

Abstract
Neurotransmitter release is mediated by the SNARE proteins synaptobrevin II (sybII, also known as VAMP2), syntaxin, and SNAP-25, generating a force transfer to the membranes and inducing fusion pore formation. However, the molecular mechanism by which this force leads to opening of a fusion pore remains elusive. Here we show that the ability of sybII to support exocytosis is inhibited by addition of one or two residues to the sybII C terminus depending on their energy of transfer from water to the membrane interface, following a Boltzmann distribution. These results suggest that following stimulation, the SNARE complex pulls the C terminus of sybII deeper into the vesicle membrane. We propose that this movement disrupts the vesicular membrane continuity leading to fusion pore formation. In contrast to current models, the experiments suggest that fusion pore formation begins with molecular rearrangements at the intravesicular membrane leaflet and not between the apposed cytoplasmic leaflets.
Keywords: chromaffin cell, patch clamp capacitance measurement, caged calcium, amperometry, electrochemical detector array
The SNARE proteins (1) mediate release of stored secretory products by exocytosis. In neurosecretion, the t-SNAREs syntaxin and SNAP-25 in the plasma membrane bind the v-SNARE synaptobrevin II (sybII, also known as VAMP2), which is anchored to the vesicle membrane by a single transmembrane (TM) domain. Upon stimulation, the SNARE complex is thought to zip up more tightly proceeding in a vectorial manner from the N to the C terminus, toward the TM domains of sybII and syntaxin (2–5), thereby transferring a force to the membranes (6). However, the molecular mechanism by which this force leads to opening of the fusion pore has not been determined (7). Several models have been proposed to explain the mechanism of fusion pore formation. In the lipid-stalk-hemifusion hypothesis, the outer and the inner leaflets of the two membranes merge via formation of a hemifusion intermediate in response to forces exerted by proteins surrounding the fusion site (8). In an alternative proteinaceous fusion pore model, the fusion pore is lined by the TM domain of syntaxin (9) and possibly synaptobrevin (10). However, it is not immediately evident how the hydrophobic transmembrane domains can line an aqueous fusion pore that allows for ion permeation by electrodiffusion (11). When the C-terminal SNARE domain interactions are reduced by mutating or deleting the C terminus of SNAP-25, or when flexible linkers are introduced between the sybII TM domain and its SNARE domain, the rate of exocytosis is reduced (12–16) and the flux of transmitter through the early fusion pore is decreased (16, 17), consistent with a structural change in the fusion pore. In an attempt to interpret these findings, a proteolipidic fusion pore model has been proposed, in which the fusion pore is formed by a molecular complex of both lipids and SNARE proteins (17). However, even this model does not explain the molecular mechanism by which the N-to C-terminal zipping of the SNARE complex leads to the formation of a fusion pore, a step that must somehow disrupt membrane continuity.
Results
Addition of Polar Amino Acids to the Sybii C Terminus Inhibits Fusion.
To determine a possible role of the sybII TM domain in fusion pore formation, sybII constructs were generated in which two charged residues (lysine or glutamate) were added to the C terminus of sybII. To determine whether these C-terminal mutants are robustly expressed and localize to secretory vesicles, synaptobrevin constructs were generated that carried a GFP tag at their N terminus. Wild-type GFP-sybII, GFP-sybII-KK, and GFP-sybII-EE were expressed in mouse chromaffin cells and imaged by total internal reflection fluorescence (TIRF) microscopy (Fig. 1 A–C). Although a considerable fraction of the sybII constructs is localized in the plasma membrane as expected (18) and generates diffuse fluorescence, punctate fluorescence is clearly visible, indicating vesicular localization. Further evidence for vesicular localization comes from observation of vesicular movement (Movies S1–S3).
Fig. 1.

Vesicular localization of GFP-sybII, GFP-sybII-KK, and GFP-sybII-EE in mouse chromaffin cells. (A–C) Simultaneous ECD array amperometry and TIRF microscopy imaging of wild-type cells expressing GFP-SybII (A), GFP-SybII-KK (B), and GFP-SybII-EE (C). Scale bars are 2 μm. Colors of amperometric traces (top panels) correspond to the respective electrodes as indicated by colored dots in images below. The traces are displayed with an offset for clarity. Frame acquisition timing of the image sequences is indicated below the amperometric traces. In a GFP-SybII cell (A), two amperometric events detected by multiple electrodes of the ECD array are associated with loss of fluorescence puncta between frames 539–540 (red arrow) and 541–542 (blue arrow). The area in the yellow rectangle is enlarged in the bottom panel. In a GFP-sybII-KK cell (B) and a GFP-sybII-EE cell (C) amperometric events (Top) are not associated with loss of fluorescence puncta in TIRF images (Bottom). (D) Overexpression of SybII-EE or SybII-KK in wild-type cells reduces the exocytotic response compared to cells overexpressing wild-type sybII. (E) Normalization of capacitance increase 1.2 s after the flash shows no obvious change in exoycotic burst kinetics in SybII-EE and SybII-KK overexpressing cells.
To determine if the vesicles carrying these constructs are fusion competent, simultaneous TIRF microscopy and recording of catecholamine release from the cell membrane in contact with the coverslip was performed using microfabricated electrochemical detector arrays (19). This method allows determination of time and location of a fusion event by amperometric recordings with multiple electrodes. It can then be determined if a fusion event is accompanied by loss of a fluorescent granule in the corresponding region of the plasma membrane. Fig. 1A shows two events where loss of a fluorescent vesicle carrying a construct with the unmodified sybII TM domain was correlated with a catecholamine release event from the same area where the vesicle disappeared. This correlation suggests that such an event likely reflects exocytosis of the marked fluorescent granule and that a granule carrying sybII fluorescently tagged with GFP at its N terminus is fusion competent. In contrast, no such correlated events were observed for GFP-sybII-KK or GFP-sybII-EE fluorescence puncta, suggesting that the modification of the sybII C terminus by addition of these charged residues renders the vesicles fusion incompetent. However, those cells did show quantal release events (Fig. 1 B and C), presumably from vesicles carrying mainly wild-type sybII with undetectable amounts of GFP-sybII-KK or GFP-sybII-EE. These wild-type sybII granules were presumably generated in the cell before the GFP construct was expressed. In cells expressing the GFP-tagged wild-type sybII, only 5 out of 124 events that showed amperometric signals in at least two electrodes and had thus likely occurred in the visible space between the electrodes were accompanied by loss of a fluorescent granule at the corresponding position. However, because even in these cells most release events come from unlabeled vesicles carrying background wild-type sybII, the significance of the lack of release events from GFP-sybII-KK (0 out of 54, p = 0.32) and GFP-sybII-EE (0 out of 121, p = 0.06) granules is uncertain.
To better quantify the inhibition of exocytosis, chromaffin cells overexpressing a sybII construct and identified by GFP fluorescence were stimulated by flash photolysis of caged calcium (NP/o-nitrophenyl-EGTA) (20), and exocytosis was monitored by whole-cell capacitance measurements (21), whereas the associated transmitter release was monitored by carbon fiber amperometry (22). The exocytotic response in cells overexpressing WT sybII consists of an exocytotic burst on the millisecond time scale followed by a sustained phase on a time scale of seconds (Fig. 1D, black trace), which is indistinguishable from the response of noninfected wild-type chromaffin cells. When sybII-EE or sybII-KK was expressed in wild-type chromaffin cells and exocytosis stimulated by photorelease of caged calcium (Fig. 1D, green and red traces), the burst amplitude and the sustained phase were moderately reduced. Expression of sybII-EE reduced the burst amplitude and the sustained phase by 35% (p = 0.05) and 27% (p = 0.07), respectively, indicating that the inhibition is likely significant and providing further evidence that the mutant protein is correctly sorted to secretory vesicles. For sybII-KK the recordings were not paired with control cells overexpressing wild-type sybII such that the significance of the sybII-KK inhibition in wild-type cells is uncertain. With both constructs the exocytotic burst kinetics was unchanged (Fig. 1E), consistent with the conclusion that the population of vesicles that carry the sybII-EE or sybII-KK contribute little, if any, to the exocytosis response.
Demonstration of colocalization of the sybII constructs with chromaffin granule markers is difficult because it is well known that virally expressed sybII constructs are also abundant on the plasma membrane and elsewhere in the cell (5). Immunolabeling of chromogranin in fixed mouse embryonal chromaffin cells expressing GFP-sybII, GFP-sybII-EE, or GFP-sybII-KK showed rather clear granular localization of chromogranin, but GFP fluorescence was generally more widely distributed, as expected. Nevertheless, in about half of the cells vesicular colocalization of the sybII constructs with chromgranin was clearly evident (Fig. 2 A–C), further supporting the notion that addition of two lysine or glutamate residues to the sybII C terminus does not prevent the vesicular localization of the construct.
Fig. 2.

Vesicular localization of sybII constructs. Mouse embryonal chromaffin cells expressing GFP-sybII (A), GFP-sybII-EE (B), and GFP-sybII-KK (C) were fixed, immunolabeled for chromogranin, and imaged using TIRF microscopy. Bright chromogranin puncta (red, Middle) also appear as bright puncta in the GFP images (green, Left) as indicated by yellow color in the overlays (Right). Expression of GFP-sybII (D), GFP-sybII-EE (E), and GFP-sybII-KK (F) in sybII/ceb DKO embryonal mouse chromaffin cells also reveals vesicular localization of all three constructs in the absence of wild-type sybII.
To eliminate the contribution of wild-type sybII to the exocytotic response, experiments were performed using double knock-out (DKO) embryonic mouse chromaffin cells deficient in sybII and cellubrevin (ceb, also known as VAMP3) (23–25). The use of DKO embryonal mouse chromaffin cells is necessary because sybII KO chromaffin cells show rather normal exocytosis, which is supported by endogenous cellubrevin (24). As in WT chromaffin cells, robust expression and vesicular localization of GFP-sybII, GFP-sybII-KK, and GFP-sybII-EE are clearly evident in DKO embryonic chromaffin cells by TIRF microscopy (Fig. 2 D–F).
To assess what properties determine the ability of the C-terminally modified constructs to support vesicle fusion, several additional constructs were generated where one or two polar or nonpolar residues were added at the C terminus of the protein (Fig. 3A) without the N terminal GFP tag. These constructs were expressed in DKO embryonal mouse chromaffin cells together with coexpressed GFP, and exocytosis was stimulated by flash photolysis of NP-EGTA. The amplitude and kinetics of exocytosis were quantified by capacitance measurements and compared to those recorded in cells in which wild-type sybII was expressed using the same viral expression method. The additional constructs were also expressed at very similar levels as shown by quantitative immunofluorescence analysis (Fig. S1).
Fig. 3.

Addition of charged amino acids to the C terminus of sybII inhibits exocytosis. (A) SybII C-terminal mutations. X indicates added residues: single lysine (K) or glutamic acid (E), or double lysine (KK), glutamic acid (EE), glutamine (QQ), histidine (HH), valine (VV), or glycine (GG). (B) Exocytosis was stimulated with intracellular release of caged calcium (top panel) using UV light at t = 0.5 s and monitored by patch clamp capacitance measurement (middle panel) and carbon fiber amperometry (lower panel). Exocytosis is virtually absent in DKO cells (blue traces) but is rescued by viral expression of wild-type sybII (black traces). Exocytosis is not rescued by expression of sybII-KK (red traces). Traces are averages from 8 to 17 cells. The exocytotic burst amplitude (C) was taken as the capacitance increase at 0.5 s after stimulation and the amplitude of the sustained phase (D) as the capacitance increase over the next 5 s. sybII-KK (n = 8), SybII-K (n = 17), SybII-EE (n = 12), SybII-E (n = 17), and SybII-HH (n = 11). (∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 student’s t test).
As previously reported (16, 24), DKO chromaffin cells did not support exocytosis (Fig. 3B blue traces), whereas viral expression of wild-type sybII restored a biphasic increase in capacitance (Fig. 3B, black traces), indistinguishable from that observed in embryonal chromaffin cells from normal wild-type mice. In contrast, when sybII-KK was expressed (Fig. 3B, red traces), the response was indistinguishable from DKO (Fig. 3 C and D). Although the bar diagrams indicate less exocytosis for sybII-KK than for DKO cells, the difference is not significant. This mutant protein failed to support vesicle fusion. Even addition of only a single lysine (sybII-K) reduced the exocytotic burst as well as the sustained phase by ∼80%. SybII-EE expressing cells showed also only a small response that was strongly reduced compared to wild-type sybII (Fig. 3 B, green traces). Similar reductions were observed with sybII-E, and sybII-HH) (Fig. 3 C and D).
Inhibition of Fusion Is Related to Amino Acid Polarity.
To assess whether the inhibition of exocytosis is specifically due to the presence of charged residues at the C terminus of sybII, we tested the uncharged amino acids glutamine (sybII-QQ), glycine (sybII-GG), and valine (sybII-VV) (Fig. 4). Although sybII-QQ expressing cells showed again only a very small exocytotic response, the response of cells expressing sybII-GG was not significantly different from control cells expressing wild-type sybII. The response of cells expressing sybII-VV was intermediate, showing a moderate reduction of 26% for the burst and of 40% (p = 0.07) for the sustained phase (Fig. 4B). When the exocytotic burst responses were normalized (Fig. 4C), they showed slower kinetics for sybII-VV and more so for sybII-QQ. The delay between the flash-induced calcium release and the onset of the exocytotic burst also increased from 4.3 ms for wild-type sybII to 6.7, 8.5, and 19.6 ms for sybII-GG, -VV, and -QQ, respectively (Fig. 4D). The kinetics of exocytosis is thus slowed down in parallel with the reduction in exocytosis amplitude.
Fig. 4.

Polarity of C-terminal additions determines fusion competence. (A) Average biphasic capacitance responses (middle trace) and associated catecholamine secretion (lower panel) after photorelease of caged Ca2+ (upper trace). Wild-type sybII (black, n = 14), sybII-GG (green, n = 20), sybII-VV (brown, n = 17), sybII-QQ (red, n = 12), and DKO (blue, n = 20). (B) The amplitudes of the exocytotic burst and of the sustained phase are reduced in parallel. (C) Normalization of the capacitance amplitude at 0.7 s after the flash reveals slower kinetics of exocytosis associated with reduced amplitude. (D) SybII-GG and sybII-VV show increase in delay between flash and onset of exocytosis. Smooth curves (left panel) show the initial parts of multiexponential fits. Statistical analysis of delays (right panel) determined by backextrapolation of multiexponential fits to individual responses indicate that only the increase in sybII-QQ is significant. (∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 student’s t test).
Discussion
Release of neurotransmitters from synaptic vesicles as well as exocytosis of chromaffin granules is mediated by the neuronal SNAREs sybII, syntaxin-1, and SNAP-25. The t-SNARE syntaxin and the v-SNARE synaptobrevin are anchored by a single TM domain to the plasma membrane and the vesicle membrane, respectively. The SNARE domains of these proteins can form a four-stranded helical bundle as revealed by the crystal structure (26). It has been suggested that sybII of vesicles in the readily releasable pool is bound tightly to the t-SNAREs only in the N-terminal part of the SNARE complex, zipping up fully only after stimulation (2–5). Complexin interacts with the C-terminal part of the SNARE complex and thereby activates and clamps SNARE complexes such that Ca2+ can trigger Synaptotagmin to reverse the clamping function (6). In the zipper model, this will allow N- to C-terminal zipping, which results in a force transfer to the membranes. But how does the force, which is exerted by this machinery, lead to fusion pore formation?
We have shown here that addition of polar residues to the intravesicular C terminus of the sybII TM domain inhibits fusion dramatically. The inhibition is not due to different expression levels or mistargeting (Fig. 2 and Fig. S1). This inhibition of fusion by addition of just one or two residues at the sybII C terminus is an unexpected observation, because sybII has been modified by even adding a GFP variant to its C terminus in the synapto-pHluorin construct (27) that is widely used to investigate exocytosis and recycling of secretory vesicles. However, in synapto-pHluorin a flexible linker with the sequence SGGSGGTGG was inserted between the sybII C terminus and GFP. This linker is rich in glycine residues, which did not interfere with sybII function (see Fig. 4, sybII-GG construct). It thus appears that the nature of the residues near the intravesicular membrane–water interface is of particular importance. The inhibition of fusion by addition of polar residues suggests that fusion may be the consequence of a movement of the C terminus toward the hydrophobic core of the vesicle membrane. We therefore compared the ability of the different sybII mutants to support exocytosis to the free energies of transfer from water to the membrane–water interface (ΔGwif) (28) for the residues added to the sybII C terminus.
Inhibition of Fusion Depends on Free Energy of Transfer from Water to the Membrane–Water Interface.
The transfer energies for the residues used here are given in Table S1. It has been shown that the transfer energies for multiple residues are additive (29), and we therefore assigned the value and its error given in ref. 28 for addition of a single residue and twice that value and error for the addition of two residues. For glutamate and histidine ΔGwif is uncertain as indicated by the large horizontal error bars because it strongly depends on the protonation state of these residues near the interfacial layer inside the acidic vesicle (for details, see Assignment of Free Energies of Transfer from Water to Membrane Interface in Materials and Methods). The relation between ΔGwif for the added residues and the amplitude of the exocytotic burst is shown in Fig. 5A. The ability to support exocytosis decreases with increased ΔGwif. The dashed line shows a fit of the function ΔCburst = A· exp(ΔGwif/E) + B yielding E = 0.66 ± 0.16 kcal/mol, consistent with a Boltzmann distribution (Fig. 5A, solid line) with E ∼ RT = 0.59 kcal/mol for T = 295 K. This result indicates that the ability of the different mutants to support the exocytotic burst correlates directly with the energy required to move the added residues from water into the interfacial layer. The linker in the synapto-pHluorin construct has a SGGSGGTGG sequence (27) and thus a serine is added next to the C terminus of sybII. Serine has a water–membrane interface transfer energy that is similar to that of two valines (28), which produce only limited inhibition. This suggests that only the residues next to the sybII C terminus are relevant for sybII function and explains why synapto-pHluorin is not a strong inhibitor of fusion. The synaptobrevin family of proteins includes several members with an extended intralumenal tail. Notably, Leech synaptobrevin (30) carries a lysine in the critical position where addition of a single lysine resulted in strong inhibition of sybII function (Fig. 3 C and D). However, this residue is immediately followed by a compensatory phenylalanine residue such that the net ΔGwif for the two residues (KF) combined is very small and presumably slightly negative (-0.14 ± 0.16 kcal/mol) (28).
Fig. 5.

(A) Dependence of exocytotic burst amplitude on energy of transfer from water to the membrane–water interface (ΔGwif) for the residues added to the sybII C terminus. The single exponential fit of the function ΔCburst = A exp(-ΔGwif/E) + B yielded the fit parameters A = 88 ± 6 fF, B = 4 ± 6 fF, and E = 0.66 ± 0.16 kcal/mol. The solid lines indicate a theoretical Boltzmann distribution fitting only amplitude A and baseline B of the exponential. The data points for sybII-HH and sybII-EE were excluded from the fit (see text). (B–E) Schematic model of fusion pore opening, for clarity only synaptobrevin and syntaxin are shown. Prefusion arrangement of N-terminally zipped SNARE complex (B). C-terminal zipping pulls C termini of sybII and possibly syntaxin deeper into the membrane disrupting membrane continuity (C) leading to fusion pore formation (D) and pairing up of the sybII and syntaxin TM domains (E).
The Mechanism of Fusion Pore Formation.
Although it is possible that added residues with high ΔGwif may change the position of the sybII TM domain in the membrane and could thereby compromise the ability of sybII to participate in SNARE complex formation, it appears more likely that fusion pore formation is initiated by pulling the sybII TM domain into the vesicle membrane, a movement that would translocate the C-terminally added residues from the water phase to the membrane interface. Such a mechanism is also supported by the finding that a reduction in exocytosis amplitude is accompanied by slowing of the kinetics (Fig. 4 C and D), as expected for an increased height of the energy barrier. Consistent with this model is also the finding that introduction of flexible linkers between the sybII SNARE domain and its TM domain reduces exocytosis in parallel with the length of the linkers (16).
Properties of the early fusion pore are reflected in amperometric foot signals. The sybII-VV construct was chosen for comparison with wild-type sybII because it produces limited inhibition allowing better statistical analysis of amperometric foot signals. Embryonal DKO chromaffin cells expressing sybII-VV showed no change in foot current amplitude indicating that this mutation does not affect the fusion pore structure. However, a small but significant decrease in foot duration by about 20% was observed for sybII-VV (5.27 ± 0.46 ms) compared to wild-type sybII (6.85 ± 0.56 ms), which points to a small change in the energetics of fusion pore expansion. This mutant has a slightly higher energy in the interfacial layer (∼0.14 kcal/mol for two valines), and it is thus expected to contribute some additional distortion of the vesicle membrane, thereby lowering the activation energy for fusion pore expansion. The reduction in fusion pore lifetime is thus also consistent with the model proposed here. Interestingly, using a simple activation energy model, the ∼20% reduction in fusion pore lifetime corresponds to a change in activation energy by ∼0.13 kcal/mol, which equals the transfer energy from water to the membrane interface of the two valine residues.
The C terminus of the wild-type sybII TM domain is located in the interfacial layer of the vesicle membrane (31) (Fig. 5B). The aromatic residues Y113 and F114 with their propensity for interfacial localization (28) are suitably positioned and are conserved in vertebrate sybII. Ceb, which also supports chromaffin granule exocytosis, has a tryptophan and a nonaromatic residue in the corresponding position, which yields a very similar energy minimum for interfacial localization. When the TM domain is pulled deeper into the membrane, the polar residues S115 and T116 will be pulled into the interfacial or hydrophobic layers (Fig. 5C). In some species (human, horse, and bovine) T116 is substituted by S116, but Thr and Ser have identical ΔGwif. In ceb, there is also one or more polar residues at the C terminus conserving the essential properties. As a consequence of this movement of the sybII C terminus into the hydrophobic membrane domain, the stability of the vesicle membrane should be significantly reduced followed by structural rearrangements leading to disruption of membrane continuity and formation of a proteolipidic fusion pore (Fig. 5 D and E). Consistent with this model is the recently reported X-ray structure of the neuronal SNARE complex, consisting of syntaxin 1A, SNAP-25, and sybII, including the carboxy-terminal linkers and transmembrane regions (32). In this postfusion structure (33), the C termini of syntaxin and sybII appear to be pulled toward the hydrophobic core of the membrane. In the zipper model of SNARE-mediated fusion, the movement of the sybII C terminus as a prerequisite for fusion pore formation is expected to occur as a consequence of N- to C-terminal zipping of the SNARE domains. A movement of the sybII C terminus is, however, also consistent with the recently proposed scissors mechanism involving a change of the sybII TM domain tilt with respect to the membrane normal (34). Such a tilt would likely be associated with a movement of the C terminus relative to the membrane interface.
The possibility of a proteolipidic fusion pore was originally proposed by Zimmerberg and coworkers (8), but the identity and function of the proteins participating in such a mechanism were unknown. In the model proposed here, the genesis of the pore formation starts at the inner leaflet of the vesicle membrane and is induced by movement of the C-terminal end of sybII. This movement is activated in response to calcium stimulation via synaptotagmin that leads to zippering up the SNARE domains toward the C terminus. It is possible that the TM domain of syntaxin plays a complementary role in disrupting the continuity of the plasma membrane as depicted in Fig. 5D of the model. However, we have not performed experiments with corresponding syntaxin mutants to support or discard such a role.
It has been suggested that synaptotagmin (35) and Doc2 (36) may aid fusion pore formation via insertion of their C2 domains into the target membrane, which produces an increase of the cytoplasmic leaflet’s area and thereby induces local membrane curvature (37). Pulling the sybII C terminus deeper into the membrane will induce complementary local curvature in the vesicle membrane as it reduces the area of the vesicle membrane’s inner leaflet. It appears that lipid protein interactions that favor appropriate membrane curvature are facilitating fusion. The fusion pore formation may, however, not necessarily proceed via highly curved lipid domains devoid of protein but may as well involve rearrangements within a proteolipid complex. In any case, the energy of C-terminal zipping of the SNARE complex appears to be remarkably balanced with respect to overcoming the energy barrier for fusion. It is just sufficient for the required sybII TM domain movement as the additional energy of a few kBT required for translocation of the added residues significantly inhibits fusion competence.
Materials and Methods
Cells and Viral Expression.
SybII/ceb DKO mouse E17-E18 embryos were obtained and chromaffin cells isolated and plated as described (24, 38). Recombinant Semliki Forest Virus was used to introduce sybII mutants into the cells after day 2 or 4 in culture (24, 39). Full-length rat sybII plasmid was a kind gift from Thomas Südhof (Stanford University, Palo Alto, CA). The sybII TM domain …WWKNLKMMIILGVICAIILIIIIVYFST was extended at the C-terminal end by addition of K, E, KK, EE, QQ, HH, VV, or GG amino acids. All constructs were confirmed by DNA sequencing.
SybII Localization.
To demonstrate sybII-chromogranin colocalization (Fig. 2 A–C), chromaffin cells from wild-type mouse embryos expressing GFP-sybII, GFP-sybII-EE, or GFP-sybII-KK were cultured in glass bottom dishes (•: 35/12 mm, glass thickness: 0.17 mm. Warner Instruments). Six hours after transfection, cells were fixed in 4% paraformaldehyde (PFA) solution for 30 min, permeabilized in 0.1%(vol/vol) triton-100X for 10 min and blocked with 6% bovine albumin serum (Sigma-Aldrich) for 1 h. After removing the blocking buffer, the cells were incubated with the primary chromogranin A antibody (1∶500 dilutions, Abcam, Cambridge) for 2 h, washed 5 times with PBS, incubated with Alexa 646-labeled secondary antibodies (Invitrogen, 1∶200 dilutions) for 1 h, washed and mounted or kept in PBS overnight at 4 °C. Fluorescence imaging was performed with a Nikon Ti-E/B microscope equipped with CFI APO 100x/1.45 oil TIRF objective. The GFP and Alexa 546 signals were excited with laser wavelengths of 488 nm and 561 nm, respectively, and images captured through appropriate emission filters (GFP, 535 nm; Alexa, 580 nm) by the camera (iXon+EMCCD, Andor). Alignments of the image pairs was performed manually using Image J.
Whole-Cell Capacitance Measurement.
Whole-cell patch clamp experiments and flash photolysis were performed as in ref. 5 (see SI Text).
Single-Event Amperometry.
For measurement of amperometric foot signals cells were infused in the patch clamp whole-cell configuration with a solution containing (in mM) 100 Cs-glutamate, 0.3 Na-GTP, 2 Mg-ATP, 2.5 CaCl2, 0.4 mM fura-4F, 0.4 mM furaptra, 20 DPTA, and 32 Hepes, pH 7.2. Carbon fibers were cut after each recording to reduce variability. The current traces were acquired with an EPC7 amplifier and filtered at 3 KHz. Data for the control and test samples were collected on the same day. Amperometric spikes were analyzed using Igor Pro software (WaveMetrics). If necessary, line frequency noise was removed by subtracting a sine wave fit from the recordings. The statistical analysis was performed on spikes > 10 pA with foot signal duration > 2 ms.
Electrochemical Detector Arrays.
Microfabricated platinum electrode arrays were used to amperometrically detect the release of catecholamines from the transfected chromaffin cells. Each electrochemical detector (ECD) array consists of a set of four planar Pt electrodes fabricated on a glass coverslip. The four electrodes of an ECD reside at the corners of a square sized such that a cell can be placed atop the array and viewed through the glass coverslip while amperometric currents are recorded from all four electrodes simultaneously. The ECD arrays were fabricated as described (19, 40) except for an improved insulation layer, which was created by chemical vapor deposition of approximately 300 nm of SiO2 onto the electrodes (IPE 1000 PECVD), and subsequent etching of the SiO2 layer via reactive ion etching (PlasmaLab 80Plus, Oxford Instruments) to expose the active areas of the electrodes and contact pads.
Live-Cell TIRF Microscopy and ECD Amperometry.
GFP was linked to the N terminus of sybII, sybII-KK, or sybII-EE and the constructs expressed in wild-type mouse chromaffin cells or in embryonal DKO chromaffin cells as described above. Cells expressing the construct were identified by characteristic GFP fluorescence with a concentration of the label in the endoplasmic reticulum and inspected under bright field illumination to ensure healthy appearance. TIRF microscopy was performed using a Zeiss Axiovert 135 TV microscope modified for TIRF microscopy as described (41) equipped with a plan-fluar 1.45 NA 100× oil objective. Illumination was provided by a 100-W mercury arc lamp with 480/40-nm excitation filter, 515-nm dichroic, and 520-nm long pass emission filter (Chroma Technology). For each cell, a 2,000 frame image sequence was collected using an EMCCD camera (Andor iXon) and its accompanying software. The exposure time per frame was 100–150 ms, with an interframe time interval of 1.8 ms. For simultaneous imaging and amperometry, cells were picked up and positioned on the ECD arrays using a micropipette. Amperometric currents recorded with the ECD array during fluorescence imaging (Fig. 1 A–C) were low pass filtered at 500 Hz, acquired with 5-kHz sampling rate, and analyzed as described (19). For analysis, a 10-pA minimum current peak height was used as threshold, with a minimum of four analyzable events per cell. The camera output signal was recorded in order to synchronize the timing of the fluorescence image frames with electrochemical signals.
Assignment of Free Energies of Transfer from Water to Membrane Interface.
The whole residue transfer energies from water to the membrane interface of the residues added to the C terminus were taken from ref. 28 (Table S1). For Lys the value in Table S1 was used. The double additions LysLys, GlnGln, GlyGly, and ValVal were assigned twice the values in Table S1. Assuming a pK of 4.3 for glutamic acid and an intravesicular pH of 5.5, one would expect 94% in Glu- and 6% in the Glu0 state. We thus assigned a transfer energy for single Glu addition:
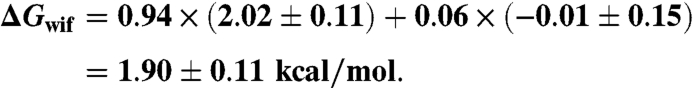 |
For the GluGlu addition, ΔGwif could be twice that value (ΔGwif = 3.8 ± 0.22). However, it appears possible that a negative charge on one Glu will significantly change the pK of the second Glu, which may thus lead to ∼50% protonation between the two added Glu residues corresponding to ΔGwif = 2.01 ± 0.26. For GluGlu we thus estimate a range ΔGwif = 1.75 - 4.0 or 2.9 ± 1.1 kcal/mol.
Assuming a pK of 6.0 for histidine one would expect 76% in His+ and 24% in His0 state. We thus assigned a transfer energy for a single His addition:
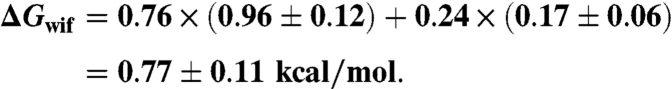 |
For the HisHis addition, ΔGwif could be twice that value (ΔGwif = 1.54 ± 0.22). However, protonation of the first His may shift the pK of the second His such that there could again be an average ∼50% protonation giving ΔGwif = 1.13 ± 0.18. For HisHis we thus estimate a range ΔGwif = 0.95 - 1.76 = 1.35 ± 0.41. Because of these uncertainties the data points for sybII-HH and sybII-EE were not included in the fits of Fig. 5A.
Supplementary Material
Acknowledgments.
We thank Dirk Reuter, Joan Lenz, and Ina Herfort for expert technical assistance and Stephan Weiss for the fusion pore artwork. We are grateful to Dr. Erwin Neher for critical reading of the manuscript. This work has been supported by the National Institutes of Health Grants R01NS38200, R01GM085808, and T32GM008267, the Nanobiotechnology Center (a National Science Foundation Science and Technology Center, Agreement ECS-9876771), the Deutsche Forschungsgemeinschaft (SFB523-B16 to J.B.S. and SFB530-C10 to D.B.), the Lundbeck Foundation (Junior Group Leader Fellowship and Center for Biomembranes in Nanomedicine to J.B.S.), and the EU 7th Framework Programme (Grant HEALTH-F2-2009-242167, “SynSys” project to J.B.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006727107/-/DCSupplemental.
References
- 1.Söllner T, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–323. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 2.Chen YA, Scales SJ, Scheller RH. Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron. 2001;30:161–170. doi: 10.1016/s0896-6273(01)00270-7. [DOI] [PubMed] [Google Scholar]
- 3.Pobbati AV, Stein A, Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- 4.Sorensen JB, et al. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sorensen JB. Synaptobrevin N-terminally bound to syntaxin-SNAP-25 defines the primed vesicle state in regulated exocytosis. J Cell Biol. 2010;188:401–413. doi: 10.1083/jcb.200907018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rizo J, Dai H. How much can SNAREs flex their muscles? Nat Struct Mol Biol. 2007;14:880–882. doi: 10.1038/nsmb1007-880. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerberg J, Vogel SS, Chernomordik LV. Mechanisms of membrane fusion. Annu Rev Biophys Biomol Struct. 1993;22:433–466. doi: 10.1146/annurev.bb.22.060193.002245. [DOI] [PubMed] [Google Scholar]
- 9.Han X, Wang CT, Bai J, Chapman ER, Jackson MB. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science. 2004;304:289–292. doi: 10.1126/science.1095801. [DOI] [PubMed] [Google Scholar]
- 10.Jackson MB, Chapman ER. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu Rev Biophys Biomol Struct. 2006;35:135–160. doi: 10.1146/annurev.biophys.35.040405.101958. [DOI] [PubMed] [Google Scholar]
- 11.Gong LW, de Toledo GA, Lindau M. Exocytotic catecholamine release is not associated with cation flux through channels in the vesicle membrane but Na+ influx through the fusion pore. Nat Cell Biol. 2007;9:915–922. doi: 10.1038/ncb1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- 13.Wei S, et al. Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J. 2000;19:1279–1289. doi: 10.1093/emboj/19.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Criado M, Gil A, Viniegra S, Gutierrez LM. A single amino acid near the C terminus of the synaptosomeassociated protein of 25 kDa (SNAP-25) is essential for exocytosis in chromaffin cells. Proc Natl Acad Sci USA. 1999;96:7256–7261. doi: 10.1073/pnas.96.13.7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deak F, Shin OH, Kavalali ET, Sudhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kesavan J, Borisovska M, Bruns D. v-SNARE actions during Ca2+-triggered exocytosis. Cell. 2007;131:351–363. doi: 10.1016/j.cell.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 17.Fang Q, et al. The role of the C terminus of the SNARE protein SNAP-25 in fusion pore opening and a model for fusion pore mechanics. Proc Natl Acad Sci USA. 2008;105:15388–15392. doi: 10.1073/pnas.0805377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wienisch M, Klingauf J. Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical. Nat Neurosci. 2006;9:1019–1027. doi: 10.1038/nn1739. [DOI] [PubMed] [Google Scholar]
- 19.Hafez I, et al. Electrochemical imaging of fusion pore openings by electrochemical detector arrays. Proc Natl Acad Sci USA. 2005;102:13879–13884. doi: 10.1073/pnas.0504098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- 21.Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflug Arch Eur J Phy. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- 22.Wightman RM, et al. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci USA. 1991;88:10754–10758. doi: 10.1073/pnas.88.23.10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoch S, et al. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- 24.Borisovska M, et al. v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 2005;24:2114–2126. doi: 10.1038/sj.emboj.7600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang C, et al. VAMP3 null mice display normal constitutive, insulin- and exercise-regulated vesicle trafficking. Mol Cell Biol. 2001;21:1573–1580. doi: 10.1128/MCB.21.5.1573-1580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 24 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 27.Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 28.White SH, Wimley WC. Membrane protein folding and stability: Physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 29.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 30.Bruns D, et al. Inhibition of transmitter release correlates with the proteolytic activity of tetanus toxin and botulinus toxin A in individual cultured synapses of Hirudo medicinalis. J Neurosci. 1997;17:1898–1910. doi: 10.1523/JNEUROSCI.17-06-01898.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowen M, Brunger AT. Conformation of the synaptobrevin transmembrane domain. Proc Natl Acad Sci USA. 2006;103:8378–8383. doi: 10.1073/pnas.0602644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellena JF, et al. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc Natl Acad Sci USA. 2009;106:20306–20311. doi: 10.1073/pnas.0908317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong J, Borbat PP, Freed JH, Shin YK. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci USA. 2009;106:5141–5146. doi: 10.1073/pnas.0813138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynch KL, et al. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell. 2008;19:5093–5103. doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groffen AJ, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMahon HT, Kozlov MM, Martens S. Membrane curvature in synaptic vesicle fusion and beyond. Cell. 2010;140:601–605. doi: 10.1016/j.cell.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Sorensen JB, et al. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- 39.Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- 40.Dias AF, et al. An electrochemical detector array to study cell biology on the nanoscale. Nanotechnology. 2002;13:285–289. [Google Scholar]
- 41.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.