


Abstract
The properties of human DNA helicase V (HDH V) were studied in greater detail following an improved purification procedure. From 450 g of cultured cells, <0.1 mg of pure protein was isolated. HDH V unwinds DNA unidirectionally by moving in the 3′ to 5′ direction along the bound strand in an ATP- and Mg2+-dependent fashion. The enzyme is not processive and can also unwind partial RNA–RNA duplexes such as HDH IV and HDH VIII. The Mr determined by SDS–PAGE (66 kDa) corresponds to that measured under native conditions, suggesting that HDH V exists as a monomer in the nucleus. Microsequencing of the purified HDH V shows that this enzyme is identical to the far upstream element-binding protein (FBP), a protein that stimulates the activity of the c-myc gene by binding specifically to the ‘FUSE’ DNA region localized upstream of its promoter. The sequence of HDH V/FBP contains RGG motifs like HDH IV/nucleolin, HDH VIII/G3BP as well as other human RNA and DNA helicases identified by other laboratories.
INTRODUCTION
DNA and RNA helicases are a ubiquitous class of enzymes defined by their capacity to unwind and translocate along DNA or RNA in reactions that are coupled to the binding and the hydrolysis of a 5′-nucleoside triphosphate (NTP) (1). These enzymes participate in a variety of cellular processes including DNA replication, DNA repair, recombination, transcription, RNA processing and translation (2). Several human disorders are due to modifications in genes that code for helicases (3). For example, two genes, XPB and XPD, encode helicases that are defective in individuals with xeroderma pigmentosum and Cockayne’s syndromes, respectively (4). Bloom’s and Werner syndromes are two other genetic disorders that arise as a consequence of abnormalities in different members of the RecQ family of helicases (5,6).
Since the discovery of the first helicase in 1976 (7) many other similar enzymes have been isolated from both prokaryotic and eukaryotic cells. The diversity of functions in which these enzymes are involved is reflected in the increasing number of different helicases that are continuously discovered. Escherichia coli contains at least 12 different helicases (8) and yeast contains at least 41 helicase genes, based on the analysis of the Saccharomyces cerevisiae genome (3). Analogous, but clearly less complete, analysis of helicase genes in human cells reveals no less than 31 different genes (3). These estimates are based on the presence of the seven different amino acid sequence motifs that characterize many known DNA helicases (9). In our laboratory we have initiated a systematic purification of human DNA helicases (HDHs) present in the nuclear extract of HeLa cells by following the catalytic activity of the enzyme (10). This approach allowed us to isolate both canonical (with the seven motifs) and non-canonical helicases. Since no functional description of the role of all the so-called ‘helicase domains’ has been obtained so far, there is no guarantee that they represent specific recognition features for all DNA helicases. In fact, in the course of our study, we have already identified three non-canonical helicases, human helicases II, IV and VIII. HDH II turned out to be Ku, a heterodimer involved in the non-homologous recombination pathway (11,12), HDH IV corresponds to human nucleolin, a protein involved in a variety of functions such as rRNA maturation, ribosome assembly, nucleocytoplasmic transport and chromatin structure (13,14), while HDH VIII corresponds to G3BP an element of the Ras transduction pathway (15). In the present work, we have revisited the purification and characterization of another previously isolated protein, namely HDH V, a very powerful helicase present in low abundance in nuclear extracts. The analysis of the properties of this protein purified from a greater amount of cells than in our previous study, led us to identify it with a different molecular species than the one initially reported. The results of this study, with the consequent identification of HDH V with another known protein (not previously recognized as a helicase), are reported here.
MATERIALS AND METHODS
Reagents
All the salts, bovine serum albumin (BSA), dithiothreitol (DTT), phenylmethylsulfonyl fluoride (PMSF), leupeptin and pepstatin were from Sigma Chemical (St Louis, MO). The M13mp19 ssDNA plasmid, the serums for growing the HeLa cells, glutamine and gentamycin were from Gibco BRL (Gaithersburg, MD). All the resins used for the different purification steps were from Amersham Pharmacia Biotech (Uppsala, Sweden). All the ssDNA oligonucleotides used for making the DNA substrates were purchased from Sigma-Genosys Ltd (Cambridgeshire, UK). The radioactive NTPs were obtained from Amersham Pharmacia Biotech (Buckinghamshire, UK). The T4 polynucleotide kinase was from Promega (Madison, WI).
Cell cultures and buffers
HeLa cells were grown in Joklin MEM supplemented with 10% fetal calf serum, 50 µg/ml gentamycin and 2 mM glutamine, and harvested as described previously (10). All the buffers used during the purification of HDH VIII contained 1 mM DTT, 0.5 mM PMSF, 1 µM pepstatin and 1 µM leupeptin. Buffer A contained 20 mM HEPES (pH 8.0), 0.1 M NaCl, 1 mM EDTA and 20% glycerol. Buffer B contained 50 mM Tris–HCl (pH 7.5), 70 mM KCl, 1 mM EDTA and 10% glycerol. The concentration of NaCl or KCl in all the buffers was increased up to 1.0 M for eluting the proteins from the different columns. Buffer C was the same as buffer B plus 4 mM MgCl2.
Preparation of DNA helicase substrates
The DNA substrates used in the helicase assay are listed in Figure 4. They consist of different 32P-labeled oligonucleotides annealed either to M13mp19 phage ssDNA or to ssDNA fragments of different lengths to create partial duplexes. The sequence of the 99 bp oligonucleotide used for the determination of the direction of unwinding is the same as described previously (13) (Fig. 4F and G). Substrate without hanging tails: 5′-GTAAAACGACGGCCAGT-3′, complementary to nucleotides 6291–6307 of M13mp19 ssDNA (Fig. 4A); substrate with the same oligonucleotide carrying a hanging tail of 15 T residues at its 5′ end (Fig. 4C); substrate with the same hanging tail at its 3′ end (Fig. 4D); substrate with hanging tails at both the 5′ and 3′ ends (Fig. 4E). The sequence of the 29 bp oligonucleotide encompassing the coding strand of the far upstream element (FUSE) is 5′-ACAAAATAAAAAATCCCGAGGGAATATTAC-3′ (Fig. 4H). A total of 25 ng of each oligonucleotide, labeled at the 5′ end with T4 polynucleotide kinase and 0.9 MBq of [γ-32P]ATP, were subsequently annealed to M13mp19 phage ssDNA (4 µg) in 40 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 1 mM DTT and 50 mM NaCl. The mixture was heated at 95°C for 2 min, and slowly cooled to room temperature. Each substrate was purified by gel filtration through a 5 ml Sepharose 4B column.
Figure 4.

Concentration- and time-dependence of HDH V activity. (A) Increasing amounts of HDH V were incubated in the standard 10 µl reaction mixture for 30 min at 37°C. The amount of enzyme in autoradiograms (I) and (II) is indicated at the top of each lane while the substrate concentration remained constant in all the experiments (0.05 nM, 1000 c.p.m.). The right panel shows the quantitative data of (I) and (II). (B) Kinetic of unwinding performed using 100 pM HDH V and the same reaction conditions used for the concentration-dependence experiments. The reaction time (min) is indicated at the top of each lane. Lanes C and D of each autoradiogram are control assays without enzyme and heat-denatured substrate, respectively.
Preparation of RNA/RNA and RNA/DNA substrates
The RNA/RNA and RNA/DNA substrates were obtained as follows: pGEM vector (Promega) containing the Ku70 gene was linearized by EcoRI and transcribed in vitro with the SP6 RNA polymerase from the specific promoter yielding a 1.3 kb RNA (15). A 17 bp DNA or RNA oligonucleotide complementary to the same region of Ku 70 (nucleotides 1840–1856 of the Ku sequence) was synthesized and labeled at the 5′ end by T4 polynucleotide kinase with 0.9 MBq [γ-32P]ATP. A large excess of labeled oligonucleotide was mixed with 1 µM of synthesized Ku RNA, heated at 95°C for 2 min and allowed to anneal by slow cooling at room temperature. The substrates were then purified by gel filtration through a 5 ml Sepharose 4B column.
DNA helicase assay
The helicase assay measures the unwinding of a γ-32P-labeled DNA fragment from a partial duplex DNA molecule. The 10 µl reaction mixture contained 20 mM Tris–HCl (pH 9.0), 8 mM DTT, 2 mM MgCl2, 2 mM ATP, 10 mM KCl, 4% (w/v) sucrose, 80 µg/ml BSA and 32P-labeled helicase substrate (1000 c.p.m., 1 ng, ∼0.05 nM). The helicase fraction to be assayed was added to the mixture, incubated at 37°C for 30 min and the reaction was terminated by the addition of 0.3% SDS, 10 mM EDTA, 5% glycerol and 0.1% bromophenol blue. The products of the reaction were fractionated by electrophoresis on a 12% non-denaturing polyacrylamide gel. The gel was dried and the extent of DNA unwinding was quantitated by electronic autoradiography (Instant Imager, Packard Corp., Meriden, CT). One unit of DNA helicase is defined as the amount of enzyme unwinding 1% of the substrate in 1 min at 37°C (30% in 30 min) in the linear range of enzyme concentration dependence.
Affinity labeling with [α-32P]ATP
Affinity labeling with [α-32P]ATP by UV cross-linking was performed as described previously (16). Briefly, ~5 ng of HDH V was incubated on ice with 0.5 µl of [α-32P]ATP (specific activity 15 TBq/mmol) and 3 µg/ml of unlabeled DNA substrate (17mer annealed to M13 ssDNA) in a buffer containing 60 mM KCl, 12 mM HEPES (pH 7.9), 1 mM MgCl2, 6 mM DTT. The mixture was irradiated for 30 min with a 4 W, 254 nm UV light at a distance of ∼5 cm in ice and then incubated for 10 min without irradiation at room temperature. The products were separated on a 10% SDS–PAGE, the gel was washed extensively with 10% acetic acid to remove non-bound radioactivity, then dried and exposed for autoradiography.
Native molecular weight determination
The native molecular weight and the sedimentation coefficient were determined by glycerol gradient centrifugation following the procedure described by Tuteja et al. (11). More specifically, 100 µl of Fraction VI was layered on a 15–35% glycerol gradient in buffer C and centrifuged at 310 000 g for 20 h at 4°C. The standard protein markers were also run under the same conditions. The markers were catalase (250 kDa, 11.3S), aldolase (158 kDa, 7.4S), BSA (66 kDa, 4.3S) and ovalbumin (45 kDa, 3.5S). Fractions of 0.2 ml were collected from the top of the tube using HSI Auto Densi-Flow IIC (Buchler Instruments, KS) and assayed for helicase activity.
Microsequence
The sequence analysis was done by the Protein and Nucleic Acid Chemistry Laboratories of Washington University School of Medicine (St Louis, MO). The silver-stained band containing HDH V was cut and treated with bovine trypsin. The products of digestion were separated by HPLC and analyzed by tandem mass spectrometry (Finnigan LCQ, Finnigan Corp., San Jose, CA).
RESULTS
Purified HDH V was obtained from 450 g of frozen HeLa cells; the cell nuclei were isolated and salt-extracted using the procedure described by Dignam et al. (17). The substrate used to measure the unwinding activity after each purification step and for most of the characterization consisted of a 5′-32P-labeled 17mer completely annealed to M13mp19 ssDNA. All the purification steps were carried out at 4°C (Table 1). The initial steps of purification up to Fraction III were the same as described previously (16). Briefly, the nuclear extract was precipitated by slowly adding ammonium sulfate (0.35 g/ml), the precipitated proteins were collected by centrifugation at 25 000 g for 30 min in Sorvall ss34 rotor, dialyzed in buffer A (Fraction I, 550 ml) and applied onto a Bio-Rex column (2.5 cm diameter × 33 cm) equilibrated with buffer A (16). Active fractions eluting at ∼0.8 M NaCl in buffer A were pooled (Fraction II, 250 ml). This pool was again treated with ammonium sulfate as described above and the resulting pellet, after centrifugation, was re-suspended and dialyzed in buffer B (Fraction III, 60 ml). The amount of helicase activity up to this step could not be quantitated precisely due to the contamination of nucleases. All the purification steps that follow were carried out by FPLC (Pharmacia-LKB, Uppsala, Sweden). Fraction III was loaded onto a 19 ml heparin–Sepharose (1.6 × 9 cm) column equilibrated with buffer B. The proteins bound to the column were eluted using a linear gradient from 0.07 to 1.0 M KCl in buffer B. The active fractions eluting at ~0.5 M KCl in buffer B were pooled (Fraction IV, 35 ml, 260 000 U). Fraction IV was dialyzed first against buffer B without KCl and then against buffer B containing 70 mM KCl. The protein solution was loaded onto a 20 ml Q-Sepharose fast flow column (1.6 × 9 cm) and the elution was carried out with the same linear gradient used for the heparin–Sepharose. Some of the activity eluted in the flow-through fractions (Fraction V, 90 ml, 192 000 U). The concentration of MgCl2 in Fraction V was adjusted to 4 mM and the same fraction was loaded onto a 4 ml ssDNA cellulose column (1.6 × 2.5 cm) equilibrated with buffer C. The elution was carried out with a linear gradient of 5 column volumes from 0.07 to 1.0 M KCl in buffer C (Fig. 1A). The chromatographic profile shows a major peak eluting at ∼0.24 M KCl. On the other hand, the maximum helicase activity was measured for Fractions 16, 17 and 18 that immediately follow this peak. SDS–PAGE stained with Coomassie blue indicates that a protein of a molecular weight of ~92 kDa is eluted at ∼0.24 M KCl, whereas no bands are visible in the most active fractions (Fig. 1B). Our previous work had brought us to erroneously identify HDH V with the protein of Mr ∼92 kDa (16). In the present work, after ssDNA purification, it was clear that the helicase activity is not associated with the 92 kDa protein, but with another molecule present in a very low concentration and eluting from the ssDNA column at ∼0.3 M NaCl. Fraction 17 was concentrated 10-fold by Speed Vac and, only after loading this concentrated material on a silver-stained gel, was a protein of molecular weight ∼66 kDa visible (Fig. 1C). The same band was also present in Fractions 16 and 18. These three fractions containing pure HDH V were pooled and used for further experiments (Fraction VI, 91 500 U). After all the purification steps, <0.1 mg of pure protein was isolated. Photoaffinity cross-linking of Fraction VI with [α-32P]ATP also showed a single radioactive band at ~66 kDa (Fig. 2) confirming the fact that this protein is able to bind ATP. The native molecular weight of HDH V was determined by glycerol gradient as described previously (18). This experiment demonstrates that HDH V has a native Mr of ∼66 kDa with a sedimentation coefficient of 5 ± 0.5 (Fig. 3). Titration of HDH V under optimal assay conditions showed a maximum value of unwinding of 95% in 30 min with 200 pM (130 pg) of enzyme (Fig. 4A). The sigmoidal shape of the titration curve is indicative of a cooperative behavior suggesting that more than one molecule of HDH V could be involved in DNA unwinding, as seen in the case of other helicases (1). On the other hand, kinetic measurements carried out in the presence of 150 pM (100 pg) of enzyme showed that the unwinding rate was linear up to 5 min and deviated from linearity with longer incubation times (Fig. 4B).
Table 1. Purification of HDH V .
| Fraction | Step | Volume (ml) | Protein (mg) | DNA helicase activity | |
| |
|
|
|
Total (U) |
Specific (U/mg) |
| Nuclear extract | 750 | 3800 | N.D. | ||
| I | Ammonium sulfate precipitate | 550 | 2100 | N.D. | |
| II | Bio-Rex 70 | 250 | 90 | N.D. | |
| III | Ammonium sulfate precipitate | 60 | 50 | N.D. | |
| IV | Heparin–Sepharose | 35 | 6 | 260 000 | 43 000 |
| V | Q-Sepharose | 90 | 1.5 | 192 000 | 128 000 |
| VI | ssDNA Sepharose | 15 | <0.1 | 91 500 | >1 000 000 |
N.D., not determined.
Figure 1.
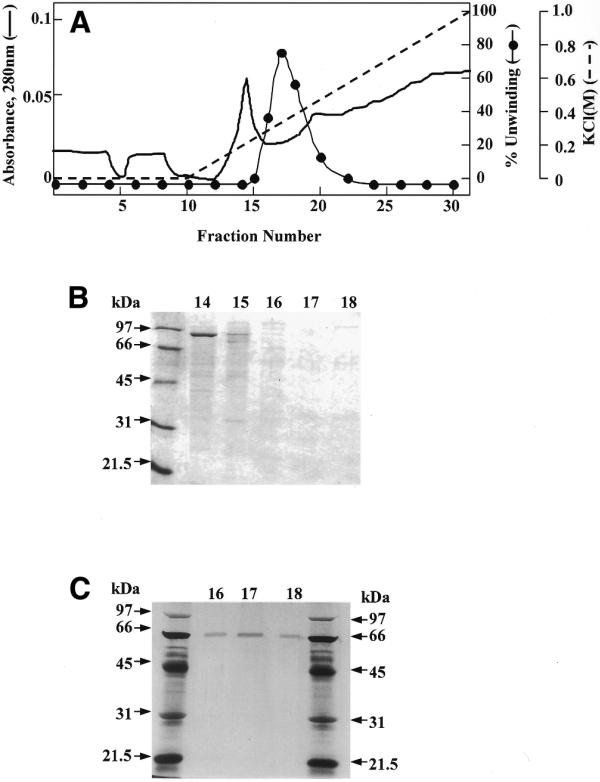
(A) ssDNA–cellulose purification of HDH V. The detailed description of the chromatographic procedure is given in the text. (B) SDS–PAGE stained by Coomassie blue of Fractions 14–18. (C) SDS–PAGE with silver staining of Fractions 16–18 concentrated (10×) before loading.
Figure 2.
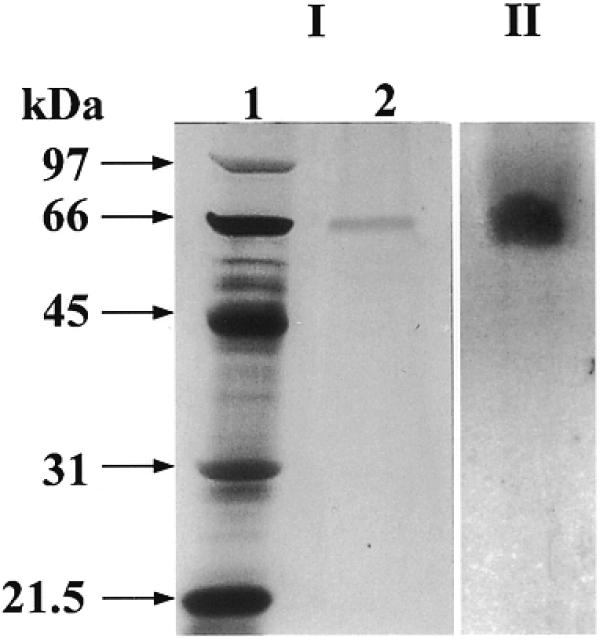
(I) SDS–PAGE with silver staining of HDH V. (II) Autoradiogram of photo-affinity labeled HDH V with [α-32P]ATP.
Figure 3.

Glycerol gradient (15–35%) sedimentation analysis of 100 µl of Fraction 310 000 g for 20 h at 4°C). The distribution of helicase activity and the position of sedimentation and Mr markers are shown. The markers were catalase (250 kDa, 11.3S), aldolase (158 kDa, 7.4S), BSA (66 kDa, 4.3S) and ovalbumin (45 kDa, 3.5S).
In order to identify the sequence of the protein that corresponds to HDH V, the silver-stained band containing HDH V was cut and trypsinized. The products of this enzymatic digestion were separated by RP-HPLC and their sequence determined by tandem mass spectrometry. Seven of the identified peptides have an amino acid sequence that exactly matches different regions of the sequence of the far upstream element-binding protein (FBP; GenBank accession no. U05040.1) (Table 2). The FBP binds to a specific ssDNA region called FUSE and, in this way, stimulates the activity of the promoter that controls the expression of human c-myc. Three variants of this protein are known so far, FBP, FBP2 and FPB3 (19). From the analysis of the sequence of the peptides identified by mass spectrometry, we were able to establish that HDH V corresponds to the first of these variants (FBP) (Fig. 5).
Table 2. Amino acid sequence of the peptides identified by mass spectrometry present in FBP1.
| Position |
Sequence |
| 45–64 | IGGDAGTSLNSNDYGYGGQK |
| 134–146 | IQIAPDSGGLPER |
| 272–284 | IGGNEGIDVPIPR |
| 309–321 | IQFKPDDGTTPER |
| 380–387 | NFIVPTGK |
| 415–425 | NPPPNADPNMK |
| 431–440 | GTPQQIDYAR |
Figure 5.
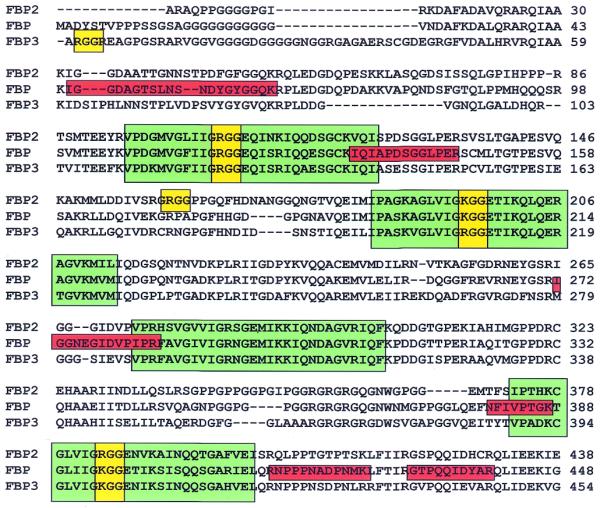
Sequence alignment of the human FBP variants. Four KH repeats recurrent in hnRNP K and involved in DNA binding and destabilization are boxed in green. The RGG or KGG motifs are highlighted in yellow, while the six peptides obtained from the mass spectrometry analysis are boxed in red.
HDH V/FBP unwinds a 17mer annealed to M13 ssDNA, regardless of the presence or absence of mismatched hanging tails at either the 5′end, the 3′end or both (Fig. 6A and C–E). However, HDH V/FBP is unable to unwind the substrate where the duplex region has been increased to 25 bp (Fig. 6B), meaning that this enzyme is a non-processive helicase. All these unwinding reactions occur only in the presence of ATP and Mg2+ as has already been shown in our laboratory (16). The substrates specificity and the unwinding activity are not modified by the addition of replication protein A (RPA) (data not shown). Two linear substrates with a ssDNA tail of 82 amino acids were constructed in order to determine the direction of unwinding (Fig. 6G and H). The results clearly confirm that FBP moves in the 3′ to 5′ direction along the bound strand since it is able to unwind only the 3′ to 5′ direction specific substrate. In analogy with nucleolin and G3BP, two other helicases that were previously purified in our laboratory, FBP is able to unwind both DNA–DNA and RNA–RNA duplexes (Fig. 6L). A blunt-end duplex substrate of 29 bp, obtained by annealing the FUSE coding and non-coding strand (NCS) oligonucleotides, was tested to see whether HDH V/FBP is able to unwind a substrate containing the same sequence that FBP binds in vivo (19) (Fig. 6H). The unwinding assay shows that HDH V/FBP is not able to unwind this substrate, alone or when a tail of 50 pT is added at the 3′end of the coding strand oligonucleotide (Fig. 6I). This result is in agreement with the previous observation that HDH V is not able to unwind duplex regions longer than 17 bp and with previous studies showing that FBP is able to bind only the single-stranded NCS of FUSE, but not the double-stranded FUSE (20).
Figure 6.
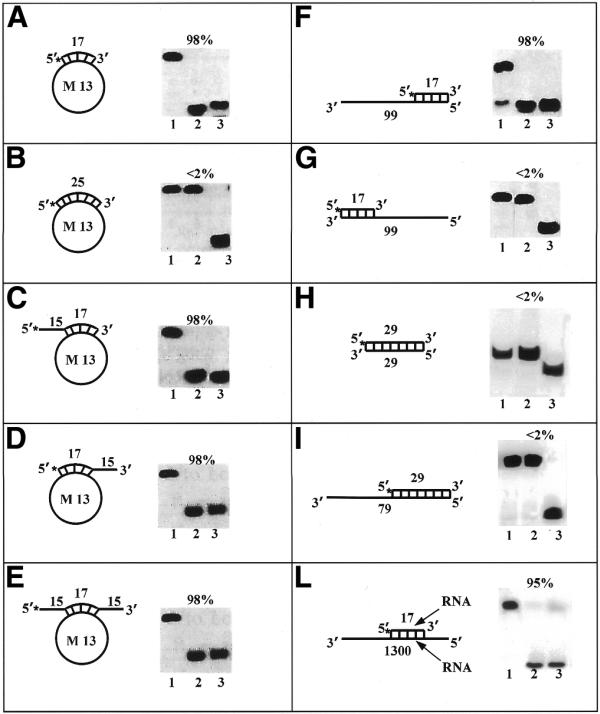
Helicase activity with various substrates. Each panel shows the structure of the substrate used and the autoradiogram of the gel. Asterisks denote the 32P-labeled end. Percent unwinding is shown above each autoradiogram. Lanes 1 and 3 correspond to control reactions without enzyme and heat-denatured substrate, respectively. Lane 2 corresponds to the reaction with pure HDH V (∼0.4 nM). Direction of unwinding is analyzed in (F) and (G). Substrate H corresponds to the double strand FUSE obtained by annealing the FUSE coding and NCS oligonucleotides. Substrate I corresponds to substrate H with the addition of a 50 pT tail at the 3′ end of the coding strand oligonucleotide.
DISCUSSION
According to the amino acid sequence, more then 30 putative DNA helicases containing the canonical DEAD-box or DEXH-box motif have been identified in human cells (3). This motif is involved in the binding of ATP, but it is not specific for helicases alone since it is also present in proteins that simply bind NTPs and do not unwind DNA (9). The other six motifs that characterize E.coli helicases are less conserved among human helicases and the functional role of most of them is still unclear. In addition, some candidate helicases containing the seven motifs fail to possess strand-displacement activity (21,22) and, on the other hand, there are proteins that behave as helicases and do not contain all the seven canonical motifs (3,11,23). For these reasons, the identification of novel HDHs cannot be based solely on the sequence homology that these enzymes may have with their cognate from E.coli. In this view, the systematic purification of HDHs present in the nuclear extract of HeLa cells by following the catalytic activity of the enzyme represents a better strategy for the identification of both canonical and non-canonical helicases. The danger of this approach is that a protein can be erroneously thought to possess ATP-dependent DNA unwinding activity if there is a helicase present as an impurity at very low concentration. For this reason, extreme care has to be taken during the purification procedure and the homogeneity of the final product has to be carefully verified.
In the present work, we describe the purification of a novel human helicase (HDH V) from the nuclear extract of HeLa cells. HDH V had been previously purified in our laboratory and was thought to correspond to a protein of molecular weight ∼92 kDa, clearly visible on an SDS–PAGE gel stained with Coomassie blue (16). On the other hand, after the last step of purification, we were able to establish that the 92 kDa molecule is not responsible for the helicase activity, although it is the major product eluting from the ssDNA–cellulose column. In fact, the helicase activity is associated with human FBP, a protein present in much lower concentration and eluting just after the 92 kDa component. Human FBP is a ssDNA binding protein that binds specifically to the NCS of FUSE and, in this way, stimulates the activity of the promoter that controls the expression of c-myc (19). The c-myc proto-oncogene encodes an important member of the basic helix–loop–helix leucine zipper family of transcription factors and is involved in cell growth, proliferation (24), differentiation and apoptosis (25).
By looking at the sequence of HDH V/FBP, we cannot find any of the seven motifs that characterize several helicases. On the other hand, the central domain contains four KH repeats recurrent in human ribonucleoprotein K (hnRNP K) and involved in DNA binding (26), as well as one RGG box and two KGG domains that are conserved in all the three FBP variants (Fig. 5). This RGG motif is present in many RNA binding proteins and is involved in RNA binding and destabilization (27). The RGG motif is also present in three other human helicases identified in our laboratory, nucleolin (14), G3BP (15) and human ribonucleoprotein D0 (M.Costa, G.Triolo, A.Vindigni, A.Ochem and A.Falaschi manuscript in preparation). Several other RNA, DNA and putative helicases identified by other groups, such as human DDX9 (28), human p68 (29), murine PL10 (30), SKI2 from S.cerevisiae (31) and VASA from Drosophila (32), also contain RGG repeats in their sequence. The fact that all these helicases contain the RGG motifs in their sequence suggests that this motif may be an important structural requirement for their mechanism of RNA or DNA recognition and unwinding.
HDH V/FBP unwinds DNA with a 3′ to 5′ polarity in a strictly ATP-dependent fashion. Its activity is not stimulated by RPA and glycerol gradient experiments suggest that this helicase exists as a monomer, although the sigmoidal curve of enzyme concentration response suggests the possibility of a cooperative interaction when bound to the substrate. Complete unwinding is observable only with short (17 bp) duplex DNA molecules, indicating that HDH V is a non-processive enzyme. Its specific activity (>1 000 000 U/mg) makes this enzyme one of the most active helicases purified so far in our laboratory. In addition, HDH V is the only one, among the helicases purified from HeLa cells, that does not require a ssDNA tail longer than 82 bp for unwinding the substrate. On the other hand, as with nucleolin and G3BP, two other human helicases identified in our laboratory, FBP works both as a DNA and an RNA helicase. The RNA unwinding ability is not a universal property of all DNA helicases and vice versa. Only a few other helicases, such as the SV40T antigen possess both properties (33).
The discovery that FBP is a helicase is not surprising in view of the physiological role of this protein. The FBP interacts specifically with the FUSE NCS and, in this way, stimulates the expression of the c-myc gene. Since FBP does not bind dsDNA, the formation of the complex between FBP and FUSE in vivo will require the unwinding of the DNA helix. One possibility is that the FUSE may be partially unwound in torsionally strained DNA since it is localized in a region of helical instability. On the other hand, it was shown that FBP induces strand separation of short stretches of dsDNA and it was suggested that this property of FBP might favor its binding to the FUSE NCS (20). Our observation that FBP is a helicase and that it requires a ssDNA tail to unwind the substrate fits very well with the following scenario: FBP binds to partially unwound DNA formed in the region of helical instability of FUSE and then further opens the double helix thanks to its ATP-dependent DNA unwinding properties.
Acknowledgments
ACKNOWLEDGEMENTS
The assistance of Ms Maria Elena Lopez in the preparation of HeLa cells is gratefully acknowledged. This work was supported by grant no. 99.00549.PF33 from Consiglio Nazionale delle Ricerche, Roma, Italy.
References
- 1.Lohman T.M. and Bjornson,K.P. (1996) Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- 2.Matson S.W., Bean,D.W. and George,J.W. (1994) DNA helicases: enzymes with essential roles in all aspects of DNA metabolism. Bioessays, 16, 13–22. [DOI] [PubMed] [Google Scholar]
- 3.Ellis N.A. (1997) DNA helicases in inherited human disorders. Curr. Opin. Genet. Dev., 7, 354–363. [DOI] [PubMed] [Google Scholar]
- 4.Weeda G., van Ham,R.C., Vermeulen,W., Bootsma,D., van der Eb,A.J. and Hoeijmakers,J.H. (1990) A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne’s syndrome. Cell, 62, 777–791. [DOI] [PubMed] [Google Scholar]
- 5.Ellis N.A., Groden,J., Ye,T.Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 6.Yu C.E., Oshima,J., Fu,Y.H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Positional cloning of the Werner’s syndrome gene. Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 7.Abdel-Monem M., Durwald,H. and Hoffmann-Berling,H. (1976) Enzymic unwinding of DNA. 2. Chain separation by an ATP-dependent DNA unwinding enzyme. Eur. J. Biochem., 65, 441–449. [DOI] [PubMed] [Google Scholar]
- 8.West S.C. (1996) DNA helicases: new breeds of translocating motors and molecular pumps. Cell, 86, 177–180. [DOI] [PubMed] [Google Scholar]
- 9.Gorbalenya A.E., Koonin,E.V., Donchenko,A.P. and Blinov,V.M. (1989) Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res., 17, 4713–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuteja N., Tuteja,R., Rahman,K., Kang,L.Y. and Falaschi,A. (1990) A DNA helicase from human cells. Nucleic Acids Res., 18, 6785–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuteja N., Tuteja,R., Ochem,A., Taneja,P., Huang,N.W., Simoncsits,A., Susic,S., Rahman,K., Marusic,L., Chen,J. et al. (1994) Human DNA helicase II: a novel DNA unwinding enzyme identified as the Ku autoantigen. EMBO J., 13, 4991–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochem A.E., Skopac,D., Costa,M., Rabilloud,T., Vuillard,L., Simoncsits,A., Giacca,M. and Falaschi,A. (1997) Functional properties of the separate subunits of human DNA helicase II/Ku autoantigen. J. Biol. Chem., 272, 29919–29926. [DOI] [PubMed] [Google Scholar]
- 13.Tuteja N., Rahman,K., Tuteja,R. and Falaschi,A. (1991) DNA helicase IV from HeLa cells. Nucleic Acids Res., 19, 3613–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuteja N., Huang,N.W., Skopac,D., Tuteja,R., Hrvatic,S., Zhang,J., Pongor,S., Joseph,G., Faucher,C., Amalric,F. et al. (1995) Human DNA helicase IV is nucleolin, an RNA helicase modulated by phosphorylation. Gene, 160, 143–148. [DOI] [PubMed] [Google Scholar]
- 15.Costa M., Ochem,A., Staub,A. and Falaschi,A. (1999) Human DNA helicase VIII: a DNA and RNA helicase corresponding to the G3BP protein, an element of the ras transduction pathway. Nucleic Acids Res., 27, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuteja N., Rahman,K., Tuteja,R. and Falaschi,A. (1993) Human DNA helicase V, a novel DNA unwinding enzyme from HeLa cells. Nucleic Acids Res., 21, 2323–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuteja N., Ochem,A., Taneja,P., Tuteja,R., Skopac,D. and Falaschi,A. (1995) Purification and properties of human DNA helicase VI. Nucleic Acids Res., 23, 2457–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L., Liu,J., Collins,I., Sanford,S., O’Connell,B., Benham,C.J. and Levens,D. (2000) Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J., 19, 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bazar L., Meighen,D., Harris,V., Duncan,R., Levens,D. and Avigan,M. (1995) Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J. Biol. Chem., 270, 8241–8248. [DOI] [PubMed] [Google Scholar]
- 21.Selby C.P. and Sancar,A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem., 272, 1885–1890. [DOI] [PubMed] [Google Scholar]
- 22.Cote J., Quinn,J., Workman,J.L. and Peterson,C.L. (1994) Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science, 265, 53–60. [DOI] [PubMed] [Google Scholar]
- 23.Applequist S.E., Selg,M., Raman,C. and Jack,H.M. (1997) Cloning and characterization of HUPF1, a human homolog of the Saccharomyces cerevisiae nonsense mRNA-reducing UPF1 protein. Nucleic Acids Res., 25, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spencer C.A. and Groudine,M. (1991) Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res., 56, 1–48. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y., Glynn,J.M., Guilbert,L.J., Cotter,T.G., Bissonnette,R.P. and Green,D.R. (1992) Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science, 257, 212–214. [DOI] [PubMed] [Google Scholar]
- 26.Tomonaga T. and Levens,D. (1995) Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J. Biol. Chem., 270, 4875–4881. [DOI] [PubMed] [Google Scholar]
- 27.Ghisolfi L., Joseph,G., Amalric,F. and Erard,M. (1992) The glycine-rich domain of nucleolin has an unusual supersecondary structure responsible for its RNA-helix-destabilizing properties. J. Biol. Chem., 267, 2955–2959. [PubMed] [Google Scholar]
- 28.Zhang S. and Grosse,F. (1997) Domain structure of human nuclear DNA helicase II (RNA helicase A). J. Biol. Chem., 272, 11487–11494. [DOI] [PubMed] [Google Scholar]
- 29.Hirling H., Scheffner,M., Restle,T. and Stahl,H. (1989) RNA helicase activity associated with the human p68 protein. Nature, 339, 562–564. [DOI] [PubMed] [Google Scholar]
- 30.Leroy P., Alzari,P., Sassoon,D., Wolgemuth,D. and Fellous,M. (1989) The protein encoded by a murine male germ cell-specific transcript is a putative ATP-dependent RNA helicase. Cell, 57, 549–559. [DOI] [PubMed] [Google Scholar]
- 31.Widner W.R. and Wickner,R.B. (1993) Evidence that the SKI antiviral system of Saccharomyces cerevisiae acts by blocking expression of viral mRNA. Mol. Cell. Biol., 13, 4331–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hay B., Jan,L.Y. and Jan,Y.N. (1988) A protein component of Drosophila polar granules is encoded by vasa and has extensive sequence similarity to ATP-dependent helicases. Cell, 55, 577–587. [DOI] [PubMed] [Google Scholar]
- 33.SenGupta D.J. and Borowiec,J.A. (1992) Strand-specific recognition of a synthetic DNA replication fork by the SV40 large tumor antigen. Science, 256, 1656–1661. [DOI] [PubMed] [Google Scholar]