

Abstract
Brain-derived neurotrophic factor (BDNF) facilitates the formation of long-term potentiation (LTP) in hippocampus, but whether this involves release from presynaptic versus postsynaptic pools is unclear. We therefore tested whether BDNF is essential for LTP in dorsal striatum, a structure in which the neurotrophin is present only in afferent terminals. Whole-cell recordings were collected from medium spiny neurons in striatal slices prepared from adult mice. High-frequency stimulation (HFS) of neocortical afferents produced a rapid and stable NMDA receptor-dependent potentiation. The ratio of AMPA to NMDA receptor-mediated components of the EPSPs was substantially increased after inducing potentiation, suggesting that the response enhancement involved postsynaptic changes. In accord with this, paired-pulse response ratios, a measure of transmitter release kinetics, were reduced by elevated calcium but not by LTP. Infusion of the BDNF scavenger TrkB-Fc blocked the formation of potentiation, beginning with the second minute after HFS, without reducing responses to HFS. These results suggest that presynaptic pools of BDNF can act within 2 min of HFS to support the formation of a postsynaptic form of LTP in striatum.
Introduction
Results of electrophysiological studies using neutralizing antisera, genetic manipulation, or chimeric ligand-scavenger proteins (Korte et al., 1998; Minichiello, 2009) indicate that endogenous brain-derived neurotrophic factor (BDNF), acting through its TrkB receptor, positively modulates processes leading to stable long-term potentiation (LTP) in hippocampus. How the neurotrophin produces these effects has been the subject of considerable research. BDNF can enhance neurotransmitter release during trains of afferent stimulation (Figurov et al., 1996; Gottschalk et al., 1998), but there is also evidence that it facilitates the calcium modulation (Kovalchuk et al., 2002), postsynaptic signaling, and cytoskeletal remodeling (Messaoudi et al., 2007; Rex et al., 2007) required for the production of stable potentiation. BDNF's contribution to these events thus provides a likely explanation for its potent influence on LTP.
The location of the BDNF pool most pertinent to LTP has not been determined. BDNF is anterogradely transported to both axon terminals (Goodman et al., 1996; Altar and DiStefano, 1998; Conner et al., 1998; Haubensak et al., 1998) and dendritic arbors (Adachi et al., 2005; Matsuda et al., 2009) and is released from cultured neurons by high-frequency stimulation (HFS) or theta burst stimulation (TBS) (Balkowiec and Katz, 2002; Gärtner and Staiger, 2002). TBS reportedly increases local, extracellular concentrations of BDNF for ∼10 min in brain slices (Aicardi et al., 2004). Cultured neuron studies have shown that depolarization, afferent stimulation, and back-propagating action potentials elicit BDNF release from dendrites (Kuczewski et al., 2008; Lessmann and Brigadski, 2009) with lesser depletion of vesicular BDNF from axons (Matsuda et al., 2009). Thus, the induction of LTP may cause dendritic BDNF release followed by autocrine signaling through postsynaptic TrkB receptors. However, the relative contributions of secretion from axonal versus dendritic compartments to LTP has not been evaluated for adult neurons expressing native BDNF in situ.
The present studies investigated the potential role of presynaptic BDNF pools in LTP using the corticostriatal projection system. The medium spiny cells that comprise the great majority of neurons in striatum do not express BDNF at detectable levels (Conner et al., 1997; Fumagalli et al., 2007), and the sizable concentrations of BDNF in the structure are largely located within cortical afferents (Altar et al., 1997). The corticostriatal system is thus particularly appropriate for testing whether presynaptic BDNF supports the production of LTP.
Materials and Methods
Animals were anesthetized with fluorothane and killed by decapitation in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with protocols approved by the Institutional Animal Care and Use Committee of the University of California at Irvine.
Slice preparation.
Parahorizontal corticostriatal slices (270 μm thick) were prepared from 2-month-old male CB57BL/6J mice using a Leica Vibroslicer and maintained at 30–32°C for ≥1 h in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 3.4 CaCl2, 2.5 MgSO4, 26 NaHCO3, and 10 d-glucose, pH 7.3, and oxygenated with 95% O2 and 5% CO2.
Electrophysiological recordings.
Slices were placed in a recording chamber, submerged, and continuously perfused at 2–3 ml/min with oxygenated modified ACSF (0 mm Mg2+, 5 mm Ca2+) at 32°C unless otherwise indicated. Whole-cell current-clamp recordings were made with an Axopatch 200A amplifier (Molecular Devices). Data were filtered at 2 kHz, digitized at 1–5 kHz, stored on a computer, and analyzed off-line using Mini Analysis Program (Synaptosoft), Origin (OriginLab), and pCLAMP 7 (Molecular Devices). Medium-sized spiny neurons (MSNs) in dorsolateral striatum were visualized using an 60× water-immersion objective and infrared differential interference optics video microscopy, and identified by morphology (small somata, post hoc analysis of Neurobiotin or AlexaFluor488 filling revealed multiple primary dendrites with a high spine density in their oblique branches). MSNs were additionally identified by their intrinsic membrane properties (e.g., resting membrane potential typically more negative than −80 mV, inward and outward rectification in response to somatic current injection, and a long depolarizing ramp to the action potential threshold leading to a delayed spike discharge) (Kawaguchi et al., 1989). Borosilicate glass patch electrodes had a resistance of 3–8 MΩ after being filled with pipette solution (130 K gluconate, 10 KCl, 0.2 EGTA, 8 NaCl, 2 ATP, 0.3 GTP, and 10 HEPES, pH 7.35, 290–300 mOsm). For voltage-clamp recordings, K gluconate was replaced by cesium methane-sulfonate.
For most experiments, cells were filled with Neurobiotin (Vector Laboratories) or AlexaFluor 488 (Invitrogen) (3 mg/ml or 60 μg/ml in the patch-clamp pipette, respectively) during recordings. Neurobiotin was visualized as described previously (Brunson et al., 2005). For AlexaFluor 488 fills, slice whole mounts were viewed using epifluorescence illumination.
Corticostriatal pathway stimulation.
Responses were evoked in MSNs by extracellular stimulation (0.01 ms, 0.05 Hz) with bipolar electrodes (FHC) placed in cortical layer VI close to the white matter. Recordings were rejected if the initial Vm was more positive than −78 mV or if Vm changed by ≥10% and input resistance changed by ≥30% during recordings. After 10 min of recording stable responses, HFS (four 1 s, 100 Hz trains at 10 s intervals) was delivered. The relative EPSP change was determined by comparing the normalized mean response from 25 to 30 min after HFS with the mean EPSP over the 10 min immediately before HFS. The criterion used to determine the induction of LTP was a 20% increase in evoked responses relative to baseline as assessed from normalized values. Data are expressed as means ± SEM, and Student's t test was used for statistical analysis, unless otherwise stated.
For comparisons of treatment effects on EPSP amplitude between slices, the values of each recording were normalized to the average amplitude for the 10 min of baseline before a compound was added. To quantify effects of compounds on EPSP amplitude, values from the last 3 min of compound application were averaged for each slice. Differences were considered significant when p < 0.05.
Receptor antagonists.
For all experiments, 50 μm picrotoxin (GABAA receptor blocker) was added to the ACSF. Where indicated, the AMPA receptor (AMPAR) antagonist CNQX (20 μm) or the NMDA receptor (NMDAR) antagonist d-2-amino-5-phosphonopentanoic acid (APV; 50 μm) was added as well. Data for APV experiments were used only if responses decreased by ≥20% within 4 min of infusion onset and remained depressed below that level.
Results
Previous work showed that LTP is more difficult to elicit in corticostriatal than in hippocampal slice preparations across stimulation protocols [e.g., spike timing (Shen et al., 2008), multiple trains, postsynaptic depolarization, etc.] but can be obtained in low-magnesium/high-calcium medium (Calabresi et al., 2007; Lovinger, 2010). Pilot experiments confirmed the latter point, and we conducted most of the following studies under these conditions. Recordings were made from MSNs, which were recognizable by morphological features and by a characteristic inward rectification (Fig. 1A). Neurons were also filled with Neurobiotin or AlexaFluor 488 for subsequent morphological confirmation (Fig. 1B). EPSPs, recorded in the presence of the GABAA receptor antagonist picrotoxin, were comparable to those described in the literature (Rohrbacher et al., 1994; Moriguchi et al., 2002; Li et al., 2009) and were notable for having a slow decay constant. Previous studies have described a significant NMDAR-mediated component to EPSPs recorded from MSNs (Li et al., 2009), and in agreement with this, we found that the AMPAR antagonist CNQX reduced the amplitude of cortically evoked responses by almost half; the remainder of the fast response was eliminated by the NMDAR antagonist APV (Fig. 1C).
Figure 1.

Characteristics of striatal neurons. A, Whole-cell recordings indicate that the neurons have a resting membrane potential of about −80 mV and emit scattered spikes at about −40 mV. The input /output curve is indicative of inward rectification. B, Intracellular fills with AlexaFluor 488 confirm that the recorded cells are medium spiny neurons (scale bar, 30 μm). C, EPSPs elicited by cortical stimulation are partially blocked by CNQX; subsequent perfusion of APV eliminates nearly all of the remaining response.
HFS trains, in accord with previous reports (Partridge et al., 2000; Fino et al., 2005; Marrone et al., 2006; Li et al., 2009), did not trigger lasting potentiation of striatal EPSPs when administered in the presence of APV (Fig. 2A), indicating that NMDARs are necessary for induction but by themselves do not express LTP. As noted, APV substantially reduced pre-HFS baseline responses. We further investigated the presynaptic versus postsynaptic locus of potentiation by comparing effects of APV on potentiated responses with those on baseline EPSPs. HFS caused a marked and stable potentiation effect that was only partially reduced by APV infusion (Fig. 2B). Direct comparison of APV's effects on baseline versus potentiated responses indicated that the antagonist had smaller effects on the latter: the percentage reduction for baseline EPSPs was 43.2 ± 7.0% (mean ± SD), whereas that for potentiated EPSPs was 34.6 ± 3.6% (p = 0.022; t test with Welch correction, two tails). The differential effect of APV strongly suggests that much of the potentiation involved selective enhancement of AMPAR-gated currents.
Figure 2.
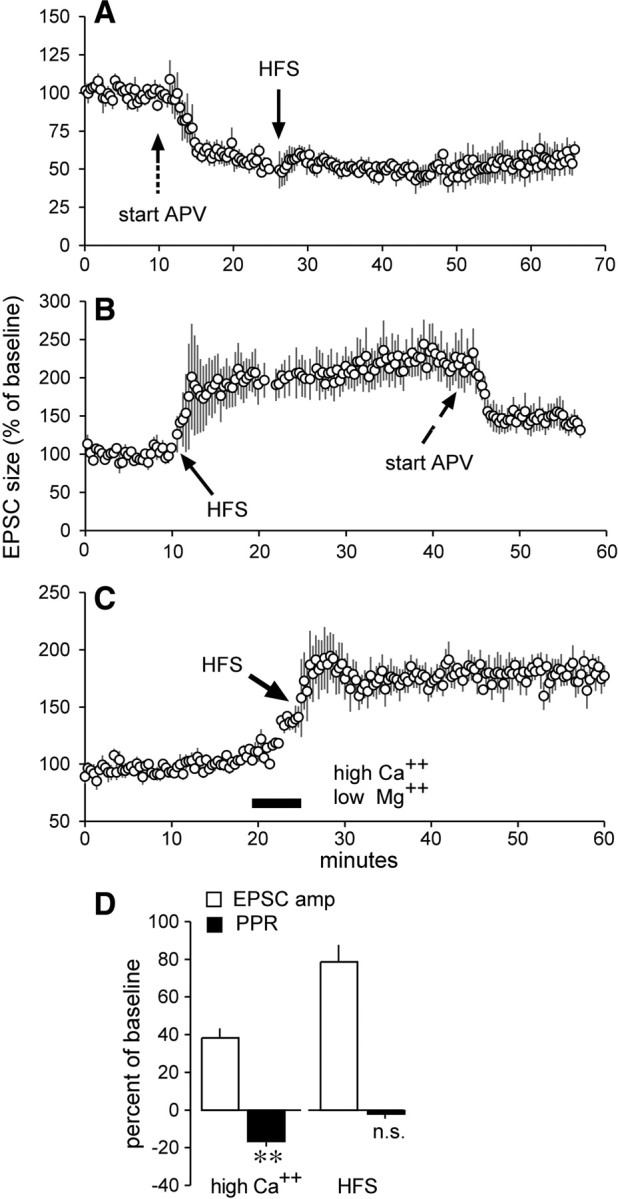
Induction of LTP reduced the NMDAR-mediated component of synaptic responses but did not affect paired-pulse ratios. A, APV infused continuously before the delivery of HFS blocked induction of LTP. B, Effects of APV when infused after HFS and the induction of LTP. C, Baseline measurements and magnitude of LTP were recorded in slices perfused with ACSF containing 2 mm Ca2+ and 2 mm Mg2+; HFS delivered at the end of 5 min infusion of 5 mm Ca2+ and 0 mm Mg2+ (black bar) produced potentiation that remained in place when lower calcium conditions were restored. D, Five minutes of high calcium increased the EPSC amplitude (amp) and reliably reduced the PPR (**p < 0.01). Applied at the end of high-calcium infusion, HFS further increased the EPSC amplitude (amp) without measurably affecting the PPR (n.s. denotes p > 0.60 vs baseline). Plots show means ± SEM for 5–7 slices/group.
We further tested the above conclusion by comparing paired-pulse response ratios (PPRs), a measure sensitive to release probability, for the corticostriatal EPSCs before and after induction of LTP. Slices were perfused with 2 mm Ca2+/2 mm Mg2+-containing ACSF, switched to 5 mm Ca2+/0 mm Mg2+ for 5 min for induction of LTP, and then returned to 2 mm Ca2+/2 mm Mg2+. Twin pulses separated by 50 ms were delivered throughout the analysis. Two of six cells exhibited paired-pulse facilitation (30%), while the remainder did not, in agreement with reports of variable PPRs at corticostriatal synapses (Cepeda et al., 2008). The switch to high calcium caused an evident increase in EPSCs (Fig. 2C); HFS delivered at the end of high-calcium infusion induced robust potentiation that remained in place following the return to low-calcium medium. The increase in EPSC amplitudes recorded at the end of the 5 min high-calcium/low-magnesium treatment (38.2 ± 12%) was accompanied by a reliable decrease in PPR (16.7 ± 5%; p < 0.01, paired t test, two tails), as expected for a measure sensitive to release kinetics (Fig. 2D, “high Ca2+”). The experimental question was whether the increase in synaptic responses that constitutes striatal LTP would similarly reduce PPR. This was not the case: a 78.1 ± 21.9% increase (measured 25–35 min after HFS) in EPSC amplitude above baseline was not accompanied by a change in PPR (−1.9 ± 6.9, p = 0.60) (Fig. 2D, “HFS”).
Having found that LTP at corticostriatal synapses shares several features with hippocampal potentiation (i.e., predominant changes in AMPAR-mediated responses; little to no change in PPR) (Kauer et al., 1988; Muller and Lynch, 1988; Muller et al., 1988), we tested whether it has similar dependency upon BDNF. The extracellular BDNF scavenger TrkB-Fc, applied at concentrations shown to block cell signaling changes induced by BDNF (Rex et al., 2007), had no evident effects on baseline responses during a 45 min infusion. This agrees with effects in hippocampus (Kovalchuk et al., 2002; Rex et al., 2007). TrkB-Fc did, however, produce a striking suppression of corticostriatal LTP. HFS delivered to control slices produced stable response facilitation, as described; the same stimulation administered in the presence of TrkB-Fc caused transient facilitation that dissipated within the first 1–2 min after induction (Fig. 3A, left). We quantified this unexpectedly rapid and complete effect of the scavenger by averaging responses for each of the first 10 min after induction. As shown in Figure 3A (right), the response facilitation produced by HFS was undetectable by the second minute after induction. Differences in percentage potentiation between control and experimental slices were highly significant (p < 0.0001 for 2–10 min after HFS). Comparable effects were obtained with the tyrosine kinase inhibitor K252a that blocks Trk autophosphorylation and signaling (Knüsel and Hefti, 1992) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Comparisons between these results and those described for the hippocampus suggest that BDNF has a substantially greater effect on synaptic plasticity in striatum.
Figure 3.

TrkB-Fc blocks LTP without changing responses to HFS. A, Left, HFS caused a long lasting increase in EPSP amplitude under control conditions but not when delivered in the presence of TrkB-Fc. Right, Effects of TrkB-Fc were evident shortly after HFS: plot shows that averaged responses for post-HFS minutes 2–10 were not elevated above baseline (dashed line) and were reduced relative to potentiated values found in control slices (***p < 0.001 for 2–10 min after HFS). B, Composite responses to 1 s HFS were comparable for control and TrkB-Fc-treated slices. Typical responses are shown at left; peak amplitudes and times to peak are summarized on the right. C, Infused BDNF increased the size of the isolated NMDAR component of the EPSP. The upper traces show that the tested responses were completely blocked by APV; lower traces illustrate the effects of BDNF. Bar graph summarizes the group effect of BDNF treatment (**p < 0.01). D, Left, Representative traces of isolated (CNQX present) NMDAR-mediated responses to 1 s HFS trains delivered before (burst #1) and 10 min after (burst #2) infusion of vehicle (“control”) or TrkB-Fc (calibration: 100 pA, 100 ms). Right, Bar graphs show peak amplitudes and time to peak for the composite responses to the first and second HFS trains for control and TrkB-Fc-treated slices. Note that TrkB-Fc infusion did not reduce either variable. Plots show mean ± SEM values for 6–8 slices/group.
We next tested whether the potent effects of TrkB-Fc were due to actions on responses to HFS. The accumulated depolarization to the 1 s trains of HFS was not different in control versus TrkB-Fc-treated slices (Fig. 3B, left); measurements indicated that the peak response amplitudes, and the within-train time to peak, were comparable for control and scavenger-treated slices (Fig. 3B, right). The possibility remains that the NMDAR component of the HFS response is enhanced to a degree that promotes potentiation but is too small to be detected in the composite responses. We investigated this by asking whether BDNF affects NMDAR-dependent EPSPs in striatum. The results were clear: 30 min infusion of BDNF in the presence of the AMPAR antagonist CNQX increased pharmacologically isolated NMDAR responses (45 ± 12%) (Fig. 3C, bottom traces).
These results led us to ask whether scavenging extracellular BDNF reduces NMDAR-gated responses to HFS. We tested this by applying an HFS train to a cell during perfusion of CNQX and then repeating it 10 min later in the presence of vehicle or TrkB-Fc. Surprisingly, given the results for infused BDNF, TrkB-Fc did not reduce the composite inward currents mediated by NMDARs during a 1 s HFS train (Fig. 3D, left). Quantitative measures across multiple slices gave no indication that NMDAR-mediated burst responses were reduced in their amplitudes or times to peak amplitude by TrkB-Fc (Fig. 3D, right). It follows from this that BDNF's contributions to LTP occur at some point after induction.
Discussion
LTP was more difficult to elicit in corticostriatal projections than is the case for intrahippocampal pathways. We found it necessary to use high-frequency stimulation trains and GABA receptor antagonists to reliably obtain potentiation in striatum, whereas only four or five theta bursts are needed to trigger robust LTP in hippocampal slices. It is thus not surprising that various groups have reported that LTP is poorly developed in striatum (Calabresi et al., 2007). The high threshold for the induction of potentiation could reflect the absence of positive modulators rather than being an indication that LTP-type plasticity is only rarely used within the basal ganglia. Dopamine, the absence of which impairs LTP in the striatum (Centonze et al., 1999; Pawlak and Kerr, 2008; Schotanus and Chergui, 2008), is an obvious candidate for an under-represented plasticity factor in our experiments. It should be noted, however, that there appear to be no reports showing that enhancing dopaminergic transmission lowers the threshold for striatal LTP. Testing this point is a reasonable first step in asking whether plasticity in the striatum occurs with a much lower probability than is the case for the cortical telencephalon.
Despite its elevated threshold, we found striatal LTP possessed a number of features that characterize potentiation in hippocampus. It is synapse specific, not accompanied by evident changes in release kinetics as assessed by PPRs, and requires NMDAR activation for induction. We also obtained evidence that corticostriatal potentiation is expressed by changes in AMPARs to a greater degree than NMDARs. EPSPs in striatum, in contrast to those in hippocampus, contain a sizable NMDAR-mediated component. This made it possible to carry out reasonably direct comparisons of the contributions of AMPA versus NMDA receptors to corticostriatal responses before and following induction of LTP. The percentage of the EPSP contributed by NMDARs, as estimated from the depression produced by APV, was significantly reduced at potentiated synapses. This result accords with measurements made of hippocampal LTP (Kauer et al., 1988; Muller and Lynch, 1988; Manabe and Nicoll, 1994) in showing that the AMPAR-mediated component of the synaptic response is differentially elevated by potentiation. Findings in both regions indicate that LTP reflects postsynaptic mechanisms as opposed to changes in presynaptic glutamate release, which would be predicted to influence AMPA and NMDA receptor-mediated responses to a comparable extent. The absence of changes to paired-pulse ratios, a measure sensitive to conditions that affect release probability (elevated ACSF calcium levels), following induction of LTP also argues against a quantitatively meaningful presynaptic contribution to corticostriatal potentiation.
The present findings constitute the first evidence that activity-induced LTP outside the cortical telencephalon is dependent upon BDNF. Infusion of the ligand scavenger TrkB-Fc completely blocked LTP of corticostriatal synapses as did the Trk signaling antagonist K252a, indicating that endogenous BDNF plays a potent and essential role in generating stable potentiation. The scavenger eliminated response enhancement by the second minute after HFS, indicating that BDNF acts on very early phases of potentiation in striatum. The mechanisms through which BDNF promotes LTP of corticostriatal synapses remain to be studied but our results suggest that postinduction processes are involved. TrkB-Fc had little or no effect on baseline EPSPs or the composite response to HFS. Thirty minute BDNF infusions enhanced NMDAR-mediated EPSPs but TrkB-Fc did not reduce the isolated NMDAR-mediated responses to HFS. Thus, it seems that prolonged exposures to infused BDNF initiate changes that are not triggered by the much briefer release events occurring during HFS. In any event, the absence of a TrkB-Fc effect on baseline and burst responses strongly suggests that endogenous BDNF promotes potentiation by transiently engaging postsynaptic mechanisms such as the signaling to actin shown to be critical for hippocampal LTP (Lynch et al., 2008; Rex et al., 2009).
The present results demonstrate that, for this particular system, release of presynaptic BDNF is fully capable of promoting the postsynaptic events responsible for the induction and expression of LTP. Although the locus of the LTP-related pools within hippocampus remain to be determined, multiple studies have shown that BDNF in striatum is predominantly contained within corticostriatal afferents as opposed to resident neurons (Altar et al., 1997; Conner et al., 1997; Fumagalli et al., 2007). Thus, there is little ambiguity regarding the origins of local extracellular BDNF contributing to striatal LTP. It remains now to isolate the mechanisms underlying neurotrophin release from cortical terminals in striatum. Evidence that such systems are required for LTP at various sites in cortical telencephalon would constitute strong evidence that conjoint release of glutamate and BDNF from terminals is a ubiquitous requirement for the potentiation effect.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS045260 to G.L. and C.M.G. We thank M. Jafari and Dr. J. C. Lauterborn for assistance with photomicroscopy, and Dr. M. S. Levine (University of California, Los Angeles) for valuable advice and discussions.
References
- Adachi N, Kohara K, Tsumoto T. Difference in trafficking of brain-derived neurotrophic factor between axons and dendrites of cortical neurons, revealed by live-cell imaging. BMC Neurosci. 2005;6:42. doi: 10.1186/1471-2202-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, Canossa M. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 2004;101:15788–15792. doi: 10.1073/pnas.0406960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, DiStefano PS. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998;21:433–437. doi: 10.1016/s0166-2236(98)01273-9. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Kramár E, Lin B, Chen Y, Colgin LL, Yanagihara TK, Lynch G, Baram TZ. Mechanisms of late-onset cognitive decline after early-life stress. J Neurosci. 2005;25:9328–9338. doi: 10.1523/JNEUROSCI.2281-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Centonze D, Gubellini P, Picconi B, Calabresi P, Giacomini P, Bernardi G. Unilateral dopamine denervation blocks corticostriatal LTP. J Neurophysiol. 1999;82:3575–3579. doi: 10.1152/jn.1999.82.6.3575. [DOI] [PubMed] [Google Scholar]
- Cepeda C, André VM, Yamazaki I, Wu N, Kleiman-Weiner M, Levine MS. Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur J Neurosci. 2008;27:671–682. doi: 10.1111/j.1460-9568.2008.06038.x. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and messenger RNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Gall CM. Anterograde transport of neurotrophin proteins in the CNS—a reassessment of the neurotrophic hypothesis. Rev Neurosci. 1998;9:91–103. doi: 10.1515/revneuro.1998.9.2.91. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Repeated exposure to cocaine differently modulates BDNF mRNA and protein levels in rat striatum and prefrontal cortex. Eur J Neurosci. 2007;26:2756–2763. doi: 10.1111/j.1460-9568.2007.05918.x. [DOI] [PubMed] [Google Scholar]
- Gärtner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci U S A. 2002;99:6386–6391. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Narz F, Heumann R, Lessmann V. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci. 1998;111:1483–1493. doi: 10.1242/jcs.111.11.1483. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Emson PC. Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs. J Neurophysiol. 1989;62:1052–1068. doi: 10.1152/jn.1989.62.5.1052. [DOI] [PubMed] [Google Scholar]
- Knüsel B, Hefti F. K-252 compounds: modulators of neurotrophin transduction. J Neurochem. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- Korte M, Kang H, Bonhoeffer T, Schuman E. A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology. 1998;37:553–559. doi: 10.1016/s0028-3908(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL. Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci. 2008;28:7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Brigadski T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res. 2009;65:11–22. doi: 10.1016/j.neures.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Li P, Li YH, Han TZ. NR2A-containing NMDA receptors are required for LTP induction in rat dorsolateral striatum in vitro. Brain Res. 2009;1274:40–46. doi: 10.1016/j.brainres.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity, and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Nicoll RA. Long-term potentiation: evidence against an increase in transmitter release probability in the CA1 region of the hippocampus. Science. 1994;265:1888–1892. doi: 10.1126/science.7916483. [DOI] [PubMed] [Google Scholar]
- Marrone MC, Marinelli S, Biamonte F, Keller F, Sgobio CA, Ammassari-Teule M, Bernardi G, Mercuri NB. Altered cortico-striatal synaptic plasticity and related behavioural impairments in reeler mice. Eur J Neurosci. 2006;24:2061–2070. doi: 10.1111/j.1460-9568.2006.05083.x. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci. 2009;29:14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Kanhema T, Soulé J, Tiron A, Dagyte G, da Silva B, Bramham CR. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Moriguchi S, Watanabe S, Kita H, Nakanishi H. Enhancement of N-methyl-D-aspartate receptor-mediated excitatory postsynaptic potentials in the neostriatum after methamphetamine sensitization. An in vitro slice study. Exp Brain Res. 2002;144:238–246. doi: 10.1007/s00221-002-1039-3. [DOI] [PubMed] [Google Scholar]
- Muller D, Lynch G. Long-term potentiation differentially affects two components of synaptic responses in hippocampus. Proc Natl Acad Sci U S A. 1988;85:9346–9350. doi: 10.1073/pnas.85.23.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- Partridge JG, Tang KC, Lovinger DM. Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol. 2000;84:1422–1429. doi: 10.1152/jn.2000.84.3.1422. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbacher J, Bijak M, Misgeld U. Suppression by memantine and amantadine of synaptic excitation intrastriatally evoked in rat neostriatal slices. Neurosci Lett. 1994;182:95–98. doi: 10.1016/0304-3940(94)90215-1. [DOI] [PubMed] [Google Scholar]
- Schotanus SM, Chergui K. Dopamine D1 receptors and group I metabotropic glutamate receptors contribute to the induction of long-term potentiation in the nucleus accumbens. Neuropharmacology. 2008;54:837–844. doi: 10.1016/j.neuropharm.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]