

Abstract
Modification of cellular proteins with O-GlcNAc (O-linked N-acetylglucosamine) competes with protein phosphorylation and regulates a plethora of cellular processes. O-GlcNAcylation is orchestrated by two opposing enzymes, O-GlcNAc transferase and OGA (O-GlcNAcase or β-N-acetylglucosaminidase), which recognize their target proteins via as yet unidentified mechanisms. In the present study, we uncovered the first insights into the mechanism of substrate recognition by human OGA. The structure of a novel bacterial OGA orthologue reveals a putative substrate-binding groove, conserved in metazoan OGAs. Guided by this structure, conserved amino acids lining this groove in human OGA were mutated and the activity on three different substrate proteins [TAB1 (transforming growth factor-β-activated protein kinase 1-binding protein 1), FoxO1 (forkhead box O1) and CREB (cAMP-response-element-binding protein)] was tested in an in vitro deglycosylation assay. The results provide the first evidence that human OGA may possess a substrate-recognition mechanism that involves interactions with O-GlcNAcylated proteins beyond the GlcNAc-binding site, with possible implications for differential regulation of cycling of O-GlcNAc on different proteins.
Keywords: β-N-acetylglucosaminidase, O-linked N-acetylglucosamine (O-GlcNAc), peptide recognition groove, protein glycosylation
Abbreviations: CpNagJ, Clostridium perfringens NagJ; CREB, cAMP-response-element-binding protein; Fmoc, fluoren-9-ylmethoxycarbonyl; FoxO1, forkhead box O1; GST, glutathione transferase; HAT, histone acetyltransferase; HEK, human embryonic kidney; LC, liquid chromatography; 4MU-GlcNAc, 4-methylumbelliferyl-β-N-acetylglucosamine; OGA, O-GlcNAcase or β-N-acetylglucosaminidase; hOGA, human OGA; OgOGA, Oceanicola granulosus OGA; OGT, O-GlcNAc transferase; O-GlcNAc, O-linked N-acetylglucosamine; pNP-GlcNAc, p-nitrophenyl-β-N-acetylglucosamine; PUGNAc, O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate; RMSD, root mean square deviation; TAB1, transforming growth factor-β-activated protein kinase 1-binding protein 1
INTRODUCTION
The O-GlcNAc (O-linked N-acetylglucosamine) modification is a dynamic and reversible form of protein glycosylation occurring on specific serine and threonine residues of intracellular proteins [1,2]. Since the initial discovery of O-GlcNAc [3], technological advances have greatly facilitated its detection, and proteomics studies [4–6] have shown that a significant proportion of cellular proteins are O-GlcNAcylated. However, the functional importance of O-GlcNAc is only just emerging, with evidence to suggest that it may regulate protein activity in a manner analogous (and complementary) to phosphorylation [7]. O-GlcNAc levels are known to respond dynamically to nutrient availability [1] and stress [8], and to undergo changes during the cell cycle [9] and development [10]. O-GlcNAc has been shown to be associated with a range of human diseases [2]. Strikingly, only two enzymes orchestrate the O-GlcNAc modification. Both the OGT (O-GlcNAc transferase) and its antagonistic OGA (O-GlcNAcase or β-N-acetylglucosaminidase) are encoded by single genes in metazoa [11,12]. Little is known about the mode of substrate recognition of either enzyme, although studies have hinted at the existence of a defined O-GlcNAc peptide sequon [2,5]. In addition, it is thought that, for a single OGT to modify specific sites on a multitude of proteins, interactions with adapter proteins may be instrumental in achieving selectivity and regulation of activity [13].
OGA was first identified as a neutral-pH hexosaminidase, which was later associated with the gene MGEA5 (meningioma-expressed antigen 5) [14]. The domain structure of hOGA (human OGA) is frequently oversimplified by designating an N-terminal hexosaminidase followed by a C-terminal domain with sequence homology with HATs (histone acetyltransferases) (referred to as the HAT domain in the present paper). A further putative domain may exist in the stretch of over 300 amino acids in between the hexosaminidase and HAT domains. hOGA (Ser405) is itself a substrate for OGT [15]. Residues 404–548 have been shown to be required for interaction with OGT [16], and it is thought that the formation of such a complex negatively regulates O-GlcNAc removal.
Pharmacological inhibition of OGA has been an invaluable tool for investigating the cellular functions of O-GlcNAc; notably the non-selective inhibitor PUGNAc [O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate] has been used extensively to raise cellular O-GlcNAc levels [17]. Advances in understanding of the OGA reaction mechanism [18], in conjunction with structures of bacterial OGAs [19,20], have informed the development of a number of highly potent and selective inhibitors for hOGA [21].
Despite these recent advances in our understanding of OGA structure, mechanism and inhibition, we do not understand how OGA interacts with the protein part of its substrates. Such interactions could constitute the basis of substrate specificity in hOGA. In the present paper, we reveal that hOGA possesses a conserved substrate-binding groove that is required for the interaction with O-GlcNAc proteins. Different substrates require distinct surface residues for binding, affording a first glimpse into the substrate selectivity of human O-GlcNAcase.
MATERIALS AND METHODS
Cloning, expression and structure determination of OgOGA (Oceanicola granulosus OGA)
The O. granulosus gene for hyaluronoglucosaminidase (ZP_01156836.1) was cloned from genomic DNA into pGEX6-P1 for expression as a GST (glutathione transferase) fusion in Escherichia coli. The protein OgOGA was purified by affinity, anion-exchange and size-exclusion chromatography (see the Supplementary material at http://www.BiochemJ.org/bj/432/bj4320001add.htm). OgOGA was crystallized at 1.9 mg/ml from PEG [poly(ethylene glycol)]/MgCl2 solutions (see the Supplementary Online Data). The structure of the apo-enzyme was solved using MAD (multiwavelength anomalous diffraction) phasing with a lead derivative and the structure of a PUGNAc complex was obtained by molecular replacement. Diffraction data and model statistics are shown in Supplementary Table S1 at http://www.BiochemJ.org/bj/432/bj4320001add.htm.
Glycopeptide synthesis and detection
A 3,4,6-triacetyl-O-GlcNAc-Fmoc (fluoren-9-ylmethoxycarbonyl)-Ser-OH derivative was synthesized as described previously [22]. Microwave-assisted solid-phase peptide synthesis was performed with a CEM Liberty instrument on low-load Rink amide MBHA (methylbenzhydrylamine) resin 100–200 mesh (Novabiochem) using standard Fmoc chemistry protocols on a 0.05 mmol scale. The structural homogeneity of the glycopeptides was verified by high-resolution mass spectra taken on Bruker micro-TOF (time-of-flight) instrument in the direct injection mode. The same instrument was used for the monitoring of the enzymatic deglycosylation in the LC (liquid chromatography)–MS mode using an Agilent 1200 binary analytical HPLC system equipped with a Waters X-Bridge C18 3 μm particles, 3.2 mm×50 mm column and diode array UV–visible detector. Typically, the reaction was diluted to 0.3 ml with water, then passed through a 10 kDa molecular-mass cut-off concentrator to remove protein. Then, 10 μl of the flow-through was taken for the analysis using gradient elution (flow rate 0.3 ml/min) with a 5–95% linear gradient of acetonitrile (0.1% ammonia) in water (0.1% ammonia).
hOGA cloning and expression
The coding sequence for full-length hOGA was cloned into the pEBG-6P vector [23] for transient expression in mammalian cells. Point mutations were introduced by site-directed mutagenesis (see Supplementary Table S2 at http://www.BiochemJ.org/bj/432/bj4320001add.htm for primers). The enzyme was expressed as an N-terminal GST fusion in HEK (human embryonic kidney)-293 cells cultivated in suspension. For transfection, 2×108 HEK-293 cells were serum-deprived for 4 h in 100 ml of Pro293sCDM medium before the addition of 10 ml of serum-free medium containing 100 μg of plasmid DNA and 600 μg of polyethylenimine. After another 4 h, the cells were diluted to 106 cells/ml, and the medium was supplemented with 1% (v/v) fetal bovine serum. Cell pellets were harvested 72 h after transfection and lysed in 50 mM Tris/HCl (pH 7.5), 270 mM sucrose, 0.1% 2-mercaptoethanol, 1 mM sodium orthovanadate, 1 mM EDTA, 1 mM EGTA, 5 mM pyrophosphate, 10 mM sodium 2-glycerophosphate, 50 mM NaF, 1% Triton X-100, 1 mM benzamidine, 0.2 mM PMSF and 5 μM leupeptin. GST–hOGA was purified from cleared lysate by pull-down with glutathione–Sepharose (GE Healthcare), followed by elution with 25 mM reduced glutathione in 25 mM Tris/HCl buffer (pH adjusted to 7.5).
Colorimetric and fluorimetric OGA assays
Initial rates of hydrolysis of pNP-GlcNAc (p-nitrophenyl-β-N-acetylglucosamine) were measured in buffer containing 25 mM Tris/HCl (pH 7.5) and 0.1 mg/ml BSA, with substrate concentrations ranging from 0 to 10 mM with a DMSO concentration of 2% throughout the assay. Reactions were performed with 5 nM enzyme in a 50 μl assay volume. After 30 min of incubation at 37 °C, the reaction was stopped by addition of 100 μl of 3 M glycine/NaOH (pH 10.3), and product concentration was measured at A405. 4MU-GlcNAc (4-methylumbelliferyl-β-N-acetylglucosamine) assays were performed in the same buffer, with substrate concentrations ranging from 0 to 8 mM, and the enzyme concentration was reduced to 0.2 nM and the assay time to 20 min. Product formation was determined fluorimetrically (λex=360 nm/λem=460 nm). Inhibition constants for OGA inhibitors were obtained with 4MU-GlcNAc with substrate concentrations equivalent to the Km. All experiments were performed in triplicate, and results were analysed and plotted using GraphPad Prism.
Kinetic parameters of 4MU-GlcNAc turnover by wild-type and mutant hOGA were determined as described above, with enzyme concentrations of 20 nM and a maximum substrate concentration of 2 mM (1% DMSO in the assay). Competition assays with peptides were performed at 200 μM 4MU-GlcNAc. Km determination for GlcNAc-modified peptides was performed following an approach using established equations for multisubstrate enzyme kinetics to derive kinetic constants of a substrate whose turnover cannot be detected directly [24]. The fluorigenic reporter substrate and the GlcNAcylated peptide are considered as competitive inhibitors of each other. From the initial hydrolysis rates of 4MU-GlcNAc in the presence and absence of the peptide, the Michaelis constant for the peptide (Km′) can be determined from eqn (1):
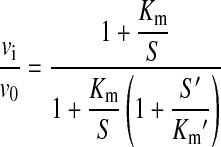 |
(1) |
where vi/v0 is the relative activity in the presence of inhibitor, Km and S are the Michaelis constant and substrate concentration of the reporter substrate respectively, and S′ is the concentration of the peptide.
Determination of hOGA in vitro activity on O-GlcNAc proteins
The known O-GlcNAc targets FoxO1 (forkhead box O1) [25,26] and CREB (cAMP-response-element-binding protein) [27] were provided by the Division of Signal Transduction and Therapy, University of Dundee, Dundee, Scotland, U.K. Both are truncated constructs and N-terminal GST-fusion proteins, GST–CREB-(1–283), and GST–FoxO1-(329–655). A GST-fusion of the TAB1 (transforming growth factor-β-activated protein kinase 1-binding protein 1) N-terminal domain and full-length hOGT were purified as described elsewhere [27a]. A truncated construct lacking the first nine TPRs (tetratricopeptide repeats) (Δ9-hOGT) was expressed in E. coli.
For GlcNAcylation reactions, target proteins were incubated with 3.75 mM UDP-GlcNAc and recombinant OGT in 50 mM Tris/HCl (pH 7.5) containing 1 mM DTT (dithiothreitol). Enzyme/substrate protein ratios were 1:20, total protein concentration was 0.5 mg/ml. The reaction was allowed to proceed for 3 h at 37 °C (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/432/bj4320001add.htm). Full-length hOGT was used to O-GlcNAcylate CREB and hOGA, whereas the truncated Δ9-hOGT was used for FoxO1 and TAB1. After the glycosylation reaction, target proteins were repurified by GST pull-down, eluted in 25 mM Tris/HCl buffer (pH 7.5) containing 25 mM reduced glutathione and spin-concentrated to approx. 5 mg/ml.
In vitro removal of O-GlcNAc was performed in a 5 μl volume in 25 mM Tris/HCl and 25 mM GST (pH 7.5), and contained 10 μM O-GlcNAcylated substrate protein and 1 μM wild-type or mutant hOGA. Reaction mixtures were incubated for 4–6 h at 37 °C, and a 1 μl aliquot was blotted for O-GlcNAc with CTD110.6 (Abcam). DM-17 rabbit anti-hOGT antibody was purchased from Sigma. GST and TAB1 antisera were obtained from the Division of Signal Transduction and Therapy [28].
RESULTS AND DISCUSSION
Recombinant hOGA is active on O-GlcNAc-modified proteins and peptides in vitro
Although hOGA has been shown to be active on a GlcNAc-modified peptide substrate [29], most studies have been restricted to measuring the enzyme activity in vitro using pseudosubstrates, or monitoring OGA activity in vivo by Western blot analysis of cellular O-GlcNAc levels after overexpression, knock down or inhibition of OGA. We aimed to demonstrate in vitro activity of hOGA on recombinant glycoproteins. Substrates were prepared by in vitro glycosylation of the previously characterized O-GlcNAc proteins TAB1 and the transcription factors FoxO1 and CREB (see Supplementary Figure S1). OGA assays were then performed with these substrates as well as a glycosylated hOGA inactive mutant, and reduction of O-GlcNAc was detected by anti-O-GlcNAc Western blot analysis, showing that hOGA is capable of removing O-GlcNAc from these glycoproteins (Figure 1A). We next sought to identify peptide substrates that would allow the determination of the Michaelis constant (Km), and also allow for varying sequence and length to probe substrate specificity. Peptides covered the O-GlcNAc sites on TAB1 (Ser395) (S. Pathak and D.M.F. van Aalten, unpublished work) and hOGA (Ser405) [4] (Table 1). hOGA activity on these peptides was confirmed by LC–MS. The affinity of hOGA for these peptides was then quantified by their ability to compete with the reporter substrate 4MU-GlcNAc (Figure 1B and Table 1). The resulting Km values for the hOGA-Ser405- and TAB1-Ser395-derived peptides are in the high-micromolar and low-millimolar range respectively, somewhat decreasing in affinity with increasing peptide length.
Figure 1. Activity of hOGA and OgOGA.

(A) The O-GlcNAc substrate proteins (10 μM) TAB1, FoxO1 and were subjected to in vitro GlcNAcylation by recombinant OGT (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/432/bj4320001add.htm). For the activity assay, these substrates, as well as GlcNAc-modified hOGA D175N, were incubated with hOGA (1 μM) at 37 °C, which led to complete deglycosylation within 6 h as detected by anti-O-GlcNAc antibody. The top panel shows the enzyme hOGA as detected through its GST tag, the middle panel shows O-GlcNAcylation using CTD110.6 anti-O-GlcNAc antibody, and the bottom panel shows all the substrate proteins through their GST tags (shorter exposure). (B) Competition of O-GlcNAc-modified peptides (tri-, penta-, hepta- and nona-peptides based on the O-GlcNAc site on hOGA Ser405) with 4MU-GlcNAc turnover by hOGA affords an estimate of the affinity of hOGA for the peptide substrates (Table 1). (C) Mass spectrum showing the deglycosylation of Val-Ala-His-Ser-(β-O-GlcNAc)-Gly-Ala (molecular mass of 785 Da) to the deglycosylated form (molecular mass of 582 Da), also see Supplementary Figure S2 at http://www.BiochemJ.org/bj/432/bj4320001add.htm.
Table 1. Michaelis constants of O-GlcNAc-modified peptides for hOGA.
Km values for peptides derived from the O-GlcNAc sites of two known substrate proteins were determined in a competitive assay with the fluorigenic substrate 4MU-GlcNAc. Ac, acetyl; amino acids in peptides are indicated using the single-letter code.
| Substrate | Km |
|---|---|
| hOGA-Ser405-derived peptide series | |
| Ac-HS(-β-O-GlcNAc)G-NH2 | 525±13 μM |
| Ac-VAHS(-β-O-GlcNAc)GA-NH2 | 1204±43 μM |
| Ac-VAHS(-β-O-GlcNAc)GAK-NH2 | 789±36 μM |
| Ac-QVAHS(-β-O-GlcNAc)GAKAS-NH2 | 528±15 μM |
| TAB1-Ser395-derived peptide series: | |
| Ac-YS(-β-O-GlcNAc)S-NH2 | 2.2±0.1 mM |
| Ac-PYS(-β-O-GlcNAc)SA-NH2 | 6.3±0.5 mM |
| Ac-VPYS(-β-O-GlcNAc)SAQ-NH2 | 4.1±0.3 mM |
| Ac-SVPYS(-β-O-GlcNAc)SAQS-NH2 | 4.8±0.3 mM |
Discovery of a bacterial OGA with improved similarity to the human enzyme
hOGA is known to possess an N-terminal catalytic domain (residues 60–366) and the C-terminal HAT domain (residues 707–916) (Figure 2A). However, sequence analysis of putative metazoan OGAs shows that they also possess a low-complexity N-terminus (residues 1–59), and a ‘middle’ domain (residues 367–707), also containing a low-complexity region (residues 396–553). Only the catalytic domain is represented in the available (bacterial) OGA structures, yet any of the additional hOGA domains could contribute to substrate recognition/specificity.
Figure 2. Structure of OgOGA and similarity to other OGAs.

(A) Domain architecture of human OGA. Predicted ordered domains are shown as coloured boxes (cyan, catalytic domain; purple, stalk domain; orange, HAT domain), predicted disordered regions are indicated by red wavy lines. (B) Sequence alignment of OgOGA and hOGA. Secondary-structure elements from the OgOGA structure are shown (red, α-helices; blue, β-strands; yellow, 310-helices), broken lines denote residues disordered in apo-OgOGA. The proposed domains are indicated by boxes coloured as in (A). (C) Cartoon view of the OgOGA apo structure and domains 2 and 3 of CpNagJ. (D) Surface view of OgOGA shaded by sequence conservation among metazoan OGAs (light blue) and metazoan OGAs+OgOGA (dark blue). The PUGNAc (green) complex is shown with additional ordered loops taken from the apo-structure. (E) Active sites of the OgOGA–PUGNAc and CpNagJ–GlcNAcstatin complexes. The ligands are represented as sticks with green carbons, an unbiased Fo−Fc;ϕc map (2.3 σ) is shown for PUGNAc. Hydrogen bonds are drawn as black broken lines. Hydrogen bonds to/from protein backbone amides are shown as originating from the Cα. Active-site residues in OgOGA are labelled in black, alongside the equivalent hOGA residues in blue.
In the absence of a hOGA structure to guide our substrate-specificity studies, we aimed to identify new bacterial OGAs with improved sequence identity and extending to beyond just the catalytic domain. A BLAST search identified a predicted protein from O. granulosus (OgOGA) that not only shows improved sequence identity (37%) with the hOGA catalytic domain without any alignment gaps (Figure 2B), but also, intriguingly, possesses a 200-residue C-terminal domain that shares approx. 20% identity with the hOGA middle domain (Figures 2A and 2B). An OgOGA structure would reveal the position of this domain with respect to the active site and serve as a guide to identifying residues or domains involved in glycoprotein substrate recognition.
To investigate the possible OGA activity of OgOGA, the gene was cloned from genomic DNA and expressed in E. coli. The enzyme shows OGA activity with pNP-GlcNAc (Km=8.5±0.6 mM, kcat=24.0±1.0 s−1) and 4MU-GlcNAc (Km=2.9±0.2 mM, kcat=8.2±0.3 s−1), with a pH optimum of 7.4–7.6 (see Supplementary Figure S2B at http://www.BiochemJ.org/bj/432/bj4320001add.htm). OgOGA is sensitive to the commonly used OGA inhibitors PUGNAc (IC50=11 μM) and GlcNAcstatin G [29a] (IC50=780 nM). OgOGA was also able to quantitatively deglycosylate a glycopentapeptide, Val-Ala-His-Ser-(β-O-GlcNAc)-Gly-Ala, derived from the reported hOGA O-GlcNAc site [4], as detected by LC–MS (Figure 1C and Supplementary Figure S2).
OgOGA possesses a conserved peptide-binding groove and ‘stalk’
We next explored the structural similarities of OgOGA with hOGA to allow exploitation of OgOGA as a structural guide for hOGA mutagenesis studies. OgOGA was crystallized and its structure solved, including a complex with PUGNAc (see Supplementary Table S1). The OgOGA structure reveals an N-terminal TIM barrel domain and a C-terminal α-helical domain (Figures 2A and 2C). The OgOGA catalytic domain is similar to that of CpNagJ (Clostridium perfringens NagJ) [RMSD (root mean square deviation)=1.5 Å (1 Å=0.1 nm) for 254 Cαs; Figure 2C]. The α4–β5, β3–α3, β4–α4 and β5–α5 loops are of different lengths and/or adopt different conformations. Strikingly, sequence alignments show that these loops match hOGA, but not CpNagJ, in length and conservation (Figure 2B). The C-terminal domain, hereafter referred to as the stalk domain, possesses good sequence similarity with hOGA (Figure 2B) and contains a core four-helical bundle (Figure 2C). Unexpectedly, this domain is structurally similar to the third domain of CpNagJ (RMSD=1.9 Å for 110 Cαs, yet <10% sequence identity; Figure 2C), although the loops connecting the helices differ in length and conformation. In particular, the α10–α11 loop is nine residues longer in CpNagJ compared with OgOGA (and, by alignment, hOGA; Figure 2B), reaching into the CpNagJ active site, which is impossible in OgOGA (and thus unlikely in hOGA). Similarly, the OgOGA structure suggests structural homology between the stalk domain of Bacteroides thetaiotaomicron OGA [20] and hOGA (see Supplementary Figure S3 at http://www.BiochemJ.org/bj/432/bj4320001add.htm).
The OgOGA active site is formed by the TIM barrel β–α loops (Figure 2C), with the catalytic loop residues Asp115/Asp116 on β4–α4 conserved with hOGA, compatible with the observed OGA activity (Figure 2C). This loop does not fully engage with the PUGNAc inhibitor, presumably related to the disordered phenylcarbamate group. Ligand-induced conformational changes in the β4–α4 loop have also been observed for CpNagJ [30]. Upon PUGNAc binding to OgOGA, the β7–η1 loop shifts by up to 4.7 Å exposing the GlcNAc-binding site, associated with small displacements of the neighbouring β8–α8 and β1–α1 loops. Residues hydrogen-bonding with the sugar hydroxy groups (Gly8, Tyr160, Asp226 and Asn254) or the N-acetyl moiety (Asn221) are conserved in hOGA (Figures 2B and 2E). Similarly, the residues that show van der Waals/stacking interactions with the sugar and the N-acetyl group (Tyr10, Tyr160 and Trp219) are conserved in hOGA. Strikingly, the surface around the active site, together with the stalk domain, forms a groove lined by side chains that are highly conserved/identical between OgOGA and hOGA and also among metazoan OGAs (Figure 2D).
Residues lining the conserved binding groove are required for hOGA–substrate interactions
We next tested the hypothesis that the conserved groove represents the binding site for O-GlcNAc glycoprotein substrates by expressing GST-fusions of 20 individual hOGA point mutants of residues lining the groove (Figures 2B and 2D). To control for effects on protein folding and/or catalysis rather than protein substrate binding, we measured 4MU-GlcNAc hydrolysis by these mutants (Figure 3A). Mutations affecting 4MU-GlcNAc hydrolysis are mostly located close to the sugar/catalytic machinery (Tyr69) or occupy positions shown to undergo a conformational change upon inhibitor binding (Tyr286 and Asp287 in the β7–η1 loop) (Figure 2). Strikingly, de-O-GlcNAcylation assays with the TAB1, FoxO1 and CREB glycoprotein substrates identified mutants of residues some distance from the active site (I176L, S190A, S190Q, T217A, E218S, F223S and K289A) with reduced activity against one or more of the protein substrates, yet unimpaired 4MU-GlcNAc hydrolysis (Figure 3).
Figure 3. Activity of hOGA groove mutants.

Twenty individual mutants of hOGA were generated based on the structural information obtained from OgOGA. (A) To evaluate the structural integrity of the hOGA mutants, the catalytic efficiency of 4MU-GlcNAc turnover was determined and compared with the wild-type (kcat/Km=7000 M−1·s−1) and the catalytically impaired D175N mutant (negative control). hOGA mutants were also incubated with O-GlcNAc substrate proteins TAB1, FoxO1 and CREB, and deglycosylation was observed by Western blotting. (B) Visualization of the correlation between the mutants' ability to hydrolyse the pseudosubstrate 4MU-GlcNAc and glycoprotein substrates. Catalytically impaired mutants (kcat/Km for 4MU-GlcNAc ≤50% of wild-type) locate in the areas shaded in red. Mutants unaffected in both 4MU-GlcNAc turnover and protein deglycosylation (cut-off 10% reduction of wild-type activity) are shown in the blue areas. Mutations reducing OGA activity on protein substrates only are depicted in the yellow areas. Location of the mutations is shown on the surface around the active site of OgOGA (coloured by domain, see Figure 2A). The mutated amino acids are shaded according to correlation plots, thus amino acids affecting protein substrate recognition are highlighted in yellow.
Conclusions
Since its discovery more than three decades ago, it has become clear that O-GlcNAc is an important post-translational modification found on hundreds of intracellular proteins and plays a role in signalling pathways. However, it is not clear what determines specificity in the targeting and cycling of O-GlcNAc on these proteins. We have investigated whether OGA specifically recognizes the peptide component of its glycoprotein substrates. Our findings show that, by similarity to the structurally related bacterial OgOGA, hOGA possesses a putative substrate-binding groove that is highly conserved within metazoan OGAs, in addition to a stalk domain in close proximity to the active site. Indeed, mutations targeting this groove identified residues that are not required for hydrolysis of the 4MU-GlcNAc pseudosubstrate, but affected hydrolysis of glycoprotein substrates. Although a limited set of glycoprotein substrates (TAB1, FoxO1 and CREB) and peptides (derived from TAB1 and hOGA itself) was tested, these mutational studies hinted at differential substrate recognition. The differences between the residues required for the turnover of these substrates may reflect differences in the molecular environment of the O-GlcNAc site(s) on these proteins/peptides, and could tune O-GlcNAc cycling rates. Together, the results of the present study constitute the first evidence for specific interactions between hOGA and its substrate proteins beyond the sugar moiety, and may form the basis of an improved understanding of the enzyme's substrate specificity and the functional implications thereof.
Online data
AUTHOR CONTRIBUTION
The scientific hypothesis was devised by Marianne Schimpl, Alexander Schüttelkopf and Daan van Aalten. The experimental design was by Marianne Schimpl, Alexander Schüttelkopf, Vladimir Borodkin and Daan van Aalten. Experimentation was by Marianne Schimpl, Alexander Schüttelkopf and Vladimir Borodkin. Interpretation of results was by Marianne Schimpl, Alexander Schüttelkopf and Daan van Aalten who also wrote the paper.
ACKNOWLEDGEMENTS
We thank Sharon Shepherd for assistance with protein production, Lindsey Gray for assistance with peptide synthesis, Adel Ibrahim for help with molecular cloning, and the European Synchrotron Radiation Facility, Grenoble, for time on BM14, ID23-2 and ID14-1.
FUNDING
This work was supported by a Wellcome Trust Senior Research Fellowship to D.M.F.v.A. and a Wellcome Trust Ph.D. Studentship to M.S.
References
- 1.Love D. C., Hanover J. A. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci. STKE. 2005;2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 2.Hart G. W., Housley M. P., Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 3.Torres C. R., Hart G. W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes: evidence for O-linked GlcNAc. J. Biol. Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- 4.Khidekel N., Ficarro S. B., Clark P. M., Bryan M. C., Swaney D. L., Rexach J. E., Sun Y. E., Coon J. J., Peters E. C., Hsieh-Wilson L. C. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 2007;3:339–348. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- 5.Vosseller K., Trinidad J. C., Chalkley R. J., Specht C. G., Thalhammer A., Lynn A. J., Snedecor J. O., Guan S., Medzihradszky K. F., Maltby D. A., et al. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol. Cell. Proteomics. 2006;5:923–934. doi: 10.1074/mcp.T500040-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z., Udeshi N. D., O'Malley M., Shabanowitz J., Hunt D. F., Hart G. W. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics. 2009;9:153–160. doi: 10.1074/mcp.M900268-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z., Udeshi N. D., Slawson C., Compton P. D., Sakabe K., Cheung W. D., Shabanowitz J., Hunt D. F., Hart G. W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signaling. 2010;3:ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zachara N. E., Hart G. W. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta. 2004;1673:13–28. doi: 10.1016/j.bbagen.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Slawson C., Zachara N. E., Vosseller K., Cheung W. D., Lane M. D., Hart G. W. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J. Biol. Chem. 2005;280:32944–32956. doi: 10.1074/jbc.M503396200. [DOI] [PubMed] [Google Scholar]
- 10.Slawson C., Shafii S., Amburgey J., Potter R. Characterization of the O-GlcNAc protein modification in Xenopus laevis oocyte during oogenesis and progesterone-stimulated maturation. Biochim. Biophys. Acta. 2002;1573:121–129. doi: 10.1016/s0304-4165(02)00369-0. [DOI] [PubMed] [Google Scholar]
- 11.Comtesse N., Maldener E., Meese E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a β-N-acetylglucosaminidase. Biochem. Biophys. Res. Commun. 2001;283:634–640. doi: 10.1006/bbrc.2001.4815. [DOI] [PubMed] [Google Scholar]
- 12.Hanover J. A., Yu S., Lubas W. B., Shin S. H., Ragano-Caracciola M., Kochran J., Love D. C. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch. Biochem. Biophys. 2003;409:287–297. doi: 10.1016/s0003-9861(02)00578-7. [DOI] [PubMed] [Google Scholar]
- 13.Cheung W. D., Sakabe K., Housley M. P., Dias W. B., Hart G. W. O-linked β-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J. Biol. Chem. 2008;283:33935–33941. doi: 10.1074/jbc.M806199200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heckel D., Comtesse N., Brass N., Blin N., Zang K. D., Meese E. Novel immunogenic antigen homologous to hyaluronidase in meningioma. Hum. Mol. Genet. 1998;7:1859–1872. doi: 10.1093/hmg/7.12.1859. [DOI] [PubMed] [Google Scholar]
- 15.Copeland R. J., Bullen J. W., Hart G. W. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am. J. Physiol. Endocrinol. Metab. 2008;295:E17–E28. doi: 10.1152/ajpendo.90281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whisenhunt T. R., Yang X., Bowe D. B., Paterson A. J., Van Tine B. A., Kudlow J. E. Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development. Glycobiology. 2006;16:551–563. doi: 10.1093/glycob/cwj096. [DOI] [PubMed] [Google Scholar]
- 17.Haltiwanger R. S., Grove K., Philipsberg G. A. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-β-N-acetylglucosaminidase inhibitor O-(2-acetamido-2deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate. J. Biol. Chem. 1998;273:3611–3617. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- 18.Macauley M. S., Stubbs K. A., Vocadlo D. J. O-GlcNAcase catalyzes cleavage of thioglycosides without general acid catalysis. J. Am. Chem. Soc. 2005;127:17202–17203. doi: 10.1021/ja0567687. [DOI] [PubMed] [Google Scholar]
- 19.Rao F. V., Dorfmueller H. C., Villa F., Allwood M., Eggleston I. M., van Aalten D. M. Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. EMBO J. 2006;25:1569–1578. doi: 10.1038/sj.emboj.7601026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dennis R. J., Taylor E. J., Macauley M. S., Stubbs K. A., Turkenburg J. P., Hart S. J., Black G. N., Vocadlo D. J., Davies G. J. Structure and mechanism of a bacterial β-glucosaminidase having O-GlcNAcase activity. Nat. Struct. Mol. Biol. 2006;13:365–371. doi: 10.1038/nsmb1079. [DOI] [PubMed] [Google Scholar]
- 21.Macauley M. S., Vocadlo D. J. Increasing O-GlcNAc levels: an overview of small-molecule inhibitors of O-GlcNAcase. Biochim. Biophys. Acta. 2010;1800:107–121. doi: 10.1016/j.bbagen.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 22.Salvador L. A., Elofsson M., Kihlberg J. Preparation of building blocks for glycopeptide synthesis by glycosylation of Fmoc amino acids having unprotected carboxyl groups. Tetrahedron. 1995;51:5643–5656. [Google Scholar]
- 23.Sanchez I., Hughes R. T., Mayer B. J., Yee K., Woodgett J. R., Avruch J., Kyriakis J. M., Zon L. I. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 24.Xie D., Suvorov L., Erickson J. W., Gulnik A. S. Real-time measurements of dark substrate catalysis. Protein Sci. 1999;8:2460–2464. doi: 10.1110/ps.8.11.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Housley M. P., Rodgers J. T., Udeshi N. D., Kelly T. J., Shabanowitz J., Hunt D. F., Puigserver P., Hart G. W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008;283:16283–16292. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo M., Zilberfarb V., Gangneux N., Christeff N., Issad T. O-glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6-phosphatase gene. FEBS Lett. 2008;582:829–834. doi: 10.1016/j.febslet.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Lamarre-Vincent N., Hsieh-Wilson L. C. Dynamic glycosylation of the transcription factor CREB: a potential role in gene regulation. J. Am. Chem. Soc. 2003;125:6612–6613. doi: 10.1021/ja028200t. [DOI] [PubMed] [Google Scholar]
- 27a.Clarke A. J., Hurtado-Guerrero R., Pathak S., Schüttelkopf A. W., Borodkin V., Shepherd S. M., Ibrahim A. F., van Aalten D. M. Structural insights into mechanism and specificity of O-GlcNAc transferase. EMBO J. 2008;27:2780–2788. doi: 10.1038/emboj.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung P. C., Campbell D. G., Nebreda A. R., Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 2003;22:5793–5805. doi: 10.1093/emboj/cdg552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao Y., Wells L., Comer F. I., Parker G. J., Hart G. W. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic β-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 29a.Dorfmueller H. C., Borodkin V. S., Schimpl M., Zheng X., Kime R., Read K. D., van Aalten D. M. F. Cell-penetrant, nanomolar O-GlcNAcase inhibitors selective against lysosomal hexosaminidases. Chem. Biol. 2010 doi: 10.1016/j.chembiol.2010.09.014. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pathak S., Dorfmueller H. C., Borodkin V. S., van Aalten D. M. F. Chemical dissection of the link between streptozotocin, O-GlcNAc, and pancreatic cell death. Chem. Biol. 2008;15:799–807. doi: 10.1016/j.chembiol.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.