

Abstract
Bacterial populations harbor vast genetic diversity that is continually shaped by abiotic and biotic selective pressures, as well as by neutral processes. Individuals coexisting in the same geographically defined population often have significantly different gene content, but whether this variation is largely adaptive or neutral remains poorly understood. Here we quantify heterogeneity in gene content for two model marine microbes, Prochlorococcus and Pelagibacter, within and between populations in the Atlantic and Pacific Oceans, to begin to understand the selective pressures that are shaping these “population genomes.” We discovered a large fraction of genes that are rare in each population, reflecting continual gene transfer and loss. Despite this high variation within each population, only a few genes significantly differ in abundance between the two biogeochemically distinct environments; nearly all of these are related to phosphorus acquisition and are enriched in the Atlantic relative to the Pacific. Moreover, P-related genes from the two sites form phylogenetically distinct clusters, whereas housekeeping genes do not, consistent with a recent spread of adaptive P-related genes in the Atlantic populations. These findings implicate phosphorus availability as the dominant selective force driving divergence between these populations, and demonstrate the promise of this approach for revealing selective agents in more complex microbial systems.
Keywords: adaptation, biogeography, microbial evolution, natural selection, phosphorus limitation
Perhaps the most surprising lesson we have learned from microbial genome sequences is that closely related isolates of the same species often harbor substantially different gene complements, owing to horizontal gene transfer and gene loss (1, 2). The set of genes unique to a particular strain or subset of strains—the flexible genome—is hypothesized to be responsible for niche-specific adaptations (2, 3). However, in any given isolate, some fraction of the flexible genome might provide no fitness benefit, making it difficult to infer ecology from individual genome sequences. Moreover, snapshots of gene content in some natural populations have revealed high levels of coexisting variation (4–7), even among individuals that appear to experience similar selection pressures, suggesting that much of this variation is neutral. Alternatively, this variation could reflect adaptation by subpopulations specialized for microenvironments (8), or could reflect predation-driven frequency-dependent or diversifying selection (9). Understanding what fraction of gene content variation is in fact acted upon by selection, and what role this variation plays in ecosystem functioning, are central questions in microbial biology (10).
In the absence of selection, genes will be lost from a bacterial genome, owing to a mutational bias that favors deletions (11, 12). Therefore, genes that persist and rise to fixation in a population are inferred to be functional and to enhance organismal fitness. Likewise, genes that are differentially maintained in two populations reveal differential selection pressures acting on the two populations. Based on these principles, quantitative population genomics—in particular, the analysis of gene frequencies and patterns of sequence variation—has the power to highlight salient genetic features amid a background of continual gene transfer and gene erosion (10, 13). Thus, we can begin to quantify the roles of horizontal gene transfer and the flexible genome in microbial adaptation across environments.
Here we apply this approach to advance our understanding of evolutionary dynamics and the selective pressures facing two model microbes, the marine cyanobacterium Prochlorococcus and the heterotroph Pelagibacter (a member of the SAR11 clade; ref. 14), in two distinct oceanic regions. These microbial groups numerically dominate the biogeochemically well-characterized North Atlantic and North Pacific subtropical gyres, hereinafter referred to by the representative long-term study stations at which we sampled, the Bermuda Atlantic Time Series (BATS) station and the Hawaii Ocean Time-Series (HOT) Station ALOHA (15). Both locations are oligotrophic with similar rates of primary production and carbon export, but BATS experiences stronger seasonal mixing and nutrient supply compared with HOT (15). BATS has lower phosphate concentrations (16) but higher fluxes of dust inputs, which bring iron and other metals (17). Thus, the two sites could potentially select for a range of distinct microbial traits. Moreover, gene content at these two sites can be framed in the context of reference genomes (12 for Prochlorococcus and 3 for Pelagibacter).
Specifically, we investigated the degree of heterogeneity in gene content among individuals in marine microbial populations, how much of the flexible genome is being maintained by selection, and what the functions of these adaptive genes tell us about ecosystem-specific selective pressures. Toward this end, we quantitatively compared gene frequencies among Prochlorococcus and Pelagibacter using pyrosequenced community DNA from HOT and BATS. Because Prochlorococcus comprises distinct high- and low-light–adapted clades (18) whose abundance varies with depth (19, 20), we sampled three analogous depths at each site to provide a more representative picture of the entire vertically integrated (meta)population (Table S1). In doing so, we captured similar abundances of high- and low-light–adapted clades at both sites, thereby minimizing the effect of clade structure on our intersite comparison (Table S2).
Results and Discussion
To begin to understand the extent of gene content variability in natural populations, and how much of this variability is likely to be adaptive, we first quantified the occurrence of each Prochlorococcus flexible gene relative to the core genome (21). Genes belonging to the core genome—the 1,221 single-copy genes shared by all 12 sequenced isolates of Prochlorococcus—appear to be shared by nearly all Prochlorococcus cells in these wild populations as well, as evidenced by the single narrow peak in the distribution of relative core gene frequencies (Fig. 1). Note that this distribution for core genes is, by definition, centered around one copy per cell, but the shape of the distribution is free to vary (see Materials and Methods). Compared with the core genome, most flexible genes (i.e., genes found in some, but not all, cultured isolates) are rare; 974 genes at HOT and 784 genes at BATS are estimated to occur at < 0.25 copy per cell (Fig. 1). A preponderance of rare flexible genes even at a given depth (Fig. S1) indicates that light adaptation cannot solely account for the distribution shown in Fig. 1. The majority of these rare flexible genes are likely neutral and transient in the population. In addition, some may have been acquired only recently, and some encode cell surface traits that may be driven by frequency-dependent selection (9). It is also possible that some of these rare flexible genes are adaptive in microenvironments within our samples, although such microenvironments are difficult to define for a planktonic organism. In contrast, about 400 flexible genes are estimated to occur at > 0.65 copy per cell at either site, and thus are “nearly core” in these particular populations (Fig. 1 and Table S3). Most of these widespread flexible genes are prevalent at both HOT and BATS, suggesting that they confer a fitness advantage in both open ocean environments, yet nearly three-quarters of these genes have no known function. One notable exception is the urease pathway; a dozen genes encoding urea transport and metabolism are found at about one copy per cell at both sites, suggesting that urea is an important nutrient source common to both these environments.
Fig. 1.

Distribution of core and flexible genes (21) among Prochlorococcus populations at HOT (A) and at BATS (B). Core genes are defined as present in all 12 sequenced cultured isolates of Prochlorococcus, and the tight single-peaked distribution around the mean implies that they also are core genes and are present in one-to-one stoichiometry in these wild populations. Most flexible genes (those present in some, but not all, genomes of cultured isolates) are rare in these populations, being present in only a small proportion of the cells. The copy number per cell for each gene was estimated as described in Materials and Methods.
To identify selective pressures affecting the HOT and BATS populations differentially, we compared relative gene frequencies between the two sites. Only 29 of 2,854 observed Prochlorococcus genes have significantly different relative frequencies between the two sites (Fig. 2A and Table S4). Nearly all of the 29 differentially maintained genes are enriched at BATS relative to HOT and have functions related to phosphorus (P) acquisition and metabolism, as inferred from homology to known P-uptake genes, up-regulation during P-starvation conditions in cultured strains MED4 (22) and MIT9301 (Fig. S2), or colocation with such genes in the genome (Fig. 2B). Many of these P-related genes are present in only a small fraction of Prochlorococcus cells at HOT but are nearly one copy per cell at BATS (Table S4); for instance, alkaline phosphatase (phoA) is estimated to be present in only 3% of Prochlorococcus cells at HOT but in 100% of these cells at BATS. A pathway for phosphonate utilization (23) is present in an estimated 13–21% of Prochlorococcus cells at BATS, but in virtually none of these cells at HOT (phnYZ; Table S4). Thus the “population genome” (i.e., the gene inventory in the Prochlorococcus collective) suggests strong selection for maintenance of accessory P- acquisition genes at BATS, but not at HOT. This difference between the two populations is consistent with exceptionally low surface water phosphate concentrations and physiological indicators of microbial phosphate limitation in the western North Atlantic (16, 24–27) relative to the Pacific. What is striking, however, is that this is the sole difference in gene content that emerged, implying that phosphate scarcity has been the most persistent and influential selective force driving diversification between the HOT and BATS populations.
Fig. 2.

Relative gene frequency among Prochlorococcus and Pelagibacter populations in the oligotrophic North Pacific (HOT) and North Atlantic (BATS) subtropical ocean gyres. (A) Detection of each Prochlorococcus gene at HOT and BATS, measured as the number of pyrosequencing reads. For each gene, the number of reads detected is proportional to the product of gene length and gene multiplicity per cell. Thus, assuming similar gene lengths at both sites, genes that fall along the diagonal trend have the same relative frequency at both sites, whereas those that fall above or below the diagonal are enriched at one site compared with the other. Squares represent significantly different frequencies between sites (G test; P < 0.01); circles represent nonsignificant differences. Colored squares represent genes whose chromosomal positions are depicted in B. (B) Genome comparison of two cultured isolates of Prochlorococcus (MED4 and MIT9301), with gray lines connecting homologous genes. Genes represented by corresponding colored squares in A are clustered together in a few distinct regions of the chromosome, including hypervariable genomic islands, in these isolates. Genes marked with an asterisk are up-regulated in response to P starvation (ref. 22 and Fig. S2). MED4 and MIT9301, isolated from the Mediterranean and the North Atlantic, respectively, are depicted because they carry the largest complements of phosphorus uptake genes among Prochlorococcus isolates (22). (C) Relative frequency of each Pelagibacter gene at HOT and BATS, as in A. (D) Genome comparison of two Pelagibacter strains, HTCC7211 and HTCC1062, as in B.
To test whether these same selective pressures have impacted the genomes of coexisting microbes in the community, we repeated the analysis with putative Pelagibacter (a heterotrophic bacterium) sequences (Fig. 2C), because this is another abundant microbe at these two sites for which reference genomes exist. Of 1,667 observed gene clusters, only 31 differ in abundance between BATS and HOT, and 29 of these are enriched at BATS (Table S5). Twenty-seven of these genes are located in one contiguous region of the genome in Pelagibacter strain HTCC7211, and nearly all are involved in phosphate or phosphonate metabolism (Fig. 2D), demonstrating that adaptation to P scarcity is a broad feature of the BATS ecosystem. Notably, however, the enriched gene set for Pelagibacter is distinct from that of Prochlorococcus. For example, genes encoding high-affinity phosphate transport (pstSCAB) and polyphosphate storage and breakdown (ppk and ppx) are represented equally among Prochlorococcus at HOT and at BATS, because they are core genes in this group (21). In contrast, the frequency of these genes varies dramatically between HOT and BATS in Pelagibacter (Table S5), in which these are not core genes in cultured isolates. The acquisition of beneficial nutrient-scavenging genes, along with their large cellular surface-to-volume ratio, helps explain the observation that Prochlorococcus and Pelagibacter-like bacteria can account for 90% of phosphate uptake in the North Atlantic (28).
Our results reveal unanticipated adaptations to phosphorus scarcity as well. Arsenate reductase and an arsenite efflux pump are enriched at BATS in Pelagibacter and Prochlorococcus, respectively (Tables S4 and S5). When cells scavenge phosphate, they sometimes take up the structurally similar arsenate ion, which is then reduced to arsenite and exported as a detoxification mechanism (29). Although average concentrations of surface inorganic arsenic (predominantly arsenate) are similar in the North Pacific and Atlantic, the arsenate:phosphate ratio is several-fold higher at BATS, where there is generally more arsenate (average, 16.3 nM) than phosphate in surface waters (30). Our population genomic analysis suggests that arsenic toxicity is an important selective force influencing Prochlorococcus and Pelagibacter at BATS, but not at HOT.
Several evolutionary scenarios could have led to this differential gene content between the two sites. If an ancestor of the BATS population had acquired genes that enhanced its fitness (e.g., alkaline phosphatase), a selective sweep could have ensued, leading to ecologically and genetically distinct HOT and BATS ecotypes (31). Alternatively, frequent gene transfer could have introduced P-acquisition genes into diverse genome backgrounds relatively recently (31). To distinguish between these scenarios, we built phylogenetic trees from both core genes and P-related flexible genes shared between the two populations and used Unifrac (32) to test whether the HOT- and BATS-derived alleles constituted distinct lineages. As exemplified by the pntA (encoding transhydrogenase) gene (Fig. 3 A and B), the housekeeping genes tested in both Prochlorococcus and Pelagibacter do not show divergence between the two populations (Table S6). Several P-uptake genes in both Prochlorococcus and Pelagibacter do show distinct HOT and BATS lineages, however (e.g., the pstB trees shown in Fig. 3 C and D). This phylogenetic divergence appears to be specific to P-uptake genes, and is not seen for the iron- and nitrogen-acquisition genes fur and amtB (Fig. 3 E and F and Table S6). This result is consistent with previous studies suggesting that the evolutionary history of the core genome backbone is decoupled from that of the P-associated regions (22, 33, 34). Together these results point to relatively recent gene acquisition, for instance, via phage-mediated gene transfer associated with genomic islands (35), combined with efficient selection in large populations, as the major processes underlying gene content variability.
Fig. 3.

Three examples of phylogenetic patterns observed in Prochlorococcus and Pelagibacter genes, from shotgun sequences sampled at HOT (orange leaf coloring) and BATS (blue leaf coloring). (A and B) pntA, a housekeeping gene found in similar abundance at HOT and at BATS, is not phylogenetically distinct between the sites. (C and D) pstB, a gene involved in phosphate transport, is phylogenetically distinct between the two sites, reflecting recent recombination and/or selection. (E and F) fur, a gene involved in regulation of iron metabolism, is not phylogenetically distinct between the sites. For clarity, sequence identifiers have been omitted except in the case of cultured isolates.
Over time, acquired genes can become core-like not only in their frequency in the population (i.e., one copy per cell), but also in their arrangement within individual genomes. For example, genes first acquired in hypervariable genomic islands can eventually migrate to more stable regions of the chromosome where insertions and deletions are less likely (36). Phosphate-acquisition genes are clustered in hypervariable regions in the genomes of isolates of both Prochlorococcus (22, 35) (Fig. 2B) and Pelagibacter (37) (Fig. 2D). To assess how conserved the gene neighborhoods surrounding accessory P-uptake genes have become within natural populations at BATS, we used small- insert clone libraries, constructed from the same DNA used for pyrosequencing (see Materials and Methods). We identified clones in which at least one read matched a Prochlorococcus or Pelagibacter BATS-enriched gene (Tables S4 and S5). In 22% of such Prochlorococcus clones (n = 307), but in 40% of such Pelagibacter clones (n = 554), the paired-end read matches another BATS-enriched gene, suggesting that these genes are more often clustered on the chromosome in Pelagibacter than in Prochlorococcus. Furthermore, we counted the number of unique clusters of orthologous groups of proteins (COGs) adjacent to each BATS-enriched gene in Prochlorococcus and Pelagibacter clones, and found a greater diversity of adjacent COGs in Prochlorococcus (Fig. S3). Notably, core genes and BATS-enriched genes in Pelagibacter show similar levels of local gene order conservation, whereas in Prochlorococcus, local gene order is significantly more variable surrounding BATS-enriched genes compared with core genes (Fig. S3). Together, these results suggest that accessory P-uptake genes have become core-like in BATS populations of Pelagibacter in both frequency and arrangement, perhaps because Pelagibacter has experienced P limitation for a longer time; indeed, despite its smaller cell and genome size, Pelagibacter likely has a higher per-cell quota for P than Prochlorococcus, owing to its dependence on phospholipids (24).
Our analysis reveals that P availability is the primary selective force driving genome divergence between these two ocean regions. It is important to note, however, that we sampled both sites only in October. Unlike HOT, where the water column is stably stratified year-round, BATS experiences a pronounced seasonal cycle. At BATS, deep mixing events every winter supply nutrients that fuel spring phytoplankton blooms, and as these blooms draw down nutrient stocks and thermal stratification intensifies, the surface layer becomes progressively more oligotrophic (38). Our October sample was collected near the end of summer stratification at BATS, when Prochlorococcus are typically most abundant (39, 40). If we sampled in winter or spring, would we still find P availability to be the predominant selective pressure distinguishing HOT and BATS? We suspect that the answer is yes, at least for Prochlorococcus, because the increased P supply at BATS is accompanied by an increased P demand from faster-growing phytoplankton groups (Synechococcus and eukaryotes); the abundance of Prochlorococcus is actually lowest at BATS in late winter and early spring and increases as conditions become more stratified and oligotrophic (39, 40). To properly compare the population genomes at HOT and BATS in winter or spring, we also would need to control for the ecotype composition of the water column. Whereas we captured similar ecotype abundance profiles at both sites in October (Table S2), we would expect to capture very different profiles in winter or spring, owing to the stronger seasonal succession of ecotypes at BATS relative to HOT (40). These dynamics, and similar ecotype oscillations within the SAR11 clade at BATS (41), underscore the need for even finer-scale phylogenetic and temporal resolution in future population genomics studies.
Here we have used a population-genomic approach, unbiased by a priori assumptions about the relative strengths of selective factors, to compare two distinct ocean regions. This approach also could reveal adaptations in response to environmental change over time in a single habitat. For instance, recently it has been suggested that the inorganic P inventory at HOT is decreasing, and if this trend toward P limitation continues, the community will likely experience a shift toward organisms with lower P requirements and alternative P-acquisition mechanisms (42, 43). Based on our results, Prochlorococcus and Pelagibacter have not yet faced prolonged P limitation at HOT, at least not relative to their Atlantic counterparts. Over time, repeated sampling of these population genomes will reveal whether selection is indeed shaping P-related gene content and gene sequence at HOT and will help constrain the time scales of evolutionary change in the oceans. Moreover, these types of analyses will likely reveal unanticipated evolutionary pressures being “felt” by the microbial community. In this way, population genomics of ocean microbes not only is a powerful tool for diagnosing environmental change, but also can illuminate the fundamental evolutionary processes underlying biological organization (44).
Materials and Methods
Seawater was collected in October 2006 from HOT Station ALOHA (22°44’N, 158°2’W) and BATS (31°40’N, 64°10’W) from three depths at each site: in the mixed layer, just below the mixed layer, and in the deep chlorophyll maximum (corresponding to 20/50/100 m at BATS and 25/75/110 m at HOT). Sampling, DNA extraction, and sequencing were performed as described previously (45). Pyrosequencing reads were mapped to taxa using BLASTX against NCBI-nr, with a minimum bit score of 40. Prochlorococcus reads were mapped to gene clusters using BLASTN against 12 reference genomes, and were assigned to a gene cluster only if the top three hits (or all hits if fewer than three total) belonged to the same gene cluster, with bit score > 40 and alignment length > 40. Pelagibacter reads were mapped to gene clusters if the top three hits (or all hits if fewer than three total) belonged to the same gene cluster with bit score > 40; if there was a large bit score dropoff (>30) between hits, then the lower hits were not considered. Gene clusters for Prochlorococcus have been described by Kettler et al. (21); clusters for Pelagibacter were constructed by reciprocal best BLASTP hits using available genomes (Pelagibacter HTCC1002, HTCC1062, and HTCC7211). Gene frequencies were compared using the G test and Bonferroni-corrected for multiple comparisons (46).
Estimating Multiplicity per Cell.
The number of DNA fragments, ni, observed for a gene i of length Li and average multiplicity per genome, mi, in a sample of NPro sequences is expected to be binomially distributed,
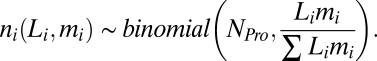 |
Thus, each sequencing read that we sample can be classified as either a “success” or “failure;” either it belongs to gene i or it does not. The probability of success (i.e., that the read came from a given gene i) depends on the length of the gene; longer genes will be detected more frequently. Indeed, using 1,221 single-copy core genes (i.e., mi = 1, defined based on isolate genomes; ref. 21), we found a tight linear relationship between gene length and the number of sequencing reads detected (r2 = 0.95 for HOT and 0.95 for BATS), and also found that the number of reads detected for nearly all core genes fell within the confidence intervals predicted by the binomial distribution assuming that mi = 1. From this relationship, based on all 1,221 known single-copy genes, we then inferred gene multiplicity per cell for the “flexible” genome (sensu ref. 21); this approach is more robust than normalizing to a single core gene. Multiplicity per cell for gene i was calculated as
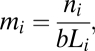 |
where ni is the number of sequence reads mapped to gene i, Li is the gene length, and b is the slope of the length-versus-reads detected relationship for core genes. Confidence intervals for mi were estimated using the binomial distribution. This approach was taken for Pelagibacter estimates of multiplicity per genome as well, using its respective core genome.
Validation of Taxon Mapping.
An obvious limitation of our approach is that we can never know whether a pyrosequencing read with strong similarity to a particular organism's genome was truly derived from a cell of that organism, or whether it resides in the genome of another organism due to horizontal gene transfer. To address this issue, we used paired-end reads from small-insert shotgun clone libraries constructed from the same DNA used for pyrosequencing. Clone libraries were constructed and sequenced by the Joint Genome Institute according to standard production protocols. We identified putative Prochlorococcus clones, in which at least one of the two paired-end reads matched Prochlorococcus as the best hit, and then identified the best-hit taxon for its paired end using BLASTN against a custom database of marine microbial genomes. If both paired-end reads match Prochlorococcus, we can be more confident that the clone came from a Prochlorococcus cell, although multigene horizontal transfer events are also possible. Only 9.3% of the putative Prochlorococcus clones at BATS and 8.6% of those at HOT matched Prochlorococcus on one end and a different taxon on the other end. We repeated the analysis for putative Pelagibacter clones and found that 18.2% of clones at BATS and 21.6% at HOT matched Pelagibacter on one end and a different taxon on the other end. Clones in which only one of two paired reads matched Prochlorococcus (or Pelagibacter) are still quite likely to have derived from a Prochlorococcus (or Pelagibacter) cell, given the high abundance of Prochlorococcus (and Pelagibacter) in these communities. There are more reference genomes available for Prochlorococcus than for Pelagibacter (12 vs. 3), providing a more comprehensive (although still incomplete) picture of the Prochlorococcus pan-genome; this larger set of reference sequences likely explains why paired-end reads were more often coidentified as Prochlorococcus than as Pelagibacter.
Synteny Analysis.
From these same clone libraries, we identified clones in which at least one read matched a BATS-enriched gene from Prochlorococcus or Pelagibacter, and clones in which at least one read matched a core gene from either taxon. We then compared the diversity of the paired-end reads associated with BATS-enriched genes and with core genes. We used rpsblast (47) to identify the best COG match for each paired-end read, and counted the number of unique COGs associated with each core gene and each BATS-enriched gene as a measure of gene order conservation (Fig. S3). We used the χ2 goodness-of-fit test to evaluate whether the BATS-enriched sample came from the null distribution specified by the core genes (Poisson).
Phylogenetic Analyses.
We constructed phylogenetic trees using small-insert shotgun clone sequences. Environmental gene sequences were aligned to the reference strain MIT9301 for Prochlorococcus and strain HTCC7211 for Pelagibacter using BlastAlignP (48), allowing a maximum of 70% gaps in the alignment due to the variable length and location of the environmental clone sequences. For Prochlorococcus, alignments were pruned to include only sequences most similar to high-light–adapted isolates, for reasons given below. Trees were constructed using PhyML (49) using the HKY85 model, with 10 different random starting trees, 4 gamma rate categories, and 0 invariant sites and parameters estimated from the data, with SH-like aLRT branch supports. Maximum likelihood is the most accurate method when using gene fragments and is the least sensitive to missing data (50). These trees were then used as input for UniFrac (32). The pruning to high-light adapted Prochlorococcus was done because long branches in the LL clades, which were detected only occasionally (the high-light–adapted clades eMIT9312 and eMED4 account for about 85% of cells at both sites; Table S2), can strongly influence the UniFrac test (e.g., if a LL clone was detected at HOT but not at BATS). Both the UniFrac significance test and the P test were performed for the pairwise HOT versus BATS comparison with 100 replicates.
Gene Expression.
To measure gene expression during P starvation, strain MIT9301 was grown to midexponential phase in Pro99 medium, harvested at 8,000 g, washed twice with −P Pro99 (no added phosphate), and resuspended in the same. Duplicate cultures, grown in continuous light at 20 μmol photon·m−2·s−1 and 21 °C, were analyzed. At each time point, an aliquot of cells was harvested at 10,000 g, resuspended in storage buffer [200 mM sucrose, 10 mM sodium acetate (pH 5.2), and 5 mM EDTA], frozen in liquid N2, and stored at −80 °C. RNA was extracted using the mirVana kit (Ambion) and Dnase-treated using Turbo DNA-free (Ambion). RNA (2–10 ng per reaction) was reverse-transcribed using SuperScript II (Invitrogen) and gene-specific primers. Transcripts of each gene were quantified using the QuantiTect SYBR Green Kit (Qiagen) and normalized to rnpB transcripts. The five genes tested in MIT9301 (P9301_12441, 12511, 12521, 12551, and 12561) were chosen because they are absent from strain MED4, for which whole genome expression data under P starvation are available (22).
Supplementary Material
Acknowledgments
We thank Stephan Schuster and Ed DeLong and members of their laboratories for pyrosequencing the BATS216 and HOT186 samples, respectively; the HOT and BATS teams and the captain and crew of the R/V Kilo Moana and R/V Atlantic Explorer; Matt Sullivan, Jay McCarren, Suzanne Kern, Sarah Bagby, and Yanmei Shi for help with sample collection and processing; Scott Chilton for laboratory assistance; Daniele Veneziano for advice on statistical analyses; Kerrie Barry and the Joint Genome Institute for shotgun sequencing; and Jake Waldbauer, Vanja Klepac-Ceraj, Jesse Shapiro, and Eric Alm for comments and discussion. This work was supported in part by the Gordon and Betty Moore Foundation, the National Science Foundation (NSF), the U.S. Department of Energy, and an NSF Graduate Research Fellowship (to M.L.C.).
Footnotes
The authors declare no conflict of interest.
Database deposition: The sequences reported in this paper have been deposited in the NCBI Sequence Read Archive, http://www.ncbi.nlm.nih.gov. For a list of accession numbers, see SI Text.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1009480107/-/DCSupplemental.
References
- 1.Welch RA, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity: A Darwinian view of the evolution of microbes. EMBO Rep. 2001;2:376–381. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simmons SL, et al. Population genomic analysis of strain variation in Leptospirillum group II bacteria involved in acid mine drainage formation. PLoS Biol. 2008;6:e177. doi: 10.1371/journal.pbio.0060177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rusch DB, et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martín-Cuadrado AB, et al. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS ONE. 2007;2:e914. doi: 10.1371/journal.pone.0000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuadros-Orellana S, et al. Genomic plasticity in prokaryotes: The case of the square haloarchaeon. ISME J. 2007;1:235–245. doi: 10.1038/ismej.2007.35. [DOI] [PubMed] [Google Scholar]
- 8.Hunt DE, et al. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science. 2008;320:1081–1085. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 9.Wildschutte H, Wolfe DM, Tamewitz A, Lawrence JG. Protozoan predation, diversifying selection, and the evolution of antigenic diversity in Salmonella. Proc Natl Acad Sci USA. 2004;101:10644–10649. doi: 10.1073/pnas.0404028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilmes P, Simmons SL, Denef VJ, Banfield JF. The dynamic genetic repertoire of microbial communities. FEMS Microbiol Rev. 2009;33:109–132. doi: 10.1111/j.1574-6976.2008.00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- 12.Kuo C-H, Ochman H. The fate of new bacterial genes. FEMS Microbiol Rev. 2009;33:38–43. doi: 10.1111/j.1574-6976.2008.00140.x. [DOI] [PubMed] [Google Scholar]
- 13.Whitaker RJ, Banfield JF. Population genomics in natural microbial communities. Trends Ecol Evol. 2006;21:508–516. doi: 10.1016/j.tree.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Morris RM, et al. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–810. doi: 10.1038/nature01240. [DOI] [PubMed] [Google Scholar]
- 15.Karl DM, et al. Building the long-term picture: The U.S. JGOFS time-series programs. Oceanography. 2001;14:6–17. [Google Scholar]
- 16.Wu JF, Sunda W, Boyle EA, Karl DM. Phosphate depletion in the western North Atlantic Ocean. Science. 2000;289:759–762. doi: 10.1126/science.289.5480.759. [DOI] [PubMed] [Google Scholar]
- 17.Jickells TD, et al. Global iron connections between desert dust, ocean biogeochemistry, and climate. Science. 2005;308:67–71. doi: 10.1126/science.1105959. [DOI] [PubMed] [Google Scholar]
- 18.Moore LR, Rocap G, Chisholm SW. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature. 1998;393:464–467. doi: 10.1038/30965. [DOI] [PubMed] [Google Scholar]
- 19.West NJ, Scanlan DJ. Niche-partitioning of Prochlorococcus populations in a stratified water column in the eastern North Atlantic Ocean. Appl Environ Microbiol. 1999;65:2585–2591. doi: 10.1128/aem.65.6.2585-2591.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zinser ER, et al. Prochlorococcus ecotype abundances in the North Atlantic Ocean as revealed by an improved quantitative PCR method. Appl Environ Microbiol. 2006;72:723–732. doi: 10.1128/AEM.72.1.723-732.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kettler GC, et al. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 2007;3:e231. doi: 10.1371/journal.pgen.0030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martiny AC, Coleman ML, Chisholm SW. Phosphate acquisition genes in Prochlorococcus ecotypes: Evidence for genome-wide adaptation. Proc Natl Acad Sci USA. 2006;103:12552–12557. doi: 10.1073/pnas.0601301103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez A, Tyson GW, Delong EF. Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses. Environ Microbiol. 2010;12:222–238. doi: 10.1111/j.1462-2920.2009.02062.x. [DOI] [PubMed] [Google Scholar]
- 24.Van Mooy BAS, et al. Phytoplankton in the ocean use non-phosphorus lipids in response to phosphorus scarcity. Nature. 2009;458:69–72. doi: 10.1038/nature07659. [DOI] [PubMed] [Google Scholar]
- 25.Ammerman JW, Hood RR, Case DA, Cotner JB. Phosphorus deficiency in the Atlantic: An emerging paradigm in oceanography. Eos Trans AGU. 2003;84 10.1029/2003EO180001. [Google Scholar]
- 26.Bjorkman K, Thomson-Bulldis AL, Karl DM. Phosphorus dynamics in the North Pacific subtropical gyre. Aquat Microb Ecol. 2000;22:185–198. [Google Scholar]
- 27.Cotner JB, Ammerman JW, Peele ER, Bentzen E. Phosphorus-limited bacterioplankton growth in the Sargasso Sea. Aquat Microb Ecol. 1997;13:141–149. [Google Scholar]
- 28.Zubkov MV, et al. Microbial control of phosphate in the nutrient-depleted North Atlantic subtropical gyre. Environ Microbiol. 2007;9:2079–2089. doi: 10.1111/j.1462-2920.2007.01324.x. [DOI] [PubMed] [Google Scholar]
- 29.Sanders JG, Windom HL. The uptake and reduction of arsenic species by marine algae. Estuar Coast Mar Sci. 1980;10:555–567. [Google Scholar]
- 30.Cutter GA, Cutter LS. Behavior of dissolved antimony, arsenic, and selenium in the Atlantic Ocean. Mar Chem. 1995;49:295–306. [Google Scholar]
- 31.Cohan FM. Towards a conceptual and operational union of bacterial systematics, ecology, and evolution. Philos Trans R Soc Lond B Biol Sci. 2006;361:1985–1996. doi: 10.1098/rstb.2006.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lozupone C, Hamady M, Knight R. UniFrac: An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-371. article no. 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martiny AC, Huang Y, Li W. Occurrence of phosphate acquisition genes in Prochlorococcus cells from different ocean regions. Environ Microbiol. 2009;11:1340–1347. doi: 10.1111/j.1462-2920.2009.01860.x. [DOI] [PubMed] [Google Scholar]
- 34.Martiny AC, Tai AP, Veneziano D, Primeau F, Chisholm SW. Taxonomic resolution, ecotypes and the biogeography of Prochlorococcus. Environ Microbiol. 2009;11:823–832. doi: 10.1111/j.1462-2920.2008.01803.x. [DOI] [PubMed] [Google Scholar]
- 35.Coleman ML, et al. Genomic islands and the ecology and evolution of Prochlorococcus. Science. 2006;311:1768–1770. doi: 10.1126/science.1122050. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence JG, Hendrickson H. Genome evolution in bacteria: Order beneath chaos. Curr Opin Microbiol. 2005;8:572–578. doi: 10.1016/j.mib.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Wilhelm LJ, Tripp HJ, Givan SA, Smith DP, Giovannoni SJ. Natural variation in SAR11 marine bacterioplankton genomes inferred from metagenomic data. Biol Direct. 2007;2 doi: 10.1186/1745-6150-2-27. article no. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinberg D, et al. Overview of the US JGOFS Bermuda Atlantic Time-series Study (BATS): A decade-scale look at ocean biology and biogeochemistry. Deep Sea Res Part II Top Stud Oceanogr. 2001;48:1405–1447. [Google Scholar]
- 39.DuRand MD, Olson RJ, Chisholm SW. Phytoplankton population dynamics at the Bermuda Atlantic Time-series station in the Sargasso Sea. Deep Sea Res Part II Top Stud Oceanogr. 2001;48:1983–2003. [Google Scholar]
- 40.Malmstrom RR, et al. Temporal dynamics of Prochlorococcus ecotypes in the Atlantic and Pacific oceans. ISME J. doi: 10.1038/ismej.2010.60. 10.1038/ismej.2010.60. [DOI] [PubMed] [Google Scholar]
- 41.Carlson CA, et al. Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J. 2009;3:283–295. doi: 10.1038/ismej.2008.117. [DOI] [PubMed] [Google Scholar]
- 42.Karl DM. In: Manual of Environmental Microbiology. Hurst CJ, editor. Washington, DC: ASM Press; 2007. pp. 523–539. [Google Scholar]
- 43.Karl DM. Microbial oceanography: Paradigms, processes and promise. Nat Rev Microbiol. 2007;5:759–769. doi: 10.1038/nrmicro1749. [DOI] [PubMed] [Google Scholar]
- 44.Woese CR, Goldenfeld N. How the microbial world saved evolution from the Scylla of molecular biology and the Charybdis of the modern synthesis. Microbiol Mol Biol Rev. 2009;73:14–21. doi: 10.1128/MMBR.00002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frias-Lopez J, et al. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA. 2008;105:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokal RR, Rohlf FJ. Biometry: The Principles and Practice of Statistics in Biological Research. New York: Freeman; 1995. [Google Scholar]
- 47.Marchler-Bauer A, Bryant SH. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32(Web Server issue):W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belshaw R, Katzourakis A. BlastAlign: A program that uses blast to align problematic nucleotide sequences. Bioinformatics. 2005;21:122–123. doi: 10.1093/bioinformatics/bth459. [DOI] [PubMed] [Google Scholar]
- 49.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 50.Hartmann S, Vision TJ. Using ESTs for phylogenomics: Can one accurately infer a phylogenetic tree from a gappy alignment? BMC Evol Biol. 2008;8:95. doi: 10.1186/1471-2148-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.