


Abstract
The RegA proteins from the bacteriophage T4 and RB69 are translational repressors that control the expression of multiple phage mRNAs. RegA proteins from the two phages share 78% sequence identity; however, in vivo expression studies have suggested that the RB69 RegA protein binds target RNAs with a higher affinity than T4 RegA protein. To study the RNA binding properties of T4 and RB69 RegA proteins more directly, the binding sites of RB69 RegA protein on synthetic RNAs corresponding to the translation initiation region of two RB69 target genes were mapped by RNase protection assays. These assays revealed that RB69 RegA protein protects nucleotides –9 to –3 (relative to the start codon) on RB69 gene 44, which contains the sequence GAAAAUU. On RB69 gene 45, the protected site (nucleotides –8 to –3) contains a similar purine-rich sequence: GAAAUA. Interestingly, T4 RegA protein protected the same nucleotides on these RNAs. To examine the specificity of RNA binding, quantitative RNA gel shift assays were performed with synthetic RNAs corresponding to recognition elements (REs) in three T4 and three RB69 mRNAs. Comparative gel shift assays demonstrated that RB69 RegA protein has an ∼7-fold higher affinity for T4 gene 44 RE RNA than T4 RegA protein. RB69 RegA protein also binds RB69 gene 44 RE RNA with a 4-fold higher affinity than T4 RegA protein. On the other hand, T4 RegA exhibited a higher affinity than RB69 RegA protein for RB69 gene 45 RE RNA. With respect to their affinities for cognate RNAs, both RegA proteins exhibited the following hierarchy of affinities: gene 44 > gene 45 > regA. Interestingly, T4 RegA exhibited the highest affinity towards RB69 gene 45 RE RNA, whereas RB69 RegA protein had the highest affinity for T4 gene 44 RE RNA. The helix–loop groove RNA binding motif of T4 RegA protein is fully conserved in RB69 RegA protein. However, homology modeling of the structure of RB69 RegA protein reveals that the divergent residues are clustered in two areas of the surface, and that there are two large areas of high conservation near the helix–loop groove, which may also play a role in RNA binding.
INTRODUCTION
The bacteriophage T4 RegA protein controls the level of expression of a group of T4 early genes at the level of translation (1). It has been shown that RegA protein inhibits translation by competing with ribosomes for binding to the translation initiation region (TIR) of specific mRNAs (1–4). The fact that RegA protein binds to 15–30 different mRNAs including its own gene, regA, and distinguishes them from the numerous other T4 RNAs concurrently present in the cell makes it an exceptional paradigm for understanding protein–RNA recognition (1,5). Comparison of 12 known RegA-regulated mRNAs indicates that the TIRs are AU-rich, but they do not contain a highly conserved nucleotide sequence nor apparent similar secondary structures (6,7). However, the finding that the target mRNAs vary in their sensitivity to RegA repression in vitro (1,3) and in vivo (8) is consistent with the nucleotide variations present in the TIRs. The studies of Brown et al. (9) in which the systematic evolution of ligands by exponential enrichment (SELEX) technique was used to identify RNAs preferentially bound by RegA protein, suggest that an optimal target for RegA protein is 5′-AAAAUUGUUAUGUAA-3′. Previous studies (6) of the RegA recognition element (RE) in T4 gene 44 indicate that in gene 44 mRNA the binding site is a single-stranded sequence of 11–12 nt, which includes the Shine–Dalgarno region and an AU-rich element, which has some similarity to the SELEX high affinity RNA (specifically, the AAAUU sequence).
RB phage are T4-related bacteriophage that contain hydroxy-methylated cytosine, which can complement some T-even phage mutations (10). To identify functional domains in RegA protein, Miller and Jozwik (11) examined the regA gene in a number of T-even and RB phages. They found that RegA protein was identical in all the phages examined (11) except for the distant phylogenetic relative RB69, where the encoded RegA protein was 22% divergent from T4 RegA protein (12). In T4 regA– infections of cells harboring plasmids carrying RB69 regA, RB69 RegA protein was found to be capable of repressing T4-encoded mRNAs, including T4 gene 44, gene 45 and rpbA mRNAs (12). In two-plasmid assays in which RB69 regA and T4 gene 44, gene 45 and rpbA were expressed in trans, RB69 RegA protein appeared to repress the three T4 genes, and protein synthesis in general, more efficiently than T4 RegA protein (12).
Although RB69 RegA represses T4 mRNAs, it is not known if T4 RegA represses homologous RB69 mRNAs. Interestingly, RB69 and T4 DNA polymerases (which are 61% identical) both function as RNA binding, autogenous translational repressors of gene 43 (13). However, T4 DNA polymerase binds only T4 gene 43 mRNA, while RB69 DNA polymerase can bind and repress operator RNAs from both phages (13). Given this difference in RNA binding specificity in the homologous T4 and RB69 DNA polymerases, we wondered whether similar differences in RNA discrimination exist in the phage RegA proteins. Also, since T4 and RB69 RegA proteins are 22% dissimilar in amino acid sequence and they appear to exhibit differences in RNA binding properties (12), the correlation of structural and functional properties of the two proteins may yield new insights into how RegA protein recognizes specific RNAs.
As a first step in probing potential structure–function differences between RB69 and T4 RegA proteins, we have examined the in vitro RNA binding properties of the two phage proteins. The RB69 RegA protein binding site has been mapped on RB69 gene 44 and gene 45 mRNAs by RNase protection assays and compared to the known T4 RegA binding sites on the respective T4 mRNAs (6,14). The affinities of the two RegA proteins for synthetic RNAs corresponding to both T4 and RB69 REs were then measured. These studies revealed that T4 and RB69 RegA protein bind to the same sites on RB69 gene 44 and gene 45 RNAs. However, the two proteins differ in their overall RNA binding affinities and in their RNA binding specificities. To explore structural relationships in the proteins, molecular modeling has been used to predict the structure of RB69 RegA protein, based on the known structure of T4 RegA protein (15). These models revealed regions of conserved and divergent residues on the surfaces of the two proteins that potentially may contribute to the observed differences in RNA binding specificities and affinities of these proteins.
MATERIALS AND METHODS
Reagents and strains
Oligodeoxyribonucleotides were synthesized on an Expedite (Model 8909) nucleic acid synthesizer by the MUSC Oligo synthesis facility. Oligoribonucleotides were synthesized by the W. M. Keck Foundation Biotechnology Resource Laboratory (Yale University, New Haven, CT). Escherichia coli AR120 (λ cI+, N+) was obtained from A.Shatzman (Smith Kline Beecham Pharmaceuticals). RNase A and RNase I were purchased from Ambion.
Cloning of RB69 RegA protein
Construction of the pAS1 vector containing T4 regA was described previously (3). The RB69 regA gene was subcloned from pEM141 (12) into pAS1 by PCR amplification of regA and ligation into BamH1/SalI cleaved pAS1. The primers used for sub-cloning RB69 regA into the pAS1 vector contained the following sequences: forward, 5′-CGC GGA TCC GGA ATG GTA AAA TGA TTG AAA TTA AAT TG-3′; reverse: 5′-CGC GTC GAC CCA TTG CTT TAA TTA CCA ATT GTA TAT TTT GC-3′. Verification that the cloned regA gene was wild-type (WT) was obtained by DNA sequence analysis of the entire regA coding region.
RegA protein purification
WT T4 RegA protein was purified from AR120 cells containing the pAS1-regA plasmid following induction of transcription from the phage λ PL promoter by nalidixic acid treatment, as described previously (16,17). Purified T4 RegA protein was used to generate concentration standard curves for quantitation of RegA protein in cell supernatants (see below). The concentrations of purified RegA protein were determined by triplicate OD280 measurements or amino acid analyses (W. M. Keck Foundation).
Preparation of induced cell supernatants
Cell supernatants were prepared by the freeze–thaw method of Johnson and Hecht (18), with slight modifications, as described previously (19). Cell supernatants were stored in 80 µl aliquots at –20°C. Evaluation of the protein content of both the supernatant and the pellet resuspension was carried out by SDS–PAGE. RegA protein concentrations in cell lysates were determined by quantitation of protein fluorescence in gels stained with Sypro Orange (Molecular Probes) compared to a standard curve of known concentrations of purified RegA protein, using a Molecular Dynamics Storm imager. Protein concentrations had an error range of 1–16% (average of 7%).
RNA purification
Synthetic oligoribonucleotides were deprotected by treatment with tetra butyl ammonium fluoride (TBAF) overnight (20) and then purified by perfusion chromatography using Poros™ HQ and R1 columns (PerSeptive Biosciences) (20). RNA purity was assessed by analysis of 32P-labeled RNA on 6% polyacrylamide gels. For RNase footprinting and gel shift assays, purified RNAs were 5′-32P-end-labeled by treatment with T4 polynucleotide kinase and [γ-32P]ATP (21).
RNase footprinting
Purified 18mer RB69 gene 44 RE RNA (5′-AUGAGGAAAAUUACAUGA-3′) and gene 45 (5′-UGAAAGGAAAUAAAAUGA-3′) RE oligomers were 5′-32P-end-labeled and then repurified by 6% polyacrylamide gel electrophoresis. 32P-RNAs (10 nM; 20 000 c.p.m.) were incubated with cell supernatants containing 20 and 50 nM T4 and RB69 RegA proteins. RNase A digestions were carried out in 10 µl of 100 mM Tris–HCl pH 6.5, 1 mM EDTA, at 4°C for 5 min at an enzyme concentration of 10–7 U/µl. RNase I digestions were performed in 10 µl of 10 mM Tris–HCl pH 8.0, 50 mM NaCl, at room temperature for 15 min at an enzyme concentration of 0.01 U/µl. Following enzymatic digestions, equal volumes (10 µl) of gel loading buffer (95% formamide, 0.05% xylene cyanol and bromophenol blue) were added to each reaction and samples were heated at 75°C for 3 min. RNAs were then electrophoresed on 15% polyacrylamide–8 M urea gels. Following electrophoresis, gels were fixed in 10% acetic acid–10% methanol for 1 h and dried. RNA fragments were visualized by phosphorimaging using a STORM™ imager (Molecular Dynamics).
RNA gel mobility shift assays
32P-labeled RNAs (10 nM) were incubated with increasing volumes of induced cell supernatants to generate titration curves. Binding was carried out in 10 mM Tris–HCl pH 7.5, 50 mM NaCl and 1 mM EDTA at 4°C for 15 min. Binding reactions were performed in 10–20 µl final volume with concentrations of 2.5–40 nM RegA protein. A freshly thawed aliquot of cell supernatant was used for each experiment and then discarded. Reaction products were analyzed by electrophoresis on a native 8% polyacrylamide gel in 0.5× TBE (89 mM Tris, 89 mM boric acid, 4 mM EDTA, pH 8.3) at 4°C. Gels were dried and analyzed by autoradiography and 32P-RNA was quantitated by phosphorimaging on a Molecular Dynamics Imager Model 425.
Apparent association constants (Kapp) for mutant proteins were determined from gel shift assays in a manner similar to that of Rebar and Pabo (22). Association constants were calculated from PhosphorImager data as follows:
Kapp = [protein:RNA complex]/([proteinf] × [RNAf])
where [protein:RNA complex] = fraction 32P-RNA bound × [total RNA]; [RNAf] = fraction 32P-RNA free × [total RNA]; and [proteinf] = [total protein] – [bound protein] (assuming one protein per bound RNA) (17).
Kapp was calculated at four points on the titration curve. The mean of the four values was calculated and Kapp values from two to four experiments were averaged. Standard deviations (calculated using the non-biased or ‘n – 1’ method using Excel) ranged from Kapp (Av)/2.9 to Kapp (Av)/9.5.
RESULTS
Overexpression of RB69 RegA protein
To facilitate in vitro studies of the RNA binding properties of RB69 RegA protein, plasmids were constructed which overexpress RB69 RegA protein. For this purpose, the RB69 regA gene was subcloned from pEM141 (12) that expresses low levels of the protein, into the pAS1 vector (23), which has been used previously for inducible high level expression of T4 regA (16). In the resulting construct, pAS1-RB69 regA, the regA gene was positioned downstream from the strong λ PL promoter and the λ cII gene AUG codon. As shown in Figure 1, in this construct the RB69 regA gene is preceded by 15 nt of upstream RB69 gene 62 sequence. Translation of the PL transcript is expected to initiate at the cII gene AUG codon, then terminate at the gene 62 termination codon, and finally, reinitiate at the regA AUG codon, through translational coupling.
Figure 1.

(A) Sequence of the TIR of RB69 regA on the pAS1-RB69 regA vector. The regA coding region is preceded by the λ cII gene ribosome binding site (underlined) and AUG start codon followed by a 15 nt ORF corresponding to the 3′-end of T4 gene 62 (lower case). Translation initiated at the cII AUG will terminate at the gene 62 stop codon (boxed), and then reinitiate at the regA AUG (italic). (B) Mutations introduced into the TIR of λ cII-RB69 regA to increase expression of regA. The ribosome binding site and start codons are in bold.
Induction of regA expression from pAS1-RB69 regA by nalidixic acid treatment resulted in an increased level of regA expression, relative to that obtained from pEM141 (12). However, as shown in Figure 2, considerably higher levels of T4 RegA protein are produced from the same vector (pAS1-T4 regA). This suggests that some autorepression of RB69 regA translation occurs with this construct, since both the T4 and RB69 regA vectors use the same promoter and the same efficient translation initiation regions. In an effort to eliminate autorepression of RB69 RegA protein, mutations were introduced into the TIR of RB69 regA on pAS1-RB69 regA. Because RB69 regA autorepression could occur through RegA protein occlusion of either the cII gene TIR or the regA TIR, mutations were introduced into both TIRs (Fig. 1B). In some mutants (e.g. G62A, G62B, CIIA and CIIB), cytosines were substituted for A or U, because RegA-sensitive mRNAs are generally deficient in cytosines. In mutant CIIB, 7 nt between the cII gene Shine–Dalgarno sequence and AUG codon were replaced by a 7 nt sequence from the TIR of T4 gene 43, which is insensitive to RegA repression. As shown in Figure 2, this mutation (CIIB; lane 6) resulted in a slightly higher level of expression of RB69 RegA than the WT (lane 7) or G62A mutant (lane 5) vectors. However, none of the pAS1-RB69 regA constructs expressed RegA to the level obtained for T4 RegA protein (Fig. 2, lane 4). This finding is consistent with the suggestion of Jozwik and Miller (12) that RB69 RegA protein generally represses translation more efficiently than T4 RegA protein.
Figure 2.
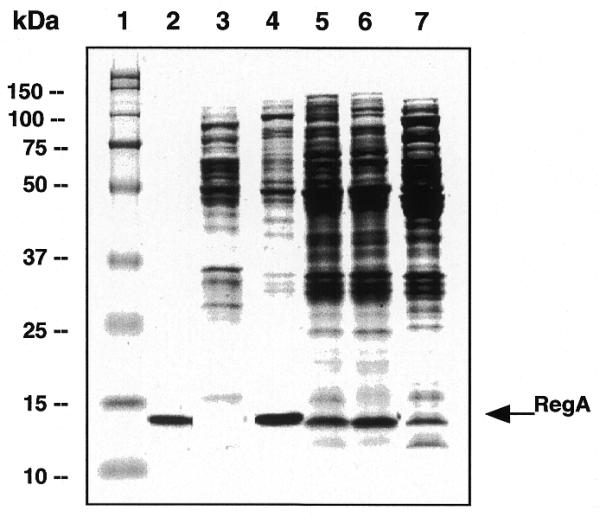
Expression of RB69 and T4 RegA proteins from pAS1 vectors. Total cell extracts from 5 µl of uninduced cell culture (lane 3), 5 µl of nalidixic acid-induced cell cultures expressing T4 RegA protein (lane 4), 10 µl of induced cell cultures expressing RB69 RegA protein from plasmids pG62A (lane 5) (see Fig. 1), pCIIB (lane 6) and WT pAS1-RB69 regA (lane 7), respectively, were applied to a 15% SDS–polyacrylamide gel. Lane 1, molecular weight markers; lane 2, purified T4 RegA protein.
Mapping of RB69 RegA protein binding sites on RB69 gene 44 and gene 45 RNAs
As noted above, T4 mRNAs vary in their sensitivities to RegA repression in vitro (1,3) and in vivo (8). Of the T4 genes examined to date, gene 44 is the most sensitive, while the regA gene itself is the least sensitive to RegA repression in vitro. Although the T4 RegA protein binding sites on five T4 mRNAs have been mapped (1,3,14), the binding sites of RB69 RegA protein on RB69 mRNAs have not been examined. Comparison of the three RegA-sensitive RB69 genes that have been sequenced to date (11,24) reveals that the sequences of the TIRs are similar, but not identical in the two phage. As shown in Figure 3, relative to the phage T4 TIRs, the RB69 TIRs have three nucleotide insertions in gene 44, one nucleotide insertion and two base substitutions in gene 45 and a single nucleotide insertion in regA mRNA. Like the T4 RegA-sensitive mRNAs, the RB69 mRNAs do not appear to contain potential secondary structures in the TIRs.
Figure 3.

Nucleotide sequence of the translation initiation regions of three RegA-sensitive mRNAs from T4 and RB69 phage. The binding site of T4 RegA protein on T4 gene 44, previously determined by RNase protection assays (3,6), is indicated by shading. The sequences of the oligonucleotides used in this study are indicated by underlining and the initiation codons are in bold type.
Given the similarities of the sequences of the RB69 TIRs, it is likely that RB69 RegA protein binds to the same regions on the homologous mRNAs as T4 RegA protein. To test this hypothesis, the ability of RB69 RegA protein to bind to a synthetic RNA corresponding to the TIR of RB69 gene 44 was examined. For this assay, an 18mer RNA (5′-AUGAGGAAAAUUACAUGA-3′) corresponding to nucleotides –14 to +4 (relative to the AUG) of RB69 gene 44 mRNA (24) was synthesized. This sequence spans the 12 nt sequence previously found to be the binding site for T4 RegA protein on T4 gene 44 mRNA (Fig. 3) (6). To detect binding, RNA gel mobility shift assays were performed using 32P-labeled RB69 gene 44 RE RNA (RB gene 44 RE RNA) as a ligand. In these assays, induced cell supernatants containing RB69 RegA protein were used to eliminate the requirements for purification of WT or mutant RB69 and T4 RegA proteins (see below). We have previously demonstrated that RNA binding affinities of WT T4 RegA measured by similar gel shift assays are equivalent to those measured by equilibrium fluorescence quenching assays using purified RegA protein (19). As shown in Figure 4, addition of cell supernatants containing RB69 RegA protein to RB gene 44 RE RNA produces a shift in the mobility of the RNA (lanes 3 and 4). Cell supernatant containing the parental plasmid without regA (pAS1) did not produce a shift in RNA mobility (Fig. 4, lane 2). Also, a similar shift was observed when RB69 RegA was incubated with a 16mer RNA corresponding to the T4 gene 44 RE (Fig. 4, lanes 6 and 7). This is consistent with the report of Allen and Miller (25) that RB69 RegA-His6 binds a 23 nt RNA containing the RegA binding site of T4 gene 44. The specificity of RNA binding was tested by incubating RB69 RegA protein with T4 gene 44 RE RNA containing a base substitution at nucleotide –9 relative to the AUG. This base substitution reduces the affinity of T4 RegA protein for gene 44 RE RNA 100-fold (6) and eliminates the ability of T4 RegA to retard the electrophoretic mobility of the RNA. As shown in Figure 4 (lanes 9 and 10), RB69 RegA protein does not bind the G–9U variant of gene 44 RE RNA, confirming that the mobility shift is specific.
Figure 4.
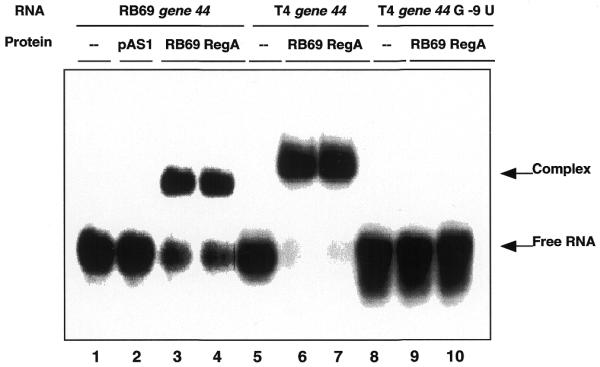
Gel mobility shift assay of RB69 RegA protein binding to RE RNAs. 32P-5′-end-labeled 18mer RB69 gene 44 RE RNA (5′-AUGAGGAAAAUUACAUGA-3′) (lanes 1–4), 16mer T4 gene 44 RE RNA (5′-AAUGAGGAAAUUAUG-3′) (lanes 5–7) and 16mer T4 gene 44 G–9U RE RNA (5′-AAUUAGGAAAUUAUG-3′) (lanes 8–10) were incubated with cell supernatants containing no RegA protein (lane 2) or 20 nM RB69 RegA protein (lanes 3, 4, 6, 7, 9 and 10). Samples were analyzed by electrophoresis on a native 6% polyacrylamide gel. Gels were dried and then analyzed on a PhosphorImager (see Materials and Methods).
The ability of RB69 RegA protein to bind to RB69 and T4 gene 44 RE RNAs and the observation that RB69 and T4 RegA proteins can both distinguish between WT and G–9U variant T4 gene 44 RE RNAs suggests that RB69 and T4 RegA proteins bind to homologous sequences on their target mRNAs. To confirm this, the binding site of RB69 RegA protein on RB69 gene 44 RE RNA was mapped by RNase protection assays. The 18mer RB69 gene 44 RE RNA (described above) was digested with E.coli RNase I and pancreatic RNase A, in the presence or absence of cell supernatants containing T4 or RB69 RegA protein. As shown in Figure 5A, when RB69 gene 44 RNA was digested with RNase I (which cleaves non-specifically), cleavages after nucleotides A–9 to A–4 were protected by both T4 (lanes 4 and 5) and RB69 (lanes 6 and 7) RegA proteins. When RNase digestion was performed in the presence of supernatants from cells lacking RB69 regA (AR120/pAS1), protection was not observed (Fig. 5A, lane 3). When the 18mer RB69 gene 44 RNA was digested with RNase A (which preferentially cleaves on the 3′ side of U and C residues in pyrimidine–adenine sequences), cleavage after nucleotides U–3 and U–4 were strongly protected, and cleavage after C–1 was moderately protected by RB69 RegA protein (data not shown). Taken together these assays map the RB69 RegA binding site to the nucleotide sequence GAAAAUU immediately upstream the AUG start codon of RB69 gene 44.
Figure 5.

RNase footprint assays of RB69 and T4 RegA protein binding to RB69 gene 44 and RB69 gene 45 TIR RNAs. (A) RB69 gene 44 RE RNA (5′-AUGAGGAAAAUUACAUGA-3′). Lane 1, RNA alone; lanes 2–7, RNA digested with RNase I. Lane 2, RNA digested in the absence of RegA protein; lane 3, RNA digested in the presence of pAS1 cell supernatant (which does not contain RegA protein); lanes 4 and 5, RNA plus cell supernatants containing 20 and 40 nM T4 RegA protein, respectively; lanes 6 and 7, RNA plus cell supernatants containing 20 and 40 nM RB69 RegA protein, respectively. (B) RB69 gene 45 TIR RNA (5′-UGAAAGGAAAUAAAAUGA-3′). Lanes 1–6, RNA digested with RNase I. Lane 1, RNA digested in absence of RegA protein; lane 2, RNA digested in the presence of pAS1 cell supernatant; lanes 3 and 4, RNA plus cell supernatants containing 20 and 40 nM T4 RegA protein, respectively; lanes 5 and 6, RNA plus cell supernatants containing 20 and 40 nM RB69 RegA protein, respectively; lane 7, RNA alone. Reactions in each panel contained 10 nM RNA and 0.01 U/ml of RNase I. RNA fragments were analyzed by electrophoresis on an 8 M urea/TBE gel and detected by PhosphorImager analysis. Nucleotides are numbered relative to the AUG start codon (in bold, above), so that U–4 is 4 nt upstream from the initiation A.
To map the RB69 RegA binding site on gene 45, an 18mer RNA corresponding to nucleotides –14 to +4 (relative to the AUG start codon, 5′-UGAAAGGAAAUAAAAUGA-3′) was digested with RNase I. As shown in Figure 5B, addition of RB69 RegA protein (lanes 5 and 6) to RNase I digestion reactions led to protection of cleavages in the region of G–8 to U–4, with weaker protection of nucleotide A–3. A similar protection pattern was observed in the presence of T4 RegA protein (Fig. 5B, lanes 3 and 4), suggesting that the two proteins bind to the same general sites on RB69 gene 45 RNA. Interestingly, the protected sequence in gene 45, GAAAUA, is similar to the purine-rich sequence protected in RB69 gene 44 (GAAAAUU) and both sequences overlap the Shine–Dalgarno sequence of the respective mRNAs.
RNA binding affinities of T4 and RB69 RegA proteins
To compare the relative RNA binding affinities and specificities of T4 and RB69 RegA proteins, quantitative RNA gel mobility shift assays (19) were performed using T4 and RB69 RE RNAs as ligands. For these assays, the concentration of recombinant RegA protein in lysed cell supernatants was determined by quantitation of Sypro Orange stained SDS gels containing known amounts of purified RegA protein as a reference (see Materials and Methods). Figure 6A shows a representative gel shift titration assay in which 2.5–20 nM RB69 (lanes 8–12) and T4 (lanes 3–7) RegA proteins were pre-incubated with 10 nM T4 gene 44 RE RNA (5′-AAUGAGGAAAUUAUGA-3′). Products of the binding reaction were separated on a native 6% polyacrylamide gel, and the fraction of 32P-labeled RNA bound by RegA protein was determined by PhosphorImager analysis (see Materials and Methods). These assays demonstrated that the affinity of RB69 and T4 RegA proteins for T4 gene 44 RE RNA are 34 × 107 M–1 and 4.9 × 107 M–1, respectively.
Figure 6.

(A) Gel shift binding titration of T4 gene 44 RE RNA with T4 and RB69 RegA proteins. Increasing concentrations of cell supernatants containing T4 or RB69 RegA proteins were incubated with 10 nM 32P-T4 gene 44 RE RNA. RB69 RegA–RNA complexes migrate faster than T4 RegA–RNA complexes, presumably due to RB69 RegA protein’s lower pI value. T4 gene 44 RE RNA: 5′-AAUGAGGAAAUUAUGA-3′. Samples were analyzed as in Figure 4. (B) Competition gel shift assay of T4 and RB69 RegA proteins for T4 gene 44 RE RNA. 32P-T4 gene 44 RE RNA (50 nM) was incubated with cell supernatants containing T4 RegA protein (lane 2), RB69 RegA protein (lane 6) or mixtures of T4 and RB69 proteins (lanes 3–5). Lane 1 in (A and B) contains RNA incubated without cell supernatant. Concentrations of RegA proteins in cell lysates were determined by quantitation of Sypro Orange stained gels using a Molecular Dynamics Storm™ imager.
Because RB69 RegA protein has a lower pI value than T4 RegA protein (7.6 versus 8.8), complexes of gene 44 RE RNA with RB69 RegA protein migrate faster than T4 RegA protein complexes in native polyacrylamide gels (Fig. 6A). This difference in migration rates enables competition assays to be performed with the two proteins. Figure 6B illustrates such a competition assay, in which varying ratios of T4 and RB69 RegA proteins were incubated with T4 gene 44 RE RNA. At a molar ratio of ∼7:1 T4 to RB69 RegA protein, approximately equal amounts of T4 RegA–RNA and RB69 RegA–RNA complexes were formed (Fig. 6B, lane 3). This is consistent with the 6.9-fold higher affinity of RB69 RegA protein relative to T4 RegA protein for T4 gene 44 RNA measured by quantitative gel shift titration assays (Table 1).
Table 1. Affinities of T4 and RB69 RegA proteins for RE RNAs.
| RNA name and sequence |
T4 RegA [Kappa ± SD (107 M–1)] |
RB69 RegA [Kappa ± SD (107 M–1)] |
| T4 gene 44 RE; 5′-AAUGAGGAAAUUAUG-3′ | 4.9 ± 1.1 | 34.0 ± 7.0 |
| RB69 gene 44 RE; 5′-AUGAGGAAAAUUACAUGA-3′ | 2.5 ± 0.4 | 9.5 ± 1.0 |
| T4 gene 45 RE; 5′-UUGAAGGAAAUUACAUGA-3′ | 3.2 ± 0.9 | 2.0 ± 0.7 |
| RB69 gene 45 RE; 5′-UGAAAGGAAAUAAAAUGA-3′ | 19.9 ± 4.0 | 1.8 ± 0.4 |
| T4 regA RE; 5′-CAUUGGAAUGGUAAAAUGAUUGAA-3′ | 0.83 ± 0.13 | 0.16 ± 0.05 |
| RB69 regA RE; 5′-CAUUGGAAUGGUAAAAAUGAUUGAA-3′ | 1.3 ± 0.3 | 0.6 ± 0.2 |
aKapp was determined by quantitative RNA mobility gel shift assays (see Materials and Methods). Binding was performed in 10 mM Tris–HCl pH 7.5, 50 mM NaCl and 1 mM EDTA. Values are the mean of two to four experiments.
To further explore the RNA binding specificity of the two RegA proteins, the affinities of T4 and RB69 RegA proteins for gene 44, gene 45 and regA RE RNAs were measured. For these assays, 18–25 nt ligands were designed that span the RegA protected regions of the TIRs of RB69 mRNAs (described above) and T4 mRNAs (14). The binding affinities of T4 and RB69 RegA proteins for RB69 gene 44 RE RNA (see above), T4 gene 45 RE RNA (5′-UUGAAGGAAAUUACAUGA-3′), RB69 gene 45 RE (see above), T4 regA RE RNA (5′-CAUUGGAAUGGUAAAAUGAUUGAA-3′) and RB69 regA RE RNA (5′-CAUUGGAAUGGUAAAAAUGAUUGAA-3′) were measured by RNA mobility gel shift assays. As illustrated in Figures 6 and 7, these assays revealed that both T4 and RB69 RegA proteins have a preference for their cognate gene 44 RE RNAs over their cognate gene 45 RE RNAs (for example, Fig. 7A and B, compare lanes 7–10). Interestingly, RB69 RegA protein has 5- and 7-fold higher affinities than T4 RegA protein for RB69 and T4 gene 44 RE RNAs, respectively (Table 1). However, T4 RegA protein exhibits a higher affinity than RB69 RegA for gene 45 RE RNAs (Table 1). Comparison of the affinities of T4 and RB69 RegA proteins for regA RE RNAs showed that both proteins have the lowest affinity for T4 regA RE RNA (Table 1). In terms of overall RNA binding specificity, T4 RegA exhibited the following hierarchy of affinities: RB69 gene 45 RE > T4 gene 44 RE > T4 gene 45 RE > RB69 gene 44 RE > RB69 regA RE > T4 regA RE (Table 2). For RB69 RegA the order of affinities was: T4 gene 44 RE > RB69 gene 44 RE > T4 gene 45 RE > RB69 gene 45 RE > RB69 regA RE > T4 regA RE.
Figure 7.

Comparative gel shift assays of T4 and RB69 RegA proteins binding to RB69 gene 44 (A), RB69 gene 45 (B), T4 gene 45 (C) and RB69 regA (D) RE RNAs. Lanes 1 and 6 in all panels contain RNA incubated without cell supernatants. Lanes 2–5 represent RNA incubated with cell supernatants containing T4 RegA at the indicated concentrations. Lanes 7–10 represent samples containing RB69 RegA at the indicated concentrations. Reactions in (A) and (B) contain 10 nM RB69 gene 44 and gene 45 RE RNAs, respectively; samples in (C) contain 20 nM T4 gene 45 RE RNA; samples in (D) contain 50 nM RB69 regA RE RNA. Binding reactions were analyzed by electrophoresis on native 6% polyacrylamide gels.
Table 2. Sequence variations in the core recognition region of RNA ligands and hierarchy of RNA affinities for T4 and RB69 regA proteins.
| T4 regA protein |
#1
#11 |
Kapp (107 M–1) |
| RB G45 | AAGGAAAUAAA | 19.9 |
| T4 G44 | GAGGAAAUUAU | 4.9 |
| T4 G45 | AAGGAAAUUAC | 3.2 |
| RB G44 | GAGGAAAAUUA | 2.5 |
| RB regA | AUGGUAAAAAU | 1.3 |
| T4 regA |
AUGGUAAAAUG |
0.8 |
| RB69 regA protein |
#1
#11 |
Kapp (107 M–1) |
| T4 G44 | GAGGAAAUUAU | 34.0 |
| RB G44 | GAGGAAAAUUA | 9.5 |
| T4 G45 | AAGGAAAUUAC | 2.0 |
| RB G45 | AAGGAAAUAAA | 1.8 |
| RB regA | AUGGUAAAAAU | 0.6 |
| T4 regA | AUGGUAAAAUG | 0.16 |
Nucleotides in bold indicate base substitutions relative to preferred, high affinity RNA.
Underlining indicates RNase protected nucleotides
Homology modeling of RB69 RegA protein structure
Using structure-guided mutagenesis, the RNA binding domain of T4 RegA protein recently was localized to a helix–loop groove motif in the protein (19). Three residues within helix A (Lys14, Thr18 and Arg21) and one residue on the loop following Helix C (Trp81) were found to play essential roles in RNA binding. Given the differences in the RNA binding affinities of the two proteins, we were interested to see where divergent amino acid residues are located in RB69 RegA protein. We expected that residues that are folded into the core of the protein would be fully conserved, to maintain the overall structure of the protein. Divergent residues would be expected to be on the surface of the protein, except in the helix–loop groove, which would also be mainly conserved. To determine the potential location of divergent residues, homology-based computer modeling of RB69 RegA protein was performed, using the program SYBYL (Tripos). Interestingly, as shown in Figure 8, the divergent residues in RB69 RegA protein cluster in two separate areas on the surface of the protein, rather than being dispersed across the surface. As illustrated in Figure 8, residues within the helix–loop groove are fully conserved in RB69 RegA protein.
Figure 8.

Space-filling models of RB69 and T4 RegA proteins, showing conserved and divergent amino acids. Divergent residues are colored, conserved residues are white. (A) front view, (B) back view, rotated 180°. Note that residues in the RNA binding groove are fully conserved in the two proteins. Arrows indicate the RNA binding pocket and the location of residue Trp81.
To confirm that the conserved helix–loop groove motif in RB69 RegA serves as the RNA binding domain, a Trp81Ala mutation was introduced in RB69 regA. This mutation in T4 RegA protein has been shown to eliminate RNA binding, without significantly disrupting RegA protein conformation (19). As shown in Figure 9, introduction of the Trp81Ala mutation (lanes 3, 6 and 9) eliminated the ability of RB69 RegA to bind any of the three RE RNAs tested.
Figure 9.
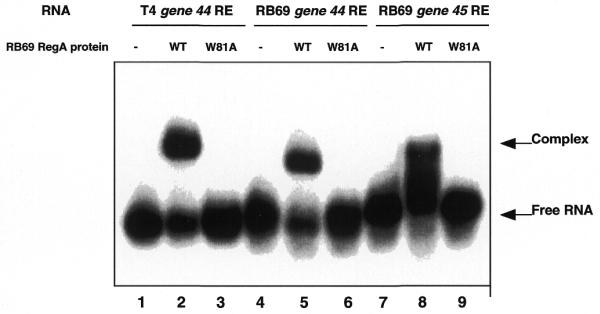
RNA gel shift assay of WT and mutant Trp81Ala RB69 RegA proteins. Lanes 1–3 and 4–6, 10 nM T4 and RB69 gene 44 RE RNAs, respectively; lanes 7–9, 50 nM RB69 gene 45 RE RNA. Lanes 2, 3, 5 and 6, 10 nM WT or W81A RB69 RegA proteins was used. Lanes 8 and 9, 50 nM RB69 RegA proteins was used. Samples were analyzed as in Figure 4.
DISCUSSION
RNA gel mobility shift assays have demonstrated that RB69 RegA protein binds to an 18mer RNA corresponding to the translation initiation region of RB69 gene 44 and to a 16mer RNA containing the translation initiation region of T4 gene 44. Interestingly, RB69 RegA protein, like T4 RegA protein, does not bind a T4 gene 44 RNA variant with a G–9U mutation. In addition, RNase protection assays have shown that T4 and RB69 RegA proteins protect the same binding sites on two RB69 RE RNAs. For RB69 gene 44 RE RNA, the protected site is GAAAAUU (Fig. 5A) which spans nucleotides –9 to –3, relative to the start codon, AUG. For RB69 gene 45 RE RNA, the protected site is GAAAUA (Fig. 5B), covering the region –8 to –3 relative to the start codon. Interestingly, the protected sequences for both RB69 gene 44 and gene 45 mRNAs overlap the Shine–Dalgarno sequence of the mRNAs. Previous studies (6) showed that in T4 gene 44 mRNA the T4 RegA binding site spans nucleotides –10 to +2, which includes the Shine–Dalgarno region and an AU-rich element.
These studies revealed that in spite of their similar RNA binding domains, T4 and RB69 RegA proteins exhibit measurable differences in their RNA binding specificities. Although the two RegA proteins exhibit the same hierarchy of affinities for their cognate RNAs (i.e. gene 44 > gene 45 > regA), the two repressors exhibit different overall RNA binding specificities (Figs 6 and 7; Table 2). It was previously proposed that RB69 RegA is a generally stronger repressor of protein synthesis than T4 RegA protein (12). However, in vivo studies of relative mRNA repression sensitivities are complicated by the fact that RegA repression involves competition between 30S ribosomal subunits and RegA protein for the translation initiation regions of mRNAs. Thus, the apparent affinity of the two RegA proteins is influenced by the relative concentrations of ribosomes, RegA protein and target mRNAs in vivo. To eliminate the effects of possible differences in substrate–ligand concentrations on RegA protein interactions with different RNAs, we have examined the affinity of RegA protein for purified target RNAs, using known concentrations of protein and RNA. These simplified assays have revealed that with respect to the six genes examined here, RB69 RegA protein exhibits a higher affinity than T4 RegA protein for two RNAs (Table 1). However, for the four other RNAs, T4 RegA protein exhibited the higher affinity (Table 1). The overall highest affinity was exhibited by RB69 RegA protein for T4 gene 44 RE RNA. Interestingly, RB69 RegA protein also exhibited a larger range between highest (T4 gene 44 RE) and lowest (T4 regA RE) affinities for the RNAs examined here (Table 1). This suggests that in vivo the range of RB69 RegA protein concentrations required to repress target mRNAs may be broader.
To better understand the nucleotide sequence preferences of the two proteins, we have examined the relationship between nucleotide variations in the target RNAs and binding affinities of both proteins. Table 2 shows an alignment of the sequences of the core recognition region (i.e. the RNase protected or analogous region) plus 1–3 nt on either side, including nucleotide –9 in T4 gene 44 (position 1 in Table 2), where base substitution has a large effect on RegA affinity (Fig. 4). The aligned REs were narrowed to 11 nt, corresponding to the site size determined for T4 RegA protein binding to T4 gene 44 RNA, to focus on the potentially most important nucleotides for protein recognition.
Examination of the hierarchy of affinities of T4 RegA protein for the six RNAs, shown in Table 2, reveals that the RNAs with the second and third highest affinities have two base substitutions (bold in Table 2) relative to the highest affinity RNA. These two base substitutions produce 4–6-fold decreases in the affinity constant. Interestingly, at position 11 in the aligned sequences, an A→U substitution (present in T4 G44) appears to be preferred over an A→C substitution (in T4 G45), which is consistent with the low C content of all RegA-regulated mRNAs. Comparison of the affinities of T4 RegA protein for T4 G44 and T4 G45 RNAs suggests that an A→G substitution at position 1 does not reduce affinity significantly, although previous studies have shown that a G→U substitution at this position in T4 G44 has a large affect (6). A third nucleotide substitution (present in RB G44) is associated with a further reduction in affinity, while the fifth and sixth lowest affinity RNAs (RB regA and T4 regA) have four and five base substitutions, respectively. Thus, decreases in the affinity of T4 RegA protein for the six RNAs correlate with progressive nucleotide substitutions within the proposed 11 nt REs. Taken together these studies suggest that the preferred sequence for T4 RegA is (A/G)AGGAAAUAAA and that individual base substitutions at six positions within this sequence produce cumulative decreases in affinity (Table 2).
A similar comparison of the effects of base substitutions on RNA binding affinities for RB69 RegA protein suggests that some bases within the RE affect affinity more than others. For example, the second highest affinity RNA (RB G44) has three base substitutions relative to the highest affinity target (T4 G44) while the third highest affinity RNA has only two base substitutions (Table 2). This suggests that RB69 RegA affinity is more sensitive to the base at position 1 than to bases at positions 8 and 10. However, additional base substitutions relative to the T4 G45 target (with third highest affinity) produces progressive decreases in RB69 RegA protein affinity. These studies indicate that the preferred sequence for RB69 RegA protein is GAGGAAAUUAU. It should be noted that all six RNAs contain the sequence NNGGNAANNNN, so the relative importance of the bases at positions 3, 4, 6 and 7 was not evaluated by these studies. However, the conservation of these bases within the six RegA-regulated mRNAs suggests they may be important for RegA protein recognition.
It has been known for some time that RegA-regulated mRNAs vary in their sensitivity to RegA repression in vivo (8) and in vitro (1,5). However, until now, it has not been clear how specific nucleotide variations between closely related RNA sequences affect RegA protein affinity. The studies reported here help to clarify the mechanism of RNA discrimination by quantifying the effects of base substitutions on RegA protein affinity. This is important because it is the differential affinity for target RNAs that allows the RegA regulatory system to exhibit a gradient of effects on gene expression, as a function of increasing RegA protein concentration during the early and middle stages of phage infection. Presumably, these differential RNA affinities enable RegA protein to turn off expression of target genes in a stepwise fashion and thus optimize the temporal expression of inter-related enzymatic activities during the viral life cycle (4).
From the point of view of understanding protein–RNA recognition, perhaps the most intriguing observation from these studies is that, although the amino acids in the proposed RNA binding domain of T4 RegA (19) are fully conserved in RB69 RegA (Fig. 8), the two repressors exhibit notable differences in RNA binding specificities (Table 2). This observation suggests that some of the divergent residues in the two RegA proteins (Fig. 8) may contribute to differences in RNA binding specificities. However, recent studies of the effects of amino acid substitutions of divergent residues do not support a role for divergent residues in RNA binding (26). Alternatively, studies of the RNA binding specificity of the MS2 coat protein by Johansson et al. (27) suggests that there may be subtle differences in the geometry of amino acid side chains in the binding pocket of the two RegA proteins (that are not apparent in the homology modeling), which enable closer fitting of certain RNAs in the groove of each protein. Ultimately, the solution of the structures of specific RegA–RNA complexes is needed in order to determine which of these possibilities is correct and to fully understand the mechanism of multiple RNA recognition and discrimination by RegA protein.
Acknowledgments
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the excellent technical assistance of Fredilyn Lipata and we thank LuAnne Harley for her careful assistance with the preparation of this manuscript. We also thank the MUSC Biotechnology Resource Laboratory for expert DNA sequence analysis. This work was generously supported by a National Science Foundation Award (MCB9727811) to E.K.S.
References
- 1.Winter R.B., Morrissey,L., Gauss,P., Gold,L., Hsu,T. and Karam,J. (1987) Bacteriophage T4 RegA protein binds to mRNAs and prevents translational initiation. Proc. Natl Acad. Sci. USA, 84, 7822–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karam J., Gold,L., Singer,B. and Dawson,M. (1981) Translational regulation: identification of the site on bacteriophage T4 rIIB mRNA recognized by the RegA gene function. Proc. Natl Acad. Sci. USA, 78, 4669–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webster K.W., Adari,H.Y. and Spicer,E.K. (1989) Bacteriophage T4 RegA protein binds to the Shine–Dalgarno sequence of gene 44 mRNA. Nucleic Acids Res., 17, 10047–10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller E.S., Karam,J.D. and Spicer,E.K. (1994) Translational initiation control: mRNA structure and protein repressors. In Karam,J.D. (ed.), Molecular Biology of Bacteriophage T4. American Society of Microbiology, Washington, DC, pp. 193–205.
- 5.Adari H. and Spicer,E.K. (1986) Translational repression in vitro by the bacteriophage T4 RegA protein. Proteins Struct. Funct. Genet., 1, 116–124. [DOI] [PubMed] [Google Scholar]
- 6.Webster K.R. and Spicer,E.K. (1990) Characterization of bacteriophage T4 RegA protein–nucleic acid interactions. J. Biol. Chem., 265, 19007–19014. [PubMed] [Google Scholar]
- 7.Szewczak A.A., Webster,K.R., Spicer,E.K. and Moore,P.B. (1991) An NMR characterization of the RegA protein-binding site of bacteriophage T4 gene 44 mRNA. J. Biol. Chem., 266, 17832–17837. [PubMed] [Google Scholar]
- 8.Miller E.S., Karam,J., Dawson,M., Trojanowska,M., Gauss,P. and Gold,L. (1987) Translational repression: biological activity of plasmid-encoded bacteriophage T4 RegA protein. J. Mol. Biol., 194, 397–410. [DOI] [PubMed] [Google Scholar]
- 9.Brown D., Brown,J., Kang,C.-H., Gold,L. and Allen,P. (1997) Single-stranded RNA recognition by the bacteriophage T4 translational repressor, RegA. J. Biol. Chem., 272, 14969–14974. [DOI] [PubMed] [Google Scholar]
- 10.Russell R.L. and Huskey,R.J. (1974) Partial exclusion between T-even bacteriophages: an incipient genetic isolation mechanism. Genetics, 78, 989–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller E.S. and Jozwik,C.E. (1990) Sequence analysis of conserved RegA and variable orf43.1 genes in T4 like bacteriophages. J. Bacteriol., 172, 5180–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jozwik C.E. and Miller,E.S. (1992) Regions of bacteriophage T4 and RB69 RegA translational repressor proteins that determine RNA binding specificity. Proc. Natl Acad. Sci. USA, 89, 5053–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C.C., Yeh,L.S. and Karam,J.D. (1995) Modular organization of T4 DNA polymerase. J. Biol. Chem., 270, 26558–26564. [DOI] [PubMed] [Google Scholar]
- 14.Unnithan S., Green,L., Morrissey,L., Binkley,J., Singer,B., Karam,J. and Gold,L. (1990) Binding of the bacteriophage T4 RegA protein to mRNA targets: an initiator AUG is required. Nucleic Acids Res., 18, 7083–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang C.-H., Chan,R., Berger,I., Lockshin,C., Green,L., Gold,L. and Rich,A. (1995) Crystal structure of the T4 RegA translational regulator protein at 1.9 Å resolution. Science, 268, 1170–1173. [DOI] [PubMed] [Google Scholar]
- 16.Adari H.Y., Rose,K., Williams,K.W., Konigsberg,W.H., Lin,T.-C. and Spicer,E.K. (1985) Cloning, nucleotide sequence, and overexpression of the bacteriophage T4 RegA gene. Proc. Natl Acad. Sci. USA, 82, 1901–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips C.A., Gordon,J. and Spicer,E.K. (1996) Bacteriophage T4 RegA protein binds RNA as a monomer, overcoming dimer interactions. Nucleic Acids Res., 24, 4319–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson B.H. and Hecht,M.H. (1994) Recombinant proteins can be isolated from E. coli cells by repeated cycles of freezing and thawing. Biotechnol., 12, 114–117. [DOI] [PubMed] [Google Scholar]
- 19.Gordon J., Sengupta,T.K., Phillips,C.A., O’Malley,S.M., Williams,K.R. and Spicer,E.K. (1999) Identification of the RNA binding domain of T4 RegA protein by structure-based mutagenesis. J. Biol. Chem., 274, 32265–32273. [DOI] [PubMed] [Google Scholar]
- 20.Webster K.R., Shamoo,Y., Konigsberg,W.H. and Spicer,E.K.(1991) A rapid method for purification of synthetic oligoribonucleotides. Biotechniques, 11, 658–661. [PubMed] [Google Scholar]
- 21.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 22.Rebar E.J. and Pabo,C.O. (1994) Zinc finger phage: affinity selection of fingers with new DNA-binding specificities. Science, 263, 671–673. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg M., Ho,Y.S. and Shatzman,A. (1983) The use of pKC-30 and its derivatives for controlled expression of genes. Methods Enzymol., 101, 123–138. [DOI] [PubMed] [Google Scholar]
- 24.Yeh L.S., Hsu,T. and Karam,J.D. (1998) Divergence of a DNA replication gene cluster in the T4-related bacteriophage. J. Bacteriol., 180, 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen S.V. and Miller,E.S. (1999). RNA-binding properties of in vitro expressed histidine tagged RB69 RegA translational repressor protein. Anal. Biochem., 269, 32–37. [DOI] [PubMed] [Google Scholar]
- 26.Gordon J. (2000) PhD thesis. Medical University of South Carolina, Charleston, SC.
- 27.Johansson H.E., Dertinger,D., LeCuyer,K.A., Behlen,L.S., Greef,C.H. and Uhlenbeck,O.C. (1999) A thermodynamic analysis of the sequence-specific binding of RNA by bacteriophage MS2 coat protein. Proc. Natl Acad. Sci. USA, 95, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]