

Abstract
MinE is required for the dynamic oscillation of Min proteins that restricts formation of the cytokinetic septum to the midpoint of the cell in gram negative bacteria. Critical for this oscillation is MinD-binding by MinE to stimulate MinD ATP hydrolysis, a function that had been assigned to the first ∼30 residues in MinE. Previous models based on the structure of an autonomously folded dimeric C-terminal fragment suggested that the N-terminal domain is freely accessible for interactions with MinD. We report here the solution NMR structure of the full-length MinE dimer from Neisseria gonorrhoeae, with two parts of the N-terminal domain forming an integral part of the dimerization interface. Unexpectedly, solvent accessibility is highly restricted for residues that were previously hypothesized to directly interact with MinD. To delineate the true MinD-binding region, in vitro assays for MinE-stimulated MinD activity were performed. The relative MinD-binding affinities obtained for full-length and N-terminal peptides from MinE demonstrated that residues that are buried in the dimeric interface nonetheless participate in direct interactions with MinD. According to results from NMR spin relaxation experiments, access to these buried residues may be facilitated by the presence of conformational exchange. We suggest that this concealment of MinD-binding residues by the MinE dimeric interface provides a mechanism for prevention of nonspecific interactions, particularly with the lipid membrane, to allow the free diffusion of MinE that is critical for Min protein oscillation.
Keywords: protein structure, conformational dynamics, protein–protein interactions, ATPase
The dynamic three-dimensional subcellular organization of proteins in bacteria is emerging as an important mechanism for the regulation of many critical processes. The bacterial MinD and MinE proteins are a well-characterized example of this type of system, oscillating in a controlled manner from pole to pole to localize MinC such that cell division at nonproductive polar sites is prevented (1–4). Underlying this oscillation is a dynamic polymeric superstructure formed by MinD that extends out from the cell pole as a helical coil (5, 6). MinE preferentially localizes to the leading edge of this coil to form a concentrated annulus of MinE (Fig. 1A). This structure, known as the E ring, drives the ordered disassembly of the polymeric structure from the midcell back to the pole (7, 8). Concomitant establishment of a new Min polymer at the opposite cell pole initiates another cycle of growth and disassembly.
Fig. 1.

(A) Model of Min protein oscillation in rod-shaped bacteria. MinD forms a coiled array (red) that extends from the cell pole and is capped by a MinE-rich region known as the E ring (blue). Disassembly of the array toward the cell pole (in the direction of the blue arrow) is accompanied by assembly of a new MinD coil at the opposing pole (red arrow). (B) Schematic diagram of the Min cycle. (1) MinD (magenta) bound to ADP (light blue) undergoes nucleotide exchange with ATP (yellow), to give rise to a membrane-bound state that can bind MinC (red), the inhibitor of cell division septum formation. (2) MinE (blue) binding to MinD displaces MinC and (3) stimulates ATP hydrolysis by MinD to release inorganic phosphate (green). MinE is released from the complex while ADP-bound MinD also dissociates from the membrane, allowing the cycle to begin again. (C) Functional domain structure of MinE. Residues 1–30 comprise the anti-MinCD (ACD) domain while the remaining C-terminal residues contain the TSD.
The energy for Min protein oscillation is provided by the MinD-catalyzed hydrolysis of ATP (9), in a reaction cycle that is stimulated by MinE (Fig. 1B). The region of MinE thought to be responsible for this stimulation of MinD ATPase activity is known as the anti-MinCD domain and encompasses the first ∼30 residues of its amino acid sequence (10). A truncated MinE sample containing the remaining C-terminal residues [31–88 in the Escherichia coli (Ec) protein], also known as the topological specificity domain (TSD), has been shown to adopt an autonomous fold as a dimeric αβ-sandwich (11, 12). The Ec-TSD can interfere with the topologically specific activity of the full-length protein, because its overexpression in a WT background gives rise to products of polar cell division known as minicells (13–15).
Current models of MinE activity are based on the Ec-TSD structure and the observation that a peptide comprised of the first 22 residues of MinE is largely unstructured (12, 16). According to this model, the anti-MinCD domain is unfolded and solvent-exposed, allowing free interactions with membrane-bound MinD. However, we have shown that in the full-length MinE protein from Neisseria gonorrhoeae (Ng) the anti-MinCD domain is stably folded, with interactions involving the topological specificity domain (17).
Since the presence of structure in this part of the protein could significantly change current models of MinD-MinE interactions, we set out to determine the structure of a full-length MinE protein by solution NMR. This structure reveals an integral part of the dimeric interface in full-length MinE being formed by two regions of the anti-MinCD domain. We show using assays based on in vitro stimulation of MinD activity that residues located at solvent-inaccessible sites in the dimeric interface of this structure nonetheless play a direct role in binding to MinD. This sequestration of a protein-binding motif away from the aqueous environment helps to explain how MinE can minimize nonspecific interactions, an event that would significantly affect Min protein oscillation.
Results
Ng-MinE Structure.
Using standard heteronuclear solution NMR techniques, an ensemble of structures was determined for a full-length Ng-MinE sample. This was done using the E46A mutant, because it exhibited more favorable solubility characteristics than the wild-type protein, and it retained structural characteristics that were highly similar to that of WT as determined by circular dichroism and backbone NMR secondary chemical shifts (17, 18). As shown in Fig. 2, E46A Ng-MinE forms a homodimer, with each subunit being comprised of a three-strand β-sheet packed against an α-helix (αB, residues 39 to 54) in an approximately parallel orientation. Flanking the other side of the β-sheet is a shorter N-terminal α-helix from the other subunit in the dimer (αA, residues 3 to 8), which is roughly perpendicular with respect to the sheet.
Fig. 2.

(A) Ribbon diagram representation of the Ng-MinE structure determined by solution NMR, with the two views related by a 180° rotation. Each subunit is shown in either blue or purple, and anti-MinCD residues 1–30 that encompass αA and β1 are darker shades of these colors. Side chains for residues corresponding to those previously identified to be important for topological specificity function (Glu46, which is mutated to Ala in this structure, and Val50) are shown as balls. (B) Detailed view of the dimeric αB interface, highlighting residues participating in interhelical interactions. (C) Electrostatic surface representation for residues 20–84, in the same orientation as that shown on the right-hand side of A. The N-terminal α-helices (ribbons) pack against small hydrophobic patches that are flanked by negatively charged residues (red) that could attract the positively charged N terminus. (D) Schematic diagram highlighting the integral role for anti-MinCD residues 19–31 in the dimeric Ng-MinE β-sheet. The orientation and color scheme is the same as in the right-hand side of A. Amino acids forming interstrand backbone hydrogen bonds are linked by a solid line, with orange lines indicating hydrogen bonds that were also observed for homologous residues in the Ec-TSD structure (12). Residues homologous to those involved in intersubunit hydrogen bonds in the Ec-TSD structure are shown in orange (Asp73 hydrogen bonded to Leu81, Leu75 to Ile79 and Leu77 to Leu77). (E) Structure of Ec-MinE residues 32–83 (PDB ID code 1EV0) with secondary structure elements homologous to those in the Ng-MinE structure indicated with labels. The pairwise Cα rmsd for a single subunit of Ec-TSD and Ng-MinE is 2.9 Å (regular secondary structure elements only). (F) Amide protons that could be observed in 1H-15N HSQC spectra of WT Ng-MinE recorded at pH 9.5 (17) are highlighted in blue, indicating residues that are protected from base-catalyzed solvent exchange due to hydrogen bonding. The region corresponding to the part of the Hp structure engaged in intermolecular backbone hydrogen-bonding interactions is shown in green for comparison.
An extensive homodimeric interface supported by intermolecular NOEs is formed by intersubunit backbone hydrogen bonding between β1 strands and interactions involving hydrophobic side chains from these strands (Leu22, Ile24, and Ile26), whereas side chains from the two αB helices (Tyr39, Thr42, Leu43, Ala46, Val50, Tyr54, and Val55) also contribute to the dimeric interface (Fig. 2B). This part of the interface buries a total of 1,200 Å2 surface area per subunit. In addition, intermolecular NOEs involving the N-terminal αA helix were also assigned, showing interactions with the β-sheet (Fig. S1). The resulting structure shows that ∼300 Å2 per subunit of the β-sheet is buried by this part of the intermolecular interaction, with Ile4, Leu7, and Phe8 from αA making specific intersubunit contacts with a conserved hydrophobic patch formed by residues Ile25, Ala27, Met72, and Val74 (Fig. 2C). This dimeric structure could be independently confirmed by paramagnetic spin relaxation enhancement of amide resonances, which showed peak broadening that was consistent with intersubunit interactions involving β1 and αA (Fig. S2).
The integral role for N-terminal anti-MinCD domain residues at the dimeric interface was unexpected, because the C-terminal fragment containing residues 31–88 of Ec-MinE (Ec-TSD) forms a dimer that does not include any of these residues (12). Although the Ec-TSD structure contains the αB helix and strands β2 and β3, with the same interstrand hydrogen bonding and side chain interactions found in the full-length Ng-MinE structure, the dimeric interface is formed by intersubunit hydrogen bonds between two antiparallel β3 strands (Fig. 2 D and E). However, there is remarkable conservation of hydrogen-bonding amino acid pairs between the two structures [i.e., for the Ng-MinE sequence Leu75 bonds to Ile79 (full-length structure) or Ile26 (TSD structure), Leu77 to Leu77/Ile24, Ile79 to Leu75/Leu22, and Leu81 to Asp73/Asp20]. Meanwhile the αB helix also participates in extensive intersubunit interactions that are similar to those in our full-length Ng-MinE structure, with residues Ala46 and Val50 forming a patch at the dimeric interface analogous to that previously identified to be important for topological specificity function (Asp45/Val49 in Ec-MinE) (12). Overall these structural similarities show how a stable dimer can be formed by the TSD in the absence of the anti-MinCD domain and raise interesting questions regarding the functional relevance of the TSD structure.
During the preparation of this manuscript, coordinates for the 2.8-Å resolution X-ray crystal structure of MinE from Helicobacter pylori (Hp) were released (19), showing the same fold for the main body of the Ng-MinE structure with a Cα rmsd of 1.7 Å for regular secondary structure elements (Figs. S3 and S4). Like the Ng-MinE structure, a portion of the anti-MinCD domain in the Hp-MinE structure forms a β-strand at the dimeric interface. However, the first 12–15 residues of Hp-MinE were not visible, preventing the observation of an intermolecular interaction involving the N-terminal α-helix. In addition, the β1 interface appears to be less extensive in the crystal structure compared to that in the solution NMR structure. Specifically, the intermolecular hydrogen-bonding network identified in the crystal structure for β1 is shorter than that inferred for Ng-MinE from NOE and solvent-exchange data (Fig. 2F and Fig. S5). These structural differences may be a consequence of the shorter loops that are characteristic of proteins from extremophiles such as H. pylori, although the potential influence of crystal packing interactions on MinE structure cannot be ruled out as a contributing factor.
Identification of Residues Important for Stimulation of MinD Activity.
One of the unexpected aspects of the full-length Ng-MinE structure was the localization of residues previously identified to be important for anti-MinCD function (residues 20 to 30) at the dimeric interface (Fig. 2A). Even though they are not at all solvent accessible, mutations made at Leu22 and Ile25 have been shown to abrogate the ability of MinE to interact with MinD and disrupt the MinCD complex (10). As has been suggested from the Hp-MinE structure (19), a reasonable explanation for this is that these residues do not directly interact with MinD, but instead are critical for maintaining a dimeric folded state. Our NMR and CD studies on L22D Ng-MinE (17) showing significant loss of structure would be consistent with this hypothesis. Therefore, if these residues do not directly participate in MinD interactions, then anti-MinCD activity must reside in the more solvent-accessible regions of the anti-MinCD domain.
To identify true MinD-binding residues in Ng-MinE, we tested the ability of a series of mutants to stimulate Ng-MinD ATPase activity in the presence of phospholipid vesicles. As shown in Fig. 3A, for most mutants in the N-terminal region of Ng-MinE, no differences in stimulation of MinD ATP hydrolysis rates from WT levels were detected. In addition, the mutant used for structure determination (E46A) also stimulated MinD activity to levels that were indistinguishable from that of WT Ng-MinE. However, two mutants previously identified to be deficient in anti-MinCD activity, namely A18D and the structurally perturbed L22D (20), showed a complete loss of MinD ATPase stimulation. R21A and K19A also showed lower activity, suggesting that the MinD interaction mainly involves the flexible loop and part of the central β-strand.
Fig. 3.

Specific activity of Ng-MinD stimulated by Ng-MinE determined in vitro. (A) Comparison of activity stimulated by either WT Ng-MinE or the indicated mutant. For most mutants there was a ∼10-fold increase in MinD ATPase activity compared to basal levels measured in the absence of MinE, similar to previous observations (9, 20). (B) Concentration dependence of in vitro Ng-MinD specific activity for full-length Ng-MinE (open squares), Ng-MinE1-22 (gray triangles), and MinE1-27 (filled circles). (C) Same as B for Ng-MinE1-27 I25R (filled circles) or I24R (open circles) mutant peptides. Lines represent best-fit curves to the Hill equation.
To determine whether any part of MinE outside of this region is also involved in MinD interactions, a peptide comprised of residues 1–22 Ng-MinE (Ng-MinE1-22) was tested for Ng-MinD ATPase activity stimulation over a range of peptide concentrations. Assuming that the rate of ATP hydrolysis is directly proportional to the amount of MinE bound, then the change in activity as a function of MinE concentration can be fit to the Hill equation. In this analysis the concentration of Ng-MinE required to reach half-maximal stimulation (K0.5) can be considered to be a measure of the relative affinity of the interaction between Ng-MinE and Ng-MinD (21). As shown in Fig. 3B, both the full-length and N-terminal Ng-MinE1-22 samples were capable of stimulating Ng-MinD ATPase activity to the same maximal level; however, the K0.5 values were significantly different between the two samples. As summarized in Table 1, K0.5 for full-length Ng-MinE was 0.11 μM, whereas approximately five times more Ng-MinE1-22 peptide was required to achieve the same level of activation. Also, the R21A mutant of this peptide, as well as a truncated version (Ng-MinE1-17), did not show any stimulation of ATPase activity, even at concentrations of 20 μM (Fig. S6). Taken together, these results show that the residues required for maximal stimulation of MinD activity are present in residues 18–22, but that residues beyond this N-terminal region also contribute to interactions with MinD.
Table 1.
Kinetic parameters for Ng-MinE-stimulated ATP hydrolysis by Ng-MinD as determined by fitting to the Hill equation*
| Ng-MinE† | K0.5, μM | Vmax, nmol/ min /mg MinD | h |
| WT | 0.11 ± 0.02 | 37.5 ± 1.5 | 2.9 ± 0.3 |
| E46A | 0.07 ± 0.01 | 31 ± 10 | 3.2 ± 0.1 |
| 1-22 | 0.5 ± 0.4 | 39.2 ± 2.3 | 2.2 ± 0.1 |
| 1-22 (L22A) | 5 ± 3 | 35.4 ± 4.9 | 1.4 ± 0.1 |
| 1-27 | 0.08 ± 0.02 | 36.2 ± 1.1 | 3.4 ± 0.7 |
| 1-27 (I25R) | 3.8 ± 0.7 | 42.5 ± 1.6 | 1.5 ± 0.1 |
| 1-27 (I24R) | 0.05 ± 0.01 | 39.9 ± 4.2 | 2.2 ± 0.7 |
| 1-27 (I25A) | 0.12 ± 0.05 | 43.1 ± 6.1 | 2.6 ± 0.1 |
| 1-27 (I24A) | 0.08 ± 0.04 | 37.5 ± 1.3 | 3.2 ± 0.5 |
*All values represent the average of kinetic parameters obtained from individual fits done for three separate experiments with different Ng-MinD sample preparations.
†Peptides are indicated with number range of residues included in the sequence, with any mutations indicated in parentheses.
Investigation of the Role of β1-Residues in Stimulation of MinD ATPase Activity.
The ability to use peptides to test stimulation of MinD ATPase activity allowed us to address the specific role of Leu22 as well as additional regions of the anti-MinCD domain in the MinD interaction. Although the L22D mutant of the full-length protein was not active, this may have been due to the significant structural perturbation that results from the introduction of a negative charge in the hydrophobic core (17). However, if Leu22 plays a direct role in MinD interactions, then an L22D version of Ng-MinE1-22 would also be expected to show reduced activity relative to the WT peptide, because the activity of the peptide does not require the globular core. This is in fact what was observed because L22D Ng-MinE1-22 failed to stimulate MinD ATPase activity over a wide range of concentrations. To evaluate whether this effect was specific for the aspartic acid mutant, we also performed this analysis with the more conservative L22A mutant of Ng-MinE1-22. In this case, although maximal MinD activity could be attained at very high concentrations of the peptide, K0.5 was almost 10-fold larger than that of the WT peptide (Table 1), providing strong evidence that Leu22 participates directly in MinD interactions.
The identification of a direct role for MinD interactions involving Leu22, a residue that is in a solvent-inaccessible position of the intersubunit interface of the Ng-MinE dimer, raised the possibility that other residues in this region of the structure may also play a similar role. For this reason, a longer N-terminal peptide, MinE1-27, was tested in the same ATPase stimulation assay. As shown in Fig. 3C, this peptide was more active than Ng-MinE1-22, with a profile that was almost the same as that of the full-length protein, including a K0.5 of 0.08 μM (Table 1). To determine whether this apparent increase in affinity provided by residues 23 to 27 was specific, we repeated the experiment using a mutant that had previously been identified to be defective for anti-MinCD activity, namely I25R (10). Although the I25R mutant of this peptide was capable of fully stimulating MinD activity, approximately 30-fold more peptide was required to reach half-maximal levels relative to the WT peptide (Fig. 3C and Table 1). In contrast, when I24R Ng-MinE1-27 was tested in the same assay, rate profiles were highly similar to those from the WT peptide. A smaller effect was observed for the more conservative I25A mutant of Ng-MinE1-27, although its apparent affinity was still weaker than that of the WT peptide. Overall these data provide strong evidence that Ile25 is one of the residues directly involved in the interaction between full-length MinE and MinD.
Evidence for Conformational Dynamics in MinE.
When the residues identified to be important for stimulation of Ng-MinD activity are mapped onto the Ng-MinE structure, it is apparent that many are in regions that cannot easily be accessed by a macromolecular binding partner (Fig. 4A). For example, although the side chains of Arg21 and Ile25 are on the same side of β1, approach to these residues would be hindered by the presence of the N-terminal helix and adjacent loop. However, if MinE undergoes dynamic exchange with a more open conformation, this could provide a mechanism for MinD to access these residues. To examine the internal dynamics of MinE, 15N spin relaxation experiments were acquired. These demonstrated that the loop connecting αA and β1 does undergo significant backbone dynamics on the nanosecond to picosecond time scale (Fig. S7).
Fig. 4.

(A) Residues important for stimulation of MinD-catalyzed hydrolysis are either partly solvent accessible (yellow side chains) or completely inaccessible (red) in the dimeric structure of full-length Ng-MinE. (B) Representative relaxation dispersion curves for backbone amide groups at 800 MHz (red) and 500 MHz (black) shown for the three residues indicated. (C) Backbone amides undergoing statistically significant exchange (> 99% confidence by F test) are shown as balls, where the diameter is linearly scaled according to the measured exchange rate (300 s-1–3100 s-1). Most exchange sites cluster to a contiguous region on each end of the dimer (yellow). A structurally distinct site of exchange was also detected for αC (white). No exchange was detected for amides in the αB helices.
A series of backbone amide relaxation dispersion measurements designed to probe the presence of motion on the microsecond to millisecond time scale also revealed motions extending to other regions of the protein. Contributions to transverse relaxation from exchange processes manifest as increased line broadening that can be compensated by a Carr–Purcell–Meiboom–Gill (CPMG) spin echo pulse train sequence (reviewed in ref. 22). Variations in the apparent transverse relaxation rate (R2,eff) as a function of CPMG pulse frequency (vCPMG) are indicative of conformational exchange. Our results (Fig. 4 B and C) show microsecond to millisecond time-scale exchange in residues from the N-terminal helix that interact with the β-sheet, and in the corresponding residues within this sheet proximal to this interaction. The localization of these residues suggests a motion that could involve dissociation of the N-terminal α-helix from the central β-sheet, a change that would significantly enhance access to Ile25 and Arg21 for MinD interactions. This model would also be consistent with the small surface area associated with this interaction that predicts a relatively weak association for the αA helix, and with the absence of electron density for this part of the Hp-MinE crystal structure (19).
In addition to the αA helix and adjacent loops, a number of residues in β1 (Asp20, Leu22, and Ile24) also showed evidence for dynamics on this time scale. This exchange may reflect the change in local chemical environment that would occur upon dissociation of the N-terminal helix. However, it is also possible that structural changes in these regions accompany this dissociation. Most significantly, conformational flexibility at the ends of β1 could provide a mechanism for MinD to access residues such as Leu22, whose side chain is completely concealed in the hydrophobic core of the dimeric interface. Some insight into the nature of this conformational change is provided by the Hp-MinE structure (19), which shows the ends of β1 diverging away from the interface (Fig. S3). Although the side chain corresponding to Leu22 is still completely buried in the Hp-MinE structure, this more open state may represent an intermediate on the path toward larger structural perturbations that might be induced by MinD binding.
Discussion
A long-standing model of the MinE-MinD interaction involving a solvent-exposed, helix-forming anti-MinCD domain (10) has been challenged by the detection of β-structure in the anti-MinCD domain (17), the partial structure of Hp-MinE (19), and now by our solution NMR structure of full-length Ng-MinE. In particular, the structures show that residues previously identified to be important for anti-MinCD function are buried in the β1 dimeric interface. Given the central role for this region in dimerization, it is likely that mutation of non-solvent-accessible residues in this region that were previously shown to abrogate anti-MinCD function (e.g., L22D, I25R) would significantly perturb this dimeric structure. However, the in vitro MinD ATPase activities obtained with anti-MinCD domain peptides suggest that the loss of binding affinity for MinD exhibited by these mutants is not a consequence of this structural change, but instead is primarily due to the elimination of large hydrophobic side chains that directly participate in the interaction. Therefore, in order for MinD to bind these residues in the full-length protein, some type of conformational change must occur to increase their accessibility. The conformational exchange we detected in the anti-MinCD domain does provide a mechanism for increased access to some of these MinD-binding residues. However, it is still not clear how MinD might simultaneously bind all of these residues in the dimer structure.
One scenario that could improve solvent accessibility for MinD-binding residues that has yet to be explored is one where the MinE dimer dissociates into a monomer prior to its association with MinD. Dissociation of the dimer would give rise to a significant increase in solvent accessibility for the anti-MinCD domain that could make the monomer the higher affinity ligand for MinD. In fact, the demonstration that the MinE1-27 fragment reconstitutes in vitro MinD activation with properties that are virtually indistinguishable from that of the full-length protein is consistent with this possibility (Fig. 3B). Previous yeast two-hybrid and gel filtration data with Ec-MinE1-31 showed that this fragment does not self-associate (13), suggesting that dimerization is not necessary to stimulate MinD activity.
A dissociation constant of 0.6 μM was previously determined for Ec-MinE dimerization (14), suggesting that a significant population of MinE should be monomeric at low μM concentrations. However, this may not be the case for Ng-MinE, because sedimentation velocity experiments showed no distinct monomer species, nor any significant shift in the molecular mass of the dimer (Fig. S8). This was the case even when sample concentrations at the limit of detection were used (3 μM), conditions that should contain ∼25% monomer if the dissociation constant were in this range. Backbone amide protection factors obtained from solvent-exchange experiments are also consistent with a strongly associated Ng-MinE dimer (Fig. S5), with the highest protection observed for central β1 residues. Nonetheless, a high affinity dimerization constant does not rule out the potential presence of low concentrations of the monomer that could be sufficient for maximal stimulation of ATP hydrolysis by MinD. Adding to the complexity is the fact that the actual association properties of MinE in vivo, and under in vitro assay conditions, are likely to be multifaceted. In particular, the higher-order structures formed by MinD give rise to elevated local concentrations of MinE that would be expected to favor its self-association. This effect should also be in operation in vitro, because dynamic mesostructures have been demonstrated for MinD on the surface of planar lipid bilayers, with a nonuniform colocalization of MinE (23, 24). In our assay we obtained Hill coefficients that are greater than two (Table 1), suggesting that a polymeric structure functionally linking multiple MinE-binding sites was also reconstituted under these conditions. However, in the absence of structural details for the MinD polymer, it is not possible to predict whether a MinE dimer or monomer would interact more favorably. Because of these considerations, in addition to other general effects that could alter the association properties of MinE (e.g., volume exclusion/molecular crowding, lipid protein interactions, etc.), the oligomerization state of MinE that is most relevant for MinD binding and E-ring formation remains an interesting question to be investigated.
Although the role of the dimeric state for MinE in the interaction with MinD is still not clear, there is one significant advantage that should be provided by the concealment of MinD-binding residues. Specifically, promiscuous interactions between MinE and other prospective binding partners should be greatly reduced by sequestering the anti-MinCD domain away from the solvent. Of particular relevance are the nonspecific interactions that could be expected to occur between hydrophobic portions of the anti-MinCD domain and the membrane. It has been shown that, in contrast with full-length MinE, the isolated anti-MinCD domain (Ec-MinE1-31) localizes to the membrane in vivo, even in the absence of MinD (10). Similarly, mutations made to full-length Ec-MinE that should disrupt the structure at the β1 interface, namely L22R, L22S, and I25R, also gave rise to this same pattern of MinD-independent membrane localization. These results show that free exposure of the full anti-MinCD domain to solvent gives rise to membrane-localized MinE, and that there is no topological specificity to this localization. This would have important functional consequences, because strong interactions with the membrane could inhibit dynamic Min protein localization by preventing the free diffusion of unbound MinE through the cytoplasm that computational models suggest is a fundamental requirement for oscillation (reviewed in ref. 25). In addition, the MinD/Par superfamily of proteins has been suggested to have a conserved mechanism for stimulation of ATPase activity (26), raising the possibility that a freely exposed anti-MinCD domain could interact with other members of this family. Whatever the potential binding partner, localization of key MinD-binding residues to the MinE dimeric interface minimizes the potential for nonspecific interactions. Sequestration of these residues away from the solvent therefore provides a mechanism to allow unhindered diffusion of MinE through the cytoplasm in regions where concentrations of membrane-bound MinD are low, allowing normal Min protein oscillation to occur.
Materials and Methods
Protein Expression, Purification, and Mutagenesis.
Plasmids used for expression of WT, E46A, L22D, and A18D Ng-MinE and WT Ng-MinD were as described previously (17, 20). All other mutants of the full-length Ng-MinE were generated by site-directed mutagenesis of the WT plasmid, using the QuikChange Site-Directed mutagenesis protocol (Stratagene). All Min protein constructs expressed Ng-Min proteins appended at the C-terminal to a sequence of eight additional residues (LEHHHHHH). Expression and purification were done as previously described (17, 20). Sedimentation velocity experiments confirmed a dimeric state for these samples (Fig. S8).
In Vitro MinD ATPase Assay.
Stimulation of Ng-MinD ATP hydrolysis by Ng-MinE was monitored using the malachite green method (27), and the MinE-dependent activation fit to the Hill equation to extract Vmax, K0.5, and h. Details for the assay and analysis are provided in SI Text.
NMR Spectroscopy.
NMR spectra were recorded at 298 K on either a 500- or an 800-MHz Varian Unity Inova spectrometer equipped with a cryoprobe. 1H, 15N, and 13C resonance assignments were obtained for E46A Ng-MinE as described elsewhere (18). NMR samples were typically 0.7–1.0 mM of uniformly labeled 15N- or 13C, 15N-labeled E46A Ng-MinE in a buffer containing 22.5 mM Tris-HCl at pH 7.2, 45 mM NaCl, 0.1 mM EDTA, 0.2 mM benzamidine, 0.02% NaN3, and 1 mM 2,2-dimethyl-2-silapentane-5-sulphonic acid. A hetero-dimeric protein sample was also prepared by mixing equimolar amounts of 15N- and 13C,15N-labeled E46A Ng-MinE in 6 M guanidinium hydrochloride. This sample was refolded on a Ni-NTA column by a stepwise buffer exchange into denaturant-free buffer and repurified by size-exclusion chromatography.
Measurement of one-bond 1H-15N residual dipolar couplings (RDCs) of backbone amides was done using a stretched 3% (wt/vol) polyacrylamide gel (28) with 15N in-phase/antiphase single-quantum coherence (IPAP-HSQC) spectra (29). Spectra were processed and analyzed using NMRPipe (30), NMRView (31), and SPARKY (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco).
Structure Calculation.
NOE distance restraints were inferred from 3D 15N-edited NOESY-HSQC (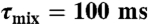 ) (32–34), aliphatic 13C-edited NOESY-HSQC (
) (32–34), aliphatic 13C-edited NOESY-HSQC (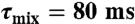 ) (35), and 3D 13C-edited methyl–methyl NOESY (τmix = 75 ms) (36) spectra, and used in the molecular simulation program XPLOR-NIH (37) to generate an initial model for E46A Ng-MinE. This model was then used as the starting structure for the torsion angle dynamics program CYANA 2.1 (38) and the automatic NOE assignment invoked to calculate 200 conformers. Based on hydrogen exchange data and the presence of characteristic secondary structure NOEs (17), 26 hydrogen bonds/subunit were also added to the restraint set. A total of 1,849 unique and nonredundant NOE distance constraints/subunit were generated from the NOESY spectra. In addition, 68 intermolecular upper-bound restraints per subunit derived from 3D F1-13C, 15N-filtered/F3-13C edited NOESY spectrum (τmix = 80 ms) (39) were added as input restraints.
) (35), and 3D 13C-edited methyl–methyl NOESY (τmix = 75 ms) (36) spectra, and used in the molecular simulation program XPLOR-NIH (37) to generate an initial model for E46A Ng-MinE. This model was then used as the starting structure for the torsion angle dynamics program CYANA 2.1 (38) and the automatic NOE assignment invoked to calculate 200 conformers. Based on hydrogen exchange data and the presence of characteristic secondary structure NOEs (17), 26 hydrogen bonds/subunit were also added to the restraint set. A total of 1,849 unique and nonredundant NOE distance constraints/subunit were generated from the NOESY spectra. In addition, 68 intermolecular upper-bound restraints per subunit derived from 3D F1-13C, 15N-filtered/F3-13C edited NOESY spectrum (τmix = 80 ms) (39) were added as input restraints.
XPLOR-NIH was subsequently used to subject the structure with the lowest target-function value from CYANA 2.1 to refinement against measured 1H-15N RDCs. Phi/Psi dihedral angles, which were derived from the program TALOS (40), were also included in the set of constraints. The 10 lowest energy structures from a calculation of 200 structures were used to represent the NMR ensemble (statistics in Table S1).
15N Relaxation Dispersion Experiments.
In order to evaluate chemical exchange, an improved version (41) of the 15N CPMG (42, 43) pulse sequence was employed at 303 K and two magnetic fields (500 and 800 MHz), using a CPMG relaxation delay time of 30 ms. Effective relaxation rates 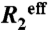 were determined from peak intensities as previously described (44). Uncertainty in effective relaxation rates were estimated from duplicates. Assuming a two-state exchange model, the relaxation rates were fitted on a per-residue basis using CATIA (available from http://pound.med.utoronto.ca/software.html). No relaxation rates were obtained for proline residues (37, 41, and 82), or overlapped and weak resonances (Met1, Ser2, Leu3, Lys13, Thr14, Ala15, Thr16, Gln23, Ile26, Tyr39, Leu40, Tyr54, Gln66, Lys68, Asp70, Asp73, Leu77, and Ile79).
were determined from peak intensities as previously described (44). Uncertainty in effective relaxation rates were estimated from duplicates. Assuming a two-state exchange model, the relaxation rates were fitted on a per-residue basis using CATIA (available from http://pound.med.utoronto.ca/software.html). No relaxation rates were obtained for proline residues (37, 41, and 82), or overlapped and weak resonances (Met1, Ser2, Leu3, Lys13, Thr14, Ala15, Thr16, Gln23, Ile26, Tyr39, Leu40, Tyr54, Gln66, Lys68, Asp70, Asp73, Leu77, and Ile79).
Supplementary Material
Acknowledgments.
For expert technical assistance we acknowledge Tara Sprules at the Quebec/Eastern Ontario High Field NMR facility in Montreal, Québec, and Kim Munro at the Protein Function Discovery facility. We are grateful to Anthony Mittermaier for helpful discussions, and Lewis Kay for critical reading of this manuscript. This work was supported by Canadian Institutes of Health Research operating Grant MOP-160206 (to N.K.G. and J.R.D.), and an Early Researcher Award (to N.K.G.). H.G. was supported by the Sweden–America Foundation and Knut and Alice Wallenberg Foundation, and C.T.H. was supported by an Ontario Graduate Scholarship. N.C. and A.A.B. are both recipients of Natural Sciences and Engineering Research Council Undergraduate Research Scholarships.
Footnotes
The authors declare no conflict of interest.
Data deposition: Atomic coordinates and NMR restraint files have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2KXO).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1007141107/-/DCSupplemental.
References
- 1.Lutkenhaus J. Min oscillation in bacteria. Adv Exp Med Biol. 2008;641:49–61. doi: 10.1007/978-0-387-09794-7_4. [DOI] [PubMed] [Google Scholar]
- 2.Hu Z, Lutkenhaus J. Topological regulation of cell division in Escherichia coli involves rapid pole to pole oscillation of the division inhibitor MinC under the control of MinD and MinE. Mol Microbiol. 1999;34:82–90. doi: 10.1046/j.1365-2958.1999.01575.x. [DOI] [PubMed] [Google Scholar]
- 3.Raskin DM, de Boer PA. MinDE-dependent pole-to-pole oscillation of division inhibitor MinC in Escherichia coli. J Bacteriol. 1999;181:6419–6424. doi: 10.1128/jb.181.20.6419-6424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raskin DM, de Boer PA. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc Natl Acad Sci USA. 1999;96:4971–4976. doi: 10.1073/pnas.96.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shih YL, Le T, Rothfield L. Division site selection in Escherichia coli involves dynamic redistribution of Min proteins within coiled structures that extend between the two cell poles. Proc Natl Acad Sci USA. 2003;100:7865–7870. doi: 10.1073/pnas.1232225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szeto J, et al. A conserved polar region in the cell division site determinant MinD is required for responding to MinE-induced oscillation but not for localization within coiled arrays. Res Microbiol. 2005;156:17–29. doi: 10.1016/j.resmic.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Fu X, Shih YL, Zhang Y, Rothfield LI. The MinE ring required for proper placement of the division site is a mobile structure that changes its cellular location during the Escherichia coli division cycle. Proc Natl Acad Sci USA. 2001;98:980–985. doi: 10.1073/pnas.031549298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hale CA, Meinhardt H, de Boer PA. Dynamic localization cycle of the cell division regulator MinE in Escherichia coli. EMBO J. 2001;20:1563–1572. doi: 10.1093/emboj/20.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Z, Lutkenhaus J. Topological regulation of cell division in E. coli. Spatiotemporal oscillation of MinD requires stimulation of its ATPase by MinE and phospholipid. Mol Cell. 2001;7:1337–1343. doi: 10.1016/s1097-2765(01)00273-8. [DOI] [PubMed] [Google Scholar]
- 10.Ma LY, King G, Rothfield L. Mapping the MinE site involved in interaction with the MinD division site selection protein of Escherichia coli. J Bacteriol. 2003;185:4948–4955. doi: 10.1128/JB.185.16.4948-4955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King GF, et al. The dimerization and topological specificity functions of MinE reside in a structurally autonomous C-terminal domain. Mol Microbiol. 1999;31:1161–1169. doi: 10.1046/j.1365-2958.1999.01256.x. [DOI] [PubMed] [Google Scholar]
- 12.King GF, et al. Structural basis topological specificity function of MinE. Nat Struct Biol. 2000;7:1013–1017. doi: 10.1038/80917. [DOI] [PubMed] [Google Scholar]
- 13.Pichoff S, Vollrath B, Touriol C, Bouché JP. Deletion analysis of gene MinE which encodes the topological specificity factor of cell division in Escherichia coli. Mol Microbiol. 1995;18:321–329. doi: 10.1111/j.1365-2958.1995.mmi_18020321.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. The relationship between hetero-oligomer formation and function of the topological specificity domain of the Escherichia coli MinE protein. Mol Microbiol. 1998;30:265–273. doi: 10.1046/j.1365-2958.1998.01059.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhao CR, de Boer PA, Rothfield LI. Proper placement of the Escherichia coli division site requires two functions that are associated with different domains of the MinE protein. Proc Natl Acad Sci USA. 1995;92:4313–4317. doi: 10.1073/pnas.92.10.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King GF, et al. The dimerization and topological specificity functions of MinE reside in a structurally autonomous C-terminal domain. Mol Microbiol. 1999;31:1161–1169. doi: 10.1046/j.1365-2958.1999.01256.x. [DOI] [PubMed] [Google Scholar]
- 17.Ramos D, et al. Conformation of the cell division regulator MinE: Evidence for interactions between the topological specificity and anti-MinCD domains. Biochemistry. 2006;45:4593–4601. doi: 10.1021/bi060022j. [DOI] [PubMed] [Google Scholar]
- 18.Ducat T, Goto NK. 1H, 13C, 15N chemical shift assignments for the Neisseria gonorrhoeae MinE regulator of cell division septum placement. Biomol NMR Assign. 2010 doi: 10.1007/s12104-010-9247-4. 10.1007/s12104-010-9247-4. [DOI] [PubMed] [Google Scholar]
- 19.Kang GB, et al. Crystal structure of Helicobacter pylori MinE, a cell division topological specificity factor. Mol Microbiol. 2010;76:1222–1231. doi: 10.1111/j.1365-2958.2010.07160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eng NF, et al. The C-terminus of MinE from Neisseria gonorrhoeae acts as a topological specificity factor by modulating MinD activity in bacterial cell division. Res Microbiol. 2006;157:333–344. doi: 10.1016/j.resmic.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Weiss JN. The Hill equation revisited: Uses and misuses. FASEB J. 1997;11:835–841. [PubMed] [Google Scholar]
- 22.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 23.Ivanov V, Mizuuchi K. Multiple modes of interconverting dynamic pattern of formation by bacterial cell division proteins. Proc Natl Acad Sci USA. 2010;107:8071–8078. doi: 10.1073/pnas.0911036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loose M, et al. Spatial regulators for bacterial cell division self-organize into surface waves in vitro. Science. 2008;320:789–792. doi: 10.1126/science.1154413. [DOI] [PubMed] [Google Scholar]
- 25.Kruse K, Howard M, Margolin W. An experimentalist’s guide to computational modelling of the Min system. Mol Microbiol. 2007;63:1279–1284. doi: 10.1111/j.1365-2958.2007.05607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leonard TA, Butler PJ, Löwe J. Bacterial chromosome segregation: Structure and DNA binding of the Soj dimer—A conserved biological switch. EMBO J. 2005;24:270–282. doi: 10.1038/sj.emboj.7600530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harder KW, et al. Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase beta (HPTP-beta) using synthetic phosphopeptides. Biochem J. 1994;298:395–401. doi: 10.1042/bj2980395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou JJ, et al. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 29.Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 30.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 32.Marion D, et al. Three-dimensional heteronuclear NMR of 15N-labeled proteins. J Am Chem Soc. 1989;111:1515–1517. [Google Scholar]
- 33.Talluri S, Wagner G. An optimized 3D NOESY-HSQC. J Magn Reson Ser B. 1996;112:200–205. doi: 10.1006/jmrb.1996.0132. [DOI] [PubMed] [Google Scholar]
- 34.Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 35.Muhandiram DR, et al. A gradient 13C NOESY-HSQC experiment for recording NOESY spectra of 13C-labeled proteins dissolved in H2O. J Magn Reson Ser B. 1993;102:317–321. [Google Scholar]
- 36.Zwahlen C, et al. An NMR experiment for measuring methyl-methyl NOEs in 13C-labeled proteins with high resolution. J Am Chem Soc. 1998;120:7617–7625. [Google Scholar]
- 37.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 38.Güntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 39.Zwahlen C, et al. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage λ N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
- 40.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 41.Hansen DF, Vallurupalli P, Kay LE. An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J Phys Chem B. 2008;112:5898–5904. doi: 10.1021/jp074793o. [DOI] [PubMed] [Google Scholar]
- 42.Carr HY, Purcell EM. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev. 1954;94:630–638. [Google Scholar]
- 43.Meiboom S, Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev Sci Instrum. 1958;29:688–691. [Google Scholar]
- 44.Tollinger M, et al. Slow dynamics in folded and unfolded states of an SH3 domain. J Am Chem Soc. 2001;123:11341–11352. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.