

Abstract
Understanding how an amino acid sequence folds into a functional, three-dimensional structure has proved to be a formidable challenge in biological research, especially for transmembrane proteins with multiple alpha helical domains. Mechanistic folding studies on helical membrane proteins have been limited to unusually stable, single domain proteins such as bacteriorhodopsin. Here, we extend such work to flexible, multidomain proteins and one of the most widespread membrane transporter families, the major facilitator superfamily, thus showing that more complex membrane proteins can be successfully refolded to recover native substrate binding. We determine the unfolding free energy of the two-domain, Escherichia coli galactose transporter, GalP; a bacterial homologue of human glucose transporters. GalP is reversibly unfolded by urea. Urea causes loss of substrate binding and a significant reduction in alpha helical content. Full recovery of helical structure and substrate binding occurs in dodecylmaltoside micelles, and the unfolding free energy can be determined. A linear dependence of this free energy on urea concentration allows the free energy of unfolding in the absence of urea to be determined as +2.5 kcal·mol-1. Urea has often been found to be a poor denaturant for transmembrane helical structures. We attribute the denaturation of GalP helices by urea to the dynamic nature of the transporter structure allowing denaturant access via the substrate binding pocket, as well as to helical structure that extends beyond the membrane. This study gives insight into the final, critical folding step involving recovery of ligand binding for a multidomain membrane transporter.
Keywords: protein folding, thermodynamic stability, linear free-energy relationship
The folding of proteins into their three-dimensional structures is of vital importance for biological activity, with incorrectly folded states increasingly being linked to disease (1). Most folding studies to date have been carried out on water-soluble proteins in vivo, in vitro, and in silico, and considerable quantitative detail is now available on the processes of their folding and unfolding, misfolding, and aggregation (2). Extending the approaches to membrane proteins has proven to be more challenging (3). Membrane proteins are frequently prone to aggregation and are unstable once removed from their native lipid bilayer environment. Many methods for examining folding mechanisms require a system of reversible unfolding, which has been established for very few membrane proteins; indeed experimental determinations of folding free energy have been made only for the α-helical proteins bacteriorhodopsin (bR), diacylglycerol kinase, and bacterial potassium channel KscA (4–6), as well as some β-barrel outer membrane proteins (7, 8). The protein folding transition state has been studied for helical bR and β-barrel PagP (9, 10). Because membrane proteins constitute about 30% of the proteome of all organisms, such detailed analyses of membrane protein folding must be expanded to include more classes of protein and thus investigate any common folding pathways. It is also important to extend the work to multidomain membrane proteins.
GalP is a proton-galactose symporter from Escherichia coli (11), a member of the major facilitator superfamily (MFS), and homologous to the physiologically important glucose family of sugar transporters in humans (12). The MFS is a large group, comprising approximately 25% of all known transport proteins in prokaryotes (13). They are predicted to have a common fold of two 6-helical bundles, with the substrate binding between these domains. GalP is thought to have a similar structure to the glycerol phosphate transporter, GlpT (14) and can be readily overexpressed in E. coli membranes providing large quantities of purified protein (15, 16) that are required for folding studies.
Here, we use GalP to demonstrate the feasibility of quantitative folding studies on MFS transporters, which have multidomain, flexible membrane-embedded structures. Not only do we accomplish reversible unfolding and determine the associated free energy, but we also reveal a linear relationship of this free energy with denaturant and find that GalP can be refolded directly into lipid vesicles.
Results
Reversible Unfolding.
A well-established method for determining the free energy of unfolding is by equilibrium chemical denaturation. GalP was unfolded by mixing protein purified in n-dodecyl-β-D-maltoside (DDM) micelles with a denaturing buffer containing urea, giving a final urea concentration of 8 M. In 8 M urea the intrinsic protein fluorescence band of GalP decreased in intensity and shifted to longer wavelengths, with a maximum at 337 nm compared to 332 nm in DDM (Fig. S1). This change in fluorescence is indicative of an increase in exposure of aromatic residues to water. CD spectroscopy showed folded GalP in DDM to have 74% helix, 6% sheet, 10% turn, and 10% disordered secondary structure (Fig. 1A). The decrease in intensity of the negative 222-nm peak, from ∼21,000 deg ·cm2 mol-1 in DDM to ∼14,500 deg ·cm2 mol-1, occurred in 8 M urea indicating a significant reduction of approximately a third of the helix content. The activity of GalP was measured by binding of the antibiotic cytochalasin B (17), which quenches GalP fluorescence (18). The decrease in GalP fluorescence intensity at different cytochalasin B concentrations fitted to a single-site saturation curve, giving a substrate dissociation constant Kd of 17 ± 4 μM (Fig. 1B). This substrate binding was abolished in 8 M urea.
Fig. 1.

GalP unfolded and refolded from urea. Changes were monitored as follows: (A) Far UV circular dichroism of 0.5 μM GalP in 1 mM DDM (solid lines), unfolded in 8 M urea (short dashes), and refolded from urea into 1 mM DDM (long dashes). (B) Binding of cytochalasin B to 0.5 μM GalP in 1 mM DDM (open circles) and GalP refolded from 8 M urea into DDM (closed circles). The solid line in B represents a fit to a one site binding model to the open circles, giving a dissociation constant, Kd of 17 ± 4 μM for GalP in DDM. The Kd for refolded GalP was 24 ± 2 μM. No binding is observed in 8 M urea (open triangles). Error bars show ± SEM.
GalP in 8 M urea was refolded by dilution into 1-mM DDM micelles, giving a final concentration of 0.8 M urea. Protein fluorescence, far UV CD spectra, and substrate binding all return to that of the original, folded GalP (Fig. 1 and Fig. S1). The residual 0.8 M urea was removed by dialysis in the CD spectrum of Fig. 1A, to enable data collection below 200 nm. The ligand-binding curve for refolded GalP in Fig. 1B gives a Kd of 24 ± 2 μM. Titration of cytochalasin B to refolded GalP at a protein concentration above this Kd showed that binding to 50 μM GalP saturated with 49 μM ligand, thus 98% of the refolded protein bound ligand (see Fig. S2). This high refolding yield was confirmed by radiolabeled ligand binding; saturation of 10 μM GalP by the ligand, tritiated forskolin, occurred at ∼9 μM forskolin for both purified GalP (that had never been denatured), as well as GalP refolded from urea into DDM, showing that in both cases ∼90% of the protein is active. The refolded protein also displayed 100% transport activity following reconstitution into lipids (see below).
Thermodynamic analysis relies on reversible, equilibrium unfolding. Equilibrium unfolding measurements were made by incubating originally folded GalP (in DDM) in a range of urea concentrations (see Fig. 2A). Equilibrium folding measurements were made by first unfolding GalP in 8 M urea, then refolding into different urea concentrations (Fig. 2B). In both cases the amount of folded protein was determined from the CD intensity at 222 nm, and the resulting values normalized between 0% and 100%. The latter represents the fully folded protein in DDM (with 74% helix) and the former 0% represents the urea-unfolded state that is partly structured having about two-thirds of the native helical content (i.e., a CD signal intensity at 222 nm of ∼14,500 deg ·cm2 mol-1 in 8 M urea; see Fig. 1A). Fig. 2B shows that the folding and unfolding curves overlay demonstrating reversibility. Both curves fit to a two-state reaction. The overall free energy of unfolding at zero denaturant was determined by fitting the unfolding data in Fig. 2A to a two-state equation (see Materials and Methods), giving an unfolded free energy in the absence of urea, 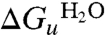 , of +2.5 ± 0.2 kcal·mol-1 (at pH 8). Free energy values at different urea concentrations for unfolding were also calculated from the separate unfolding data points. These free energy values were linear with urea concentration (Fig. 2C), allowing extrapolation to obtain the unfolding free energy in the absence of urea,
, of +2.5 ± 0.2 kcal·mol-1 (at pH 8). Free energy values at different urea concentrations for unfolding were also calculated from the separate unfolding data points. These free energy values were linear with urea concentration (Fig. 2C), allowing extrapolation to obtain the unfolding free energy in the absence of urea, 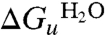 of +2.4 ± 0.2 kcal·mol-1. The linear dependence is characterized by the gradient, m, of -0.58 ± 0.04 kcal·mol-1.
of +2.4 ± 0.2 kcal·mol-1. The linear dependence is characterized by the gradient, m, of -0.58 ± 0.04 kcal·mol-1.
Fig. 2.

Free energy of GalP unfolding. (A) Equilibrium unfolding data of 15 μM GalP in different urea concentrations in the presence of 1 mM DDM micelles, at pH 8. The percentage folded protein on the y axis is determined from the CD signal intensity at 222 nm, with 100% representing the 222-nm value of fully folded GalP in DDM and 0% the partly unfolded 8 M urea state that possesses some helical content. Each data point is the average of up to 16 measurements on different samples but the same protein preparation; error bars show ± the standard error. The solid line shows a fit to a two-state unfolding curve, giving a 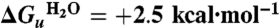 at pH 8, with a standard error of ± 0.2 kcal·mol-1. Further analysis of the best fit of the curve gives a 95% confidence interval of ± 0.3 kcal·mol-1. (B) Refolding of 1.5 μM GalP from 8 M urea into 1 mM DDM and different concentrations of urea (closed circles) overlaying the two-state unfolding curve derived in A; the solid line is the fit to the unfolding curve data in A. The tenfold lower protein concentration required in the refolding experiments of 1.5 μM in B compared to 15 μM for the unfolding data in A contributes to the larger error bars for the data in B. Unfolding data at pH 4 are also shown (open squares). (C) The free energy of unfolding was determined at different urea concentrations using equation ΔG = -RT ln([unfolded]/[folded]) and data from A; extrapolation to zero urea gives a
at pH 8, with a standard error of ± 0.2 kcal·mol-1. Further analysis of the best fit of the curve gives a 95% confidence interval of ± 0.3 kcal·mol-1. (B) Refolding of 1.5 μM GalP from 8 M urea into 1 mM DDM and different concentrations of urea (closed circles) overlaying the two-state unfolding curve derived in A; the solid line is the fit to the unfolding curve data in A. The tenfold lower protein concentration required in the refolding experiments of 1.5 μM in B compared to 15 μM for the unfolding data in A contributes to the larger error bars for the data in B. Unfolding data at pH 4 are also shown (open squares). (C) The free energy of unfolding was determined at different urea concentrations using equation ΔG = -RT ln([unfolded]/[folded]) and data from A; extrapolation to zero urea gives a 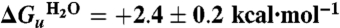 .
.
Fig. 2B also shows the unfolding curve of GalP titrated with increasing urea concentrations at a pH of 4, reflecting an increase in GalP stability at this lower pH. CD revealed no loss of secondary structure below 6 M urea at pH 4. GalP is a proton-galactose symporter, and hence the enhanced stability at pH 4 may arise from binding of protons.
Kinetic Measurements.
The kinetics of unfolding were measured by rapid, stopped-flow mixing of GalP in DDM with concentrated urea and the decrease in protein fluorescence recorded over time (Fig. 3A). The fluorescence trace fit well to the sum of two exponential components. The faster rate constant, ku, of 0.54 ± 0.01 s-1 is assigned to unfolding, whereas the slower rate of 0.09 s-1 reflects photobleaching or back mixing of product into the stopped-flow cuvette. This latter slow rate was absent when the folding was initiated by manual mixing into a standard cuvette and the sample was stirred continuously. Refolding from 8 M urea into DDM was a slower, single exponential process, giving a folding rate, kf, of 0.013 ± 0.006 s-1 (Fig. 3B). Thus, both the unfolding and refolding reactions were observed to be single exponential processes with no detectable transient intermediates.
Fig. 3.

Folding kinetics. (A) Unfolding of 0.5 μM GalP from 1 mM DDM into 8 M urea by stopped-flow mixing and measuring the decrease in GalP fluorescence. (B) Refolding from 8 M urea into 1 mM DDM, measured by the increase in protein fluorescence. Residuals for single exponential fits to the data are shown below the fluorescence curves, together with those for a double exponential fit to the unfolding data. The latter gives an unfolding rate constant ku of 0.53 s-1 and a slower rate constant of 0.09 s-1. The single exponential fit to the unfolding data resolved a rate constant of 0.47 s-1 The folding rate constant from B, kf, is 0.013 s-1.
GalP Stability.
The stability of GalP in other denaturants was also examined; SDS was ineffective (see Fig. S3), but guanidine hydrochloride (GuHCl) caused greater denaturation than urea. The CD spectrum of GalP in 7 M GuHCl shows a substantial decrease in secondary structure (Fig. 4A), with signal intensity of ∼9,000 deg ·cm2 mol-1 at 222 nm, less than that of ∼14,500 deg ·cm2 mol-1 in 8 M urea. Fig. 4B shows there may be two unfolding transitions: below and above ∼4 M GuHCl. The first transition seems to be partly reversible as GalP could be partially refolded from 3.5 M GuHCl, with some recovery of secondary structure (Fig. S4).
Fig. 4.

GalP unfolding in GuHCl. (A) CD spectrum of 5 μM GalP in 1 mM DDM (solid line) and unfolded by 7 M GuHCl (dashed line). (B) Dependence of 222-nm CD intensity as a function of GuHCl.
Refolding in Lipids.
The membrane environment has previously been shown to be important for the correct folding of secondary transporters (19, 20). The impact of the bilayer on GalP folding was therefore assessed by examining the ability of GalP to refold from 8 M urea into lipid vesicles composed of varying proportions of L-α-1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and L-α-1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE). The CD signal showed that optimal refolding occurred with 60 mol % DOPE, where the signal intensity at 222 nm increased to ∼19,000 deg ·cm2 mol-1 (see Fig. 5A and Fig. S5), compared to that of GalP in DDM of ∼21,000 deg ·cm2 mol-1. The protein was tested for transport activity by mixing lipid vesicles with buffer containing 50 mM D-galactose and the pH-sensitive dye phenol red. Substrate-driven proton uptake was followed by measuring the change in absorbance of this pH-sensitive dye. Protein-lipid vesicles showed increased proton uptake over control vesicles (Fig. 5B). Purified, functional GalP reconstituted from DDM into vesicles, as well as GalP refolded into DDM and then reconstituted into vesicles; both had initial transport rates that were 10 times faster than control vesicles with no protein. The initial rate of GalP refolded directly into lipid vesicles was only 4 times that of the control, and thus 2.5 less active than the reconstituted samples.
Fig. 5.

Refolding GalP into lipids. (A) CD spectrum of 5 μM GalP refolded from 8 M urea into 1% (wt/vol) DOPC/DOPE vesicles with 60 mol % DOPE. (Inset) CD spectrum of 5-mM folded GalP reconstituted into E. coli lipids. (B) H+ transport measured across DOPC/DOPE vesicles containing folded GalP reconstituted from DDM; i.e., GalP that was not denatured as an active control (solid black line), GalP refolded into DDM and then reconstituted into DOPC/DOPE (dotted line), GalP refolded directly into DOPC/DOPE (dashed line), and control vesicles in the absence of protein (gray line). All vesicles contain 60 mol % DOPE.
The CD spectrum of refolded protein in vesicles shows an increase in the ratio of CD signal intensity at 222 nm to 208 nm, compared to the 222∶208 nm intensity ratio of detergent solubilized GalP (compare Figs. 1A and 5A). Reconstitution of fully folded GalP into a native-like lipid environment also showed the same CD spectrum as GalP refolded into lipid vesicles, showing that the different 222∶208 nm intensity ratio is due to the lipid environment rather than the refolding process. Analysis of the CD spectra in lipids shows an apparent reduction in helicity, with 67% α-helix, 11% β-sheet, 12% β-turn, and 10% disordered secondary structure for reconstituted GalP and 61% α-helix, 15% β-sheet, 14% β-turn, and 10% disordered structure for refolded GalP; both showing lower α-helix content than the 74% in DDM. A similar effect of lipids on CD spectra has been previously noted for other membrane proteins, for example in the cases of bacteriorhodopsin or spinach aquaporin. This may result from a membrane inserted conformation slightly different to that in detergent micelles that is general to several membrane proteins, or an effect of the lipid environment on the CD spectral shape that is not accounted for in the analysis dataset used to deconvolute the CD spectra.
The CD spectra and transport activity data show that GalP refolded directly into lipids has the same helix content, but 2.5-fold lower transport activity, as folded, active GalP reconstituted into lipids. This suggests there are different helical arrangements of the protein that is refolded in lipids, possibly with all the refolded protein having suboptimal helix packing and thus transport activity. Alternatively, less than half the refolded protein could have full activity and the remainder a helix packing arrangement that gives no activity.
Discussion
The MFS transport protein, GalP, can be refolded with high efficiency into DDM micelles, from a partially unfolded state in urea. Refolding is reversible and approximates to a two-state reaction, enabling the equilibrium constant and free energy for folding to be determined. A linear free energy relationship on urea allows the thermodynamic stability of GalP to be found from the unfolding free energy in DDM micelles, in the absence of urea. This provides an excellent method to compare stabilities of dynamic multidomain membrane protein structures in different detergent conditions. Moreover, we show that it is possible to regain native-like substrate binding, with > 90% of GalP recovering binding activity and exhibiting the same transport activity as active GalP after reconstitution into lipid vesicles.
Urea Unfolding.
Urea is an effective denaturant of GalP, affecting both secondary and tertiary structure. Abolition of substrate binding and a significant reduction of ∼30% in helix content occurs in 8 M urea. Urea has been used in folding studies of E. coli diacylglycerol kinase (DGK), but causes little helical loss (21); moreover, it is a poor denaturant for bR (22), for which an SDS-denatured state is used. Urea has worked well for β-barrel membrane proteins, enabling several folding studies from extensively denatured states (e.g., refs. 23–25).
GalP seems to have helical structure outside the membrane that will be accessible to denaturation by urea. GalP is structurally similar to GlpT where several transmembrane α-helices, constituting ∼25% of total helical content, extend beyond the membrane on the cytoplasmic side (14, 26). However, as urea denaturation also results in loss of ligand binding, the GalP binding site must also be affected. Ligands bind at a site in the middle of the membrane, via a large solvent exposed cavity between the two transporter domains and this would enable small chaotropic agents such as urea and guanidine to gain access into the core of the protein embedded within the membrane and cause partial unfolding.
Linear Free Energy Relationships.
A linear dependence of unfolding free energy on denaturant enables a key parameter of interest to be found—the free energy of unfolding in the absence of denaturant, under “folding” conditions. There are only a few such observations for membrane proteins. The approach was first shown to apply to helical membrane proteins using SDS denaturation of DGK (5) and subsequently for SDS denaturation of bR (4, 5, 27). Linear relationships also occur for beta barrel protein folding in urea (7, 10), but until now had not been demonstrated for urea unfolding of a helical membrane protein. The unfolding transition of GalP in urea occurs over a wide range of urea concentrations (1–6 M urea), as opposed to a very narrow range (0.6- to 0.8-mol fraction) of SDS concentrations for bR (cf. figures 2a and 1a of ref. 4), or the transmembrane region of DGK. This means that unfolding free energies can be obtained over a broad range of urea concentrations, such that only a relatively small extrapolation to zero urea is necessary. This gives a reliable determination of the unfolding free energy in the absence of denaturant.
GalP Stability.
The free energy for unfolding of GalP in DDM, in the absence of urea, 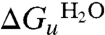 , is found to be +2.5 kcal·mol-1. This is about an order of magnitude less than the corresponding free energies determined for SDS unfolding of bR in L-α-1,2-dimyristoylphosphatidylcholine and CHAPS micelles of ∼+20 kcal·mol-1 and DGK in decylmaltoside (DM) of ∼+16 kcal·mol-1; both free energy values are those extrapolated to zero SDS. The GalP and bR unfolding reactions monitor a process where the protein loses a large proportion of the native helix content: ∼4 helices for GalP and ∼3 helices for bR, whereas only a small amount, if any, helix is lost for DGK (5). Thus the differences in unfolding free energy are not directly linked to a reduction in helicity, although GalP probably has helices that extend beyond the membrane in contrast to DGK and bR. The free energy differences are also unlikely to be due to the nature of the renaturing micelles because DDM and DM differ by only two carbons in their chains, but the free energy of unfolding GalP from DDM is less than that for DGK from DM. The differences in free energy are more likely to arise from a greater intrinsic stability of bR and DGK. bR and DGK also seem to be more thermally stable than GalP, losing activity above ∼80 °C or ∼70 °C, respectively (28, 29), whereas GalP has a broader transition and starts to unfold (as shown by decrease in protein fluorescence and reduction in secondary structure) above 40 °C (see Fig. S6).
, is found to be +2.5 kcal·mol-1. This is about an order of magnitude less than the corresponding free energies determined for SDS unfolding of bR in L-α-1,2-dimyristoylphosphatidylcholine and CHAPS micelles of ∼+20 kcal·mol-1 and DGK in decylmaltoside (DM) of ∼+16 kcal·mol-1; both free energy values are those extrapolated to zero SDS. The GalP and bR unfolding reactions monitor a process where the protein loses a large proportion of the native helix content: ∼4 helices for GalP and ∼3 helices for bR, whereas only a small amount, if any, helix is lost for DGK (5). Thus the differences in unfolding free energy are not directly linked to a reduction in helicity, although GalP probably has helices that extend beyond the membrane in contrast to DGK and bR. The free energy differences are also unlikely to be due to the nature of the renaturing micelles because DDM and DM differ by only two carbons in their chains, but the free energy of unfolding GalP from DDM is less than that for DGK from DM. The differences in free energy are more likely to arise from a greater intrinsic stability of bR and DGK. bR and DGK also seem to be more thermally stable than GalP, losing activity above ∼80 °C or ∼70 °C, respectively (28, 29), whereas GalP has a broader transition and starts to unfold (as shown by decrease in protein fluorescence and reduction in secondary structure) above 40 °C (see Fig. S6).
A lower stability of GalP, compared to bR, may reflect the flexibility of the GalP structure in terms of interactions between its two domains. An alternating access model has been proposed for MFS transport, where the hydrophilic binding pocket between the two domains opens to one side of the membrane and then the other, thus requiring dynamic interdomain contacts. The urea-induced reduction in helical structure in GalP could be due to either unfolding predominantly of one domain, or at the domain interface. Interestingly, denaturation by GuHCl seems to reveal two stages to GalP unfolding. GuHCl is a harsher denaturant than urea and the final GuHCl-unfolded state has a lower helix content than that in 8 M urea. Thus, part of GalP has high stability and, like bR, is resistant to urea denaturation. This too could partly reflect unfolding of one domain of GalP before the other in GuHCl. Another MFS transporter, LacY, has previously been shown to recover ligand binding after denaturation and refolding from intermediate concentrations of GuHCl (30).
Lipid Influence.
We have previously shown that variations in lipid composition and different bilayer forces that result can have a profound effect on folding yields (31, 32). Refolding of GalP from urea in DOPC/DOPE vesicles requires a high DOPE content of about 60 mol % DOPE. This contrasts with bR and the E. coli small multidrug transporter, EmrE, where high PE is detrimental to folding in vitro; the differences probably resulting from different folding mechanisms (31, 33). PE lipids have also been shown to be important for correct topology of other MFS transporters in E. coli membranes (19).
Conclusions
Reversible unfolding has rarely been demonstrated for α-helical membrane proteins, thus limiting measurements of thermodynamic stability. Here we show that urea denaturation leads to reversible unfolding of a multidomain, transmembrane α-helical transport protein. We report on the extent of helix reduction accompanying loss of ligand-binding activity and determine the free energy changes associated with complete recovery of this activity. This success is pertinent for many helical membrane proteins that have flexible structures and are frequently less stable in detergent micelles than lipid environments, instability that is usually accompanied by a reduction in ligand-binding affinity. Recovering correct ligand binding has often proved to be challenging. Here, we give an authoritative measure of the free energy for the final folding step, when native ligand binding is recovered, of a flexible, multidomain protein.
The ability to perform thermodynamic and kinetic measurements on folding from urea into DDM bodes well for extending our recent transition state analysis (9) to this very widespread class of MFS membrane transport proteins. Moreover, the success of refolding from urea into lipid vesicles raises the possibility of establishing reversible folding into lipid bilayers at equilibrium, which would herald a previously undescribed and much needed method for thermodynamic measurements of helical proteins in bilayers, as elegantly demonstrated for beta barrel proteins (7). Another area of future interest will be to probe the interactions between the two domains of GalP during folding, thus providing previously undescribed avenues of research into the molecular mechanisms of multidomain membrane proteins.
Materials and Methods
Protein Purification.
Histidine-tagged GalP was overexpressed in E. coli and purified as described (15). The purified protein was then exchanged into 50 mM sodium phosphate (pH 8), 1 mM DDM, and 1 mM β-mercaptoethanol (β-ME) by dialysis.
Equilibrium Unfolding.
GalP was unfolded by a 1∶5 dilution into 50 mM sodium phosphate (pH 8), 1 mM DDM, 1 mM β-ME, and 10 M urea and incubated at 20 °C for 2 min. The protein was then refolded by a tenfold dilution into buffer without denaturant and incubated for a further 10 min. Unfolding curves were constructed by the mixing of protein with a range of urea concentrations both during unfolding and refolding. GalP was also titrated with buffer containing guanidine hydrochloride or by unfolding in an alternative buffer composed of 50 mM sodium citrate (pH 4), 1 mM DDM, and 1 mM β-ME.
Circular Dichroism Spectroscopy.
CD spectra were measured using synchrotron sources and conventional spectrometers with specially adapted sample detection to eliminate scattering artefacts (see SI Text). The protein concentration was 15 μM during unfolding and 1.5 μM for refolded samples. The equilibrium unfolding curve was used for analysis to obtain a free energy value as the higher protein concentration in these experiments increased the signal-to-noise ratio. The unfolding curve was fitted to a two-state folding equation where the mean residue ellipticity θ = θF-θU(exp(m([denaturant]-Cm)/RT)/(1 + exp(m([denaturant]-Cm)/RT). θF and θU are the CD values of the folded and unfolded states, and Cm is the midpoint where there are equal amounts of folded and unfolded protein. The free energy of unfolding in the absence of denaturant is obtained from the fitted values, where 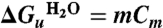 . The nonlinear regression was carried out using Grafit software (Erithacus), and the standard error of the best-fit curve was calculated from the residuals. This error was further multiplied by a t value of 2.080 to give a 95% confidence interval for the assigned free energy change.
. The nonlinear regression was carried out using Grafit software (Erithacus), and the standard error of the best-fit curve was calculated from the residuals. This error was further multiplied by a t value of 2.080 to give a 95% confidence interval for the assigned free energy change.
Fluorescence Spectroscopy.
Intrinsic protein fluorescence spectra were recorded at 20 ºC on a Fluoromax-2 fluorometer (Jobin-Yvon) in a 4-mm pathlength cell using a protein concentration of 0.5 μM. Samples were excited at 280 nm and emission spectra recorded from 300–400 nm (both 5-nm bandwidth). For ligand-binding experiments, aliquots of concentrated cytochalsin B were added to 2 mL of 0.5 μM GalP in a 1-cm pathlength cell, with 3 min between scans. Samples were excited at 280 nm (1-nm bandwidth) and the fluorescence at 330 nm (5-nm bandwidth) recorded.
Unfolding kinetics were measured using stopped-flow and steady state spectrophotometers (see SI Text). Changes in protein fluorescence could be used only to measure unfolding with high urea concentrations of ∼8 M urea. At lower urea concentrations only small changes in protein fluorescence intensity were observed, as the smaller decrease in Trp intensity during unfolding at lower urea is counteracted by loss of quenching between Trp residues (GalP contains 14 Trp residues).
Refolding into Lipids.
Large unilamellar vesicles composed of E. coli, polar lipids or varying compositions of DOPC and DOPE were prepared as previously described (34) in 50 mM sodium phosphate (pH 8), 1 mM β-ME at a concentration of 10 mg/mL and extruded to 100 nm in size. GalP was reconstituted by the detergent presaturation method (see SI Materials and Methods). For lipid folding experiments, GalP was unfolded and refolded as before, except the urea denaturation buffer contained no DDM to prevent detergent solubilization of the vesicles. Liposomes were again recovered by centrifugation, samples run out on SDS-PAGE, and stained with SYPRO red (Molecular Probes). Liposome-associated protein was calculated by densitometric analysis using AlphaEaseFC software (Innotech). Refolding yield was assayed by CD.
Transport Assay.
Proton transport was measured using the pH-sensitive dye phenol red (35). Liposomes of 100-nm diameter were prepared as above in 10 mM Hepes (pH 8), 1 mM β-ME, and GalP refolded as before. A 100-μL volume of liposomes was diluted into 2 mL 50 mM D-galactose, 1 mM β-ME, 60 μM phenol red, and the change in pH monitored by measuring the absorbance at 557 nm in a Cary spectrophotometer (Varian).
Supplementary Material
Acknowledgments.
We thank K. Bettaney, P. Sukumar, and D. Sharples for technical help, funded by the EU Grant 201924 EDICT. We are also grateful to R. Hope for growth of E. coli JM1100 (pPER3) in fermenters and preparation of subcellular membrane vesicles, D. Miller for help with data analysis, and P. Curnow for a critical appraisal of the manuscript. P.J.B. thanks the Royal Society for a Wolfson Merit Award. This research was funded by the European Membrane Protein consortium, EMeP LSHG-CT-2004-504601, and the Wellcome Trust 062164/Z/00/Z.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1005729107/-/DCSupplemental.
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Daggett V, Fersht A. The present view of the mechanism of protein folding. Nat Rev Mol Cell Biol. 2003;4:497–502. doi: 10.1038/nrm1126. [DOI] [PubMed] [Google Scholar]
- 3.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 4.Curnow P, Booth PJ. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc Natl Acad Sci USA. 2007;104:18970–18975. doi: 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 6.Barrera FN, et al. Unfolding and refolding in vitro of a tetrameric, alpha-helical membrane protein: The prokaryotic potassium channel KcsA. Biochemistry. 2005;44:14344–14352. doi: 10.1021/bi050845t. [DOI] [PubMed] [Google Scholar]
- 7.Hong H, Tamm LK. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc Natl Acad Sci USA. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisele JL, Rosenbusch JP. In vitro folding and oligomerization of a membrane protein. Transition of bacterial porin from random coil to native conformation. J Biol Chem. 1990;265:10217–10220. [PubMed] [Google Scholar]
- 9.Curnow P, Booth PJ. The transition state for integral membrane protein folding. Proc Natl Acad Sci USA. 2009;106:773–778. doi: 10.1073/pnas.0806953106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huysmans GH, Baldwin SA, Brockwell DJ, Radford SE. The transition state for folding of an outer membrane protein. Proc Natl Acad Sci USA. 2010;107:4099–4104. doi: 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson PJ, Giddens RA, Jones-Mortimer MC. Transport of galactose, glucose and their molecular analogues by Escherichia coli K12. Biochem J. 1977;162:309–320. doi: 10.1042/bj1620309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldwin SA, Henderson PJ. Homologies between sugar transporters from eukaryotes and prokaryotes. Annu Rev Physiol. 1989;51:459–471. doi: 10.1146/annurev.ph.51.030189.002331. [DOI] [PubMed] [Google Scholar]
- 13.Law CJ, Maloney PC, Wang DN. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol. 2008;62:289–305. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301:616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 15.Sanderson NM, Martin GE, Rutherford NG, Henderson PJ. Purification, reconstitution and circular dichroism of the galactose-H+ transport protein [GalP-(His)6] of Escherichia coli. Biochem Soc Trans. 1997;25:471S. doi: 10.1042/bst025471s. [DOI] [PubMed] [Google Scholar]
- 16.Patching SG, Henderson PJ, Herbert RB, Middleton DA. Solid-state NMR spectroscopy detects interactions between tryptophan residues of the E. coli sugar transporter GalP and the alpha-anomer of the D-glucose substrate. J Am Chem Soc. 2008;130:1236–1244. doi: 10.1021/ja075584k. [DOI] [PubMed] [Google Scholar]
- 17.Cairns MT, McDonald TP, Horne P, Henderson PJ, Baldwin SA. Cytochalasin B as a probe of protein structure and substrate recognition by the galactose/H+ transporter of Escherichia coli. J Biol Chem. 1991;266:8176–8183. [PubMed] [Google Scholar]
- 18.Walmsley AR, Lowe AG, Henderson PJ. The kinetics and thermodynamics of the binding of cytochalasin B to sugar transporters. Eur J Biochem. 1994;221:513–522. doi: 10.1111/j.1432-1033.1994.tb18763.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Bogdanov M, Dowhan W. Topology of polytopic membrane protein subdomains is dictated by membrane phospholipid composition. EMBO J. 2002;21:5673–5681. doi: 10.1093/emboj/cdf571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W, Bogdanov M, Pi J, Pittard AJ, Dowhan W. Reversible topological organization within a polytopic membrane protein is governed by a change in membrane phospholipid composition. J Biol Chem. 2003;278:50128–50135. doi: 10.1074/jbc.M309840200. [DOI] [PubMed] [Google Scholar]
- 21.Nagy JK, Lonzer WL, Sanders CR. Kinetic study of folding and misfolding of diacylglycerol kinase in model membranes. Biochemistry. 2001;40:8971–8980. doi: 10.1021/bi010202n. [DOI] [PubMed] [Google Scholar]
- 22.Chen GQ, Gouaux E. Probing the folding and unfolding of wild-type and mutant forms of bacteriorhodopsin in micellar solutions: Evaluation of reversible unfolding conditions. Biochemistry. 1999;38:15380–15387. doi: 10.1021/bi9909039. [DOI] [PubMed] [Google Scholar]
- 23.Klug CS, Su W, Liu J, Klebba PE, Feix JB. Denaturant unfolding of the ferric enterobactin receptor and ligand-induced stabilization studied by site-directed spin labeling. Biochemistry. 1995;34:14230–14236. doi: 10.1021/bi00043a030. [DOI] [PubMed] [Google Scholar]
- 24.Surrey T, Schmid A, Jahnig F. Folding and membrane insertion of the trimeric beta-barrel protein OmpF. Biochemistry. 1996;35:2283–2288. doi: 10.1021/bi951216u. [DOI] [PubMed] [Google Scholar]
- 25.Kleinschmidt JH, Wiener MC, Tamm LK. Outer membrane protein A of E. coli folds into detergent micelles, but not in the presence of monomeric detergent. Protein Sci. 1999;8:2065–2071. doi: 10.1110/ps.8.10.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patching SG, et al. Relative substrate affinities of wild-type and mutant forms of the Escherichia coli sugar transporter GalP determined by solid-state NMR. Mol Membr Biol. 2008;25:474–484. doi: 10.1080/09687680802371963. [DOI] [PubMed] [Google Scholar]
- 27.Faham S, et al. Side-chain contributions to membrane protein structure and stability. J Mol Biol. 2004;335:297–305. doi: 10.1016/j.jmb.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 28.Brouillette CG, Muccio DD, Finney TK. pH dependence of bacteriorhodopsin thermal unfolding. Biochemistry. 1987;26:7431–7438. doi: 10.1021/bi00397a035. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y, Bowie JU. Building a thermostable membrane protein. J Biol Chem. 2000;275:6975–6979. doi: 10.1074/jbc.275.10.6975. [DOI] [PubMed] [Google Scholar]
- 30.He MM, Kaback HR. In vitro folding of a membrane protein: Effect of denaturation and renaturation on substrate binding by the lactose permease of Escherichia coli. Mol Membr Biol. 1998;15:15–20. doi: 10.3109/09687689809027513. [DOI] [PubMed] [Google Scholar]
- 31.Allen SJ, Curran AR, Templer RH, Meijberg W, Booth PJ. Controlling the folding efficiency of an integral membrane protein. J Mol Biol. 2004;342:1293–1304. doi: 10.1016/j.jmb.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 32.Seddon AM, et al. Phosphatidylglycerol lipids enhance folding of an alpha helical membrane protein. J Mol Biol. 2008;380:548–556. doi: 10.1016/j.jmb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Miller D, et al. In vitro unfolding and refolding of the small multidrug transporter EmrE. J Mol Biol. 2009;393:815–832. doi: 10.1016/j.jmb.2009.08.039. [DOI] [PubMed] [Google Scholar]
- 34.Curran AR, Templer RH, Booth PJ. Modulation of folding and assembly of the membrane protein bacteriorhodopsin by intermolecular forces within the lipid bilayer. Biochemistry. 1999;38:9328–9336. doi: 10.1021/bi982322+. [DOI] [PubMed] [Google Scholar]
- 35.Ishmukhametov RR, Galkin MA, Vik SB. Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1Fo ATP synthase. Biochim Biophys Acta. 2005;1706:110–116. doi: 10.1016/j.bbabio.2004.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.