factor mediates migration and cell death in cerebellar granule neurons. Eur J Neurosci. 2001;13:1913–1922. doi: 10.1046/j.0953-816x.2001.01572.x. [DOI] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Bellagamba R, Corpolongo A, Salvatori MF, Visco-Comandini U, Vlassi C, Giulianelli M, Galgani S, Antinori A, Narciso P. Persistence of neuropsychologic deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors. J Acquir Immune Defic Syndr. 2007;45:174–182. doi: 10.1097/QAI.0b013e318042e1ee. [DOI] [PubMed] [Google Scholar]
- Tse W, Cersosimo MG, Gracies JM, Morgello S, Olanow CW, Koller W. Movement disorders and AIDS: a review. Parkinsonism Relat Disord. 2004;10:323–334. doi: 10.1016/j.parkreldis.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DR, Dodson S, Nath A, Booze RM. Estrogen attenuates gp120- and tat1-72-induced oxidative stress and prevents loss of dopamine transporter function. Synapse. 2006;59:51–60. doi: 10.1002/syn.20214. [DOI] [PubMed] [Google Scholar]
- Wang W, Yang Y, Ying C, Li W, Ruan H, Zhu X, You Y, Han Y, Chen R, Wang Y, Li M. Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology. 2007;52:1678–1684. doi: 10.1016/j.neuropharm.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci Lett. 2003;340:189–192. doi: 10.1016/s0304-3940(03)00097-1. [DOI] [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci U S A. 1986;83:7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Smart EJ, Weksler B, Couraud PO, Hennig B, Toborek M. Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci. 2008;28:7788–7796. doi: 10.1523/JNEUROSCI.0061-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mactutus CF, Wallace DR, Booze RM. HIV-1 Tat protein-induced rapid and reversible decrease in [3H]dopamine uptake: dissociation of [3H]dopamine uptake and [3H]2beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) binding in rat striatal synaptosomes. J Pharmacol Exp Ther. 2009;329:1071–1083. doi: 10.1124/jpet.108.150144. [DOI] [PMC free article] [PubMed] [Google Scholar]